

Abstract
This perspective provides an overview of the progress in two of the original programs in my research group focused on the biosynthesis of the antibiotics nisin, lacticin 481, fosfomycin, and bialaphos. The path from start-up funds to tenure and beyond offers insights into the opportunities realized and missed along the road.
Keywords: lantibiotics, biosynthesis, fosfomycin, bialaphos, phosphinothricin
A perspective on the research in my laboratory provided an opportunity not only to reflect on the advances and progress made in our program but also to recognize missed chances. In this perspective, I describe the often bumpy and certainly non-linear path from start-up to tenure and beyond in the nine years since our research group was established at the University of Illinois. Hopefully, this journey will provide insights into the choices made at critical junctures along the road as well as into the serendipitous and unpredictable nature of research at the interface of organic chemistry and biology.
With a Ph.D. in organic chemistry and a subsequent postdoc that introduced me to enzymology, the central theme of my research proposals written in 1996/97 was to use the synthetic tools of organic chemistry to explore novel biochemical transformations in antibiotic biosynthesis and vitamin B12 dependent reactions. At the time, the work of Khosla on polyketide biosynthesis1 was instrumental in shaping my interest in natural product biosynthesis, which was further cemented by a paper from the Walsh group2 on the post-translational maturation of microcin B17. With the advent of increasingly powerful molecular biology techniques and the first sequencing of whole bacterial genomes, it seemed that a wealth of novel chemistry and enzymology imbedded in natural products biosynthesis would soon be accessible to organic chemists. Since the fields of polyketide and non-ribosomal peptide biosynthesis already appeared well populated by scientists applying chemical approaches, our laboratory focused on two classes of antibiotics that were relatively unexplored but yet of significant commercial and therapeutic importance, the lantibiotics and phosphonate antibiotics (Figure 1).
Figure 1.
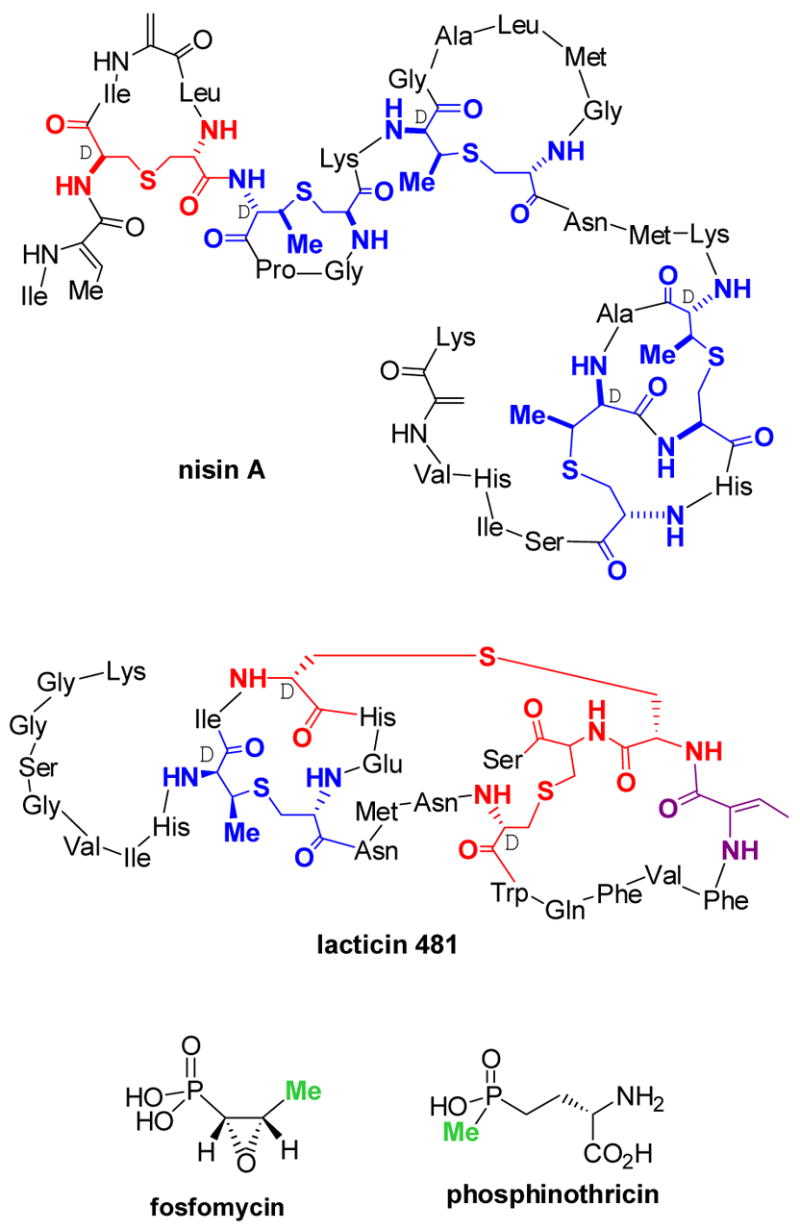
Structures of the lantibiotics nisin A and lacticin 481, the phosphonate antibiotic fosfomycin, and the phosphinate phosphinothricin. Lanthionine (Lan) residues in the lantibiotics are shown in red and methyllanthionine (MeLan) residues in blue. The two methyl groups in fosfomycin and phosphinothricin that are considered to be derived from methylcobalamin are highlighted in green.
The choice of these systems for investigation was influenced by a number of factors. As described in more detail below, at the time good evidence existed that lantibiotic synthetases would have flexible substrate specificity, which we intended to explore by altering the natural substrates using organic synthesis. Our interest in phosphonate antibiotics was provoked by two unusual transformations that had been proposed in the literature for the biosynthesis of fosfomycin and phosphinothricin (Figure 1). The methyl groups highlighted in green had been reported to be derived from methylcobalamin.3,4a To date the natural vitamin B12 derivatives adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl)5 are still the only confirmed compounds in nature containing a metal-carbon bond (Figure 2), and as such were intriguing to me based on my graduate studies in organometallic chemistry.6,7
Figure 2.
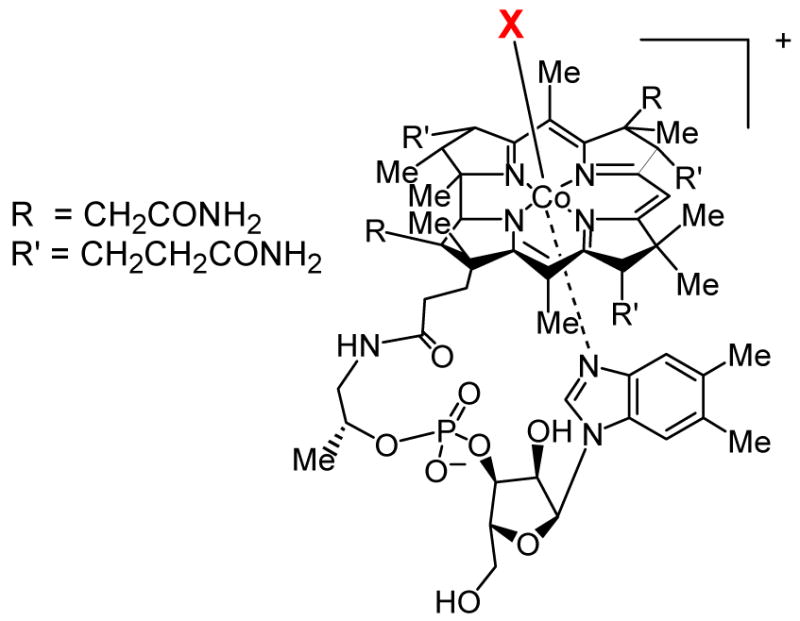
Structures of vitamin B12 (X = cyanide), and the natural cofactors methylcobalamin (X = CH3) and adenosylcobalamin (X = 5′-deoxyadenosine).
After a difficult start, we made much progress in the past two years in the project on lantibiotics biosynthesis, whereas research on the phosphonate antibiotics is still in a more preliminary stage. This perspective will present our current understanding of the biosynthetic pathways of these two classes of compounds.
Lantibiotic biosynthesis
In the past 15 years, the biosynthesis of peptide natural products, whether gene encoded or of non-ribosomal origin, has received much attention. Of special interest to us were the lantibiotics, which are extensively post-translationally modified. For some members, as many as 47% of the amino acids in the peptide undergo some form of modification.8 The unifying structural motifs, which also have given lantibiotics their name,9 are the lanthionine and methyllanthionine amino acids highlighted in red and blue, respectively, in Figure 1 for two representative members of the class I and class II lantibiotics, nisin and lacticin 481. For class I, these structures are introduced by dehydration of Ser and Thr residues in a ribosomally produced prepeptide to the corresponding dehydroalanine (Dha, green Figure 3) and Z-dehydrobutyrine (Dhb, magenta) residues by a dehydratase LanB (Lan is used as a generic label for lantibiotic biosynthetic proteins; for instance, NisB is the LanB for nisin). This step is then followed by a LanC-catalyzed intramolecular and stereoselective Michael-type addition of the thiols of cysteines to Dha or Dhb to produce lanthionine (Lan) or methyllanthionine (MeLan) thioether crosslinks, respectively. The stereochemistry at the α-carbon of the former Ser/Thr residue is inverted to the D-configuration in this process. In a final step of maturation a protease (LanP) removes a leader peptide that has not been modified (Figure 3).
Figure 3.
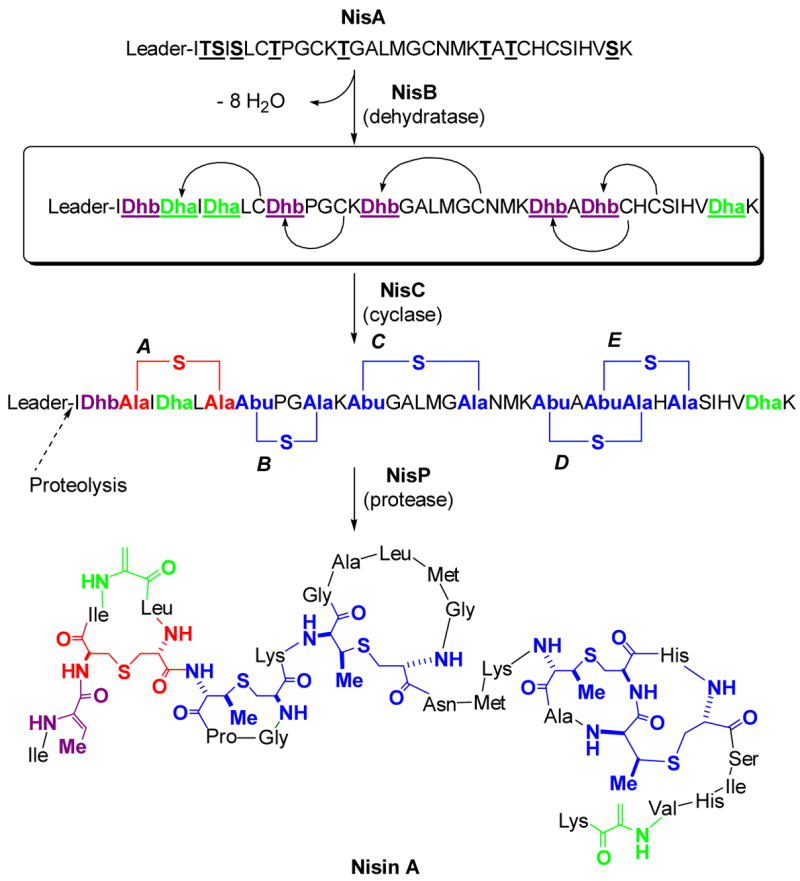
The post-translational maturation process of nisin as an example of a class I lantibiotic. The prepeptide NisA is ribosomally synthesized, followed by NisB catalyzed dehydration of underlined Ser and Thr residues of NisA. NisC catalyzes the conjugate addition of Cys residues in a regio- and stereoselective manner to five of the Dha (green) and Dhb (magenta) residues to generate five cyclic thioethers: one lanthionine (red) and four methyllanthionines (blue). After dehydration/cyclization is complete, the unmodified leader peptide is proteolytically removed by the protease NisP. The sequence of the leader peptide is MSTKDFNLDLVSVSKKDSGASPR. Abu, S-2-aminobutyric acid.
The best known lantibiotic family member is nisin, first recognized as an antimicrobial substance in 192810 but not assigned a structure until 1971.11 Nisin has been used extensively in the food industry as a safe and highly effective preservative with minimal inhibitory concentrations in the low nanomolar range against many Gram-positive bacteria including food-borne pathogens like Clostridium botulinum and Listeria monocytogenes8,12 and multi-drug resistant bacteria.13 Our interest in nisin and other lantibiotics stemmed from several in vivo site-directed mutagenesis experiments that suggested that the biosynthetic machinery was very forgiving with respect to the structure of its substrate.14 Whereas site-directed mutagenesis at the time could only replace an amino acid with one of the other 20 proteinogenic amino acids, we envisioned that a combination of organic synthesis and solid phase peptide synthesis with the biosynthetic enzymes could greatly increase the accessible structural and functional space to explore nisin analogs. This premise required us to obtain active biosynthetic enzymes, but that process proved extremely challenging. Although we ultimately succeeded,15,16 our first success came after five years of unsuccessful and mostly unpublished attempts. The major culprits in our eyes were the dehydratase enzymes. We expressed and purified these proteins from E. coli or performed activity assays with crude cell preparations of lantibiotic producing bacterial strains without even once observing dehydration of the peptide substrates.17 Without dehydration we had no substrate for the cyclase enzymes NisC (for nisin production) and SpaC (for production of subtilin, a close relative of nisin). At this time of our struggles, a high profile report showed that nisin was not only a pore former, as was generally accepted, but also interacted in a highly specific manner with the cell wall biosynthetic precursor lipid II,18 which interestingly is also the target of vancomycin and ramoplanin.19,20 By binding to lipid II, nisin inhibits cell wall biosynthesis, in itself a lethal activity, but it packs an even more powerful punch by assembling into a higher order aggregate consisting of 4 nisin molecules and 8 lipid II molecules that form extremely stable pores in the cell membrane of target bacteria.19,21 Although not directly relevant to our efforts on nisin biosynthesis, these findings strengthened our resolve to try and prepare nisin in vitro as the prospect of analogs was now even more appealing.
Since we had been unable to obtain the substrates for the cyclase enzymes through the enzymatic action of the dehydratases, we turned to organic synthesis. Our targets were the dehydrated prepeptide intermediates of nisin (box, Figure 3) and subtilin biosynthesis. Although a myriad of routes had been reported for preparation of Dha and Dhb residues, none of them were compatible with either established solid phase peptide synthesis (SPPS) protocols or with the amino acids present in our targets.22 We therefore developed new synthetic methodology that was suitable for our needs. The monomer 1, appropriately protected for Fmoc-based SPPS, was incorporated efficiently into peptides and after treatment with oxidizing agents (H2O2 or NaIO4) afforded Dha-containing peptides (eg Scheme 1). Importantly, this methodology proved compatible with unprotected side chains of the common amino acids, and hence a range of different dehydropeptides could be prepared.22 An interesting modification of this strategy was recently used by the Szostak laboratory for ribosomal incorporation of 4-selenalysine (Scheme 1), which upon treatment with oxidizing agents also resulted in the formation of Dha.23
Scheme 1.
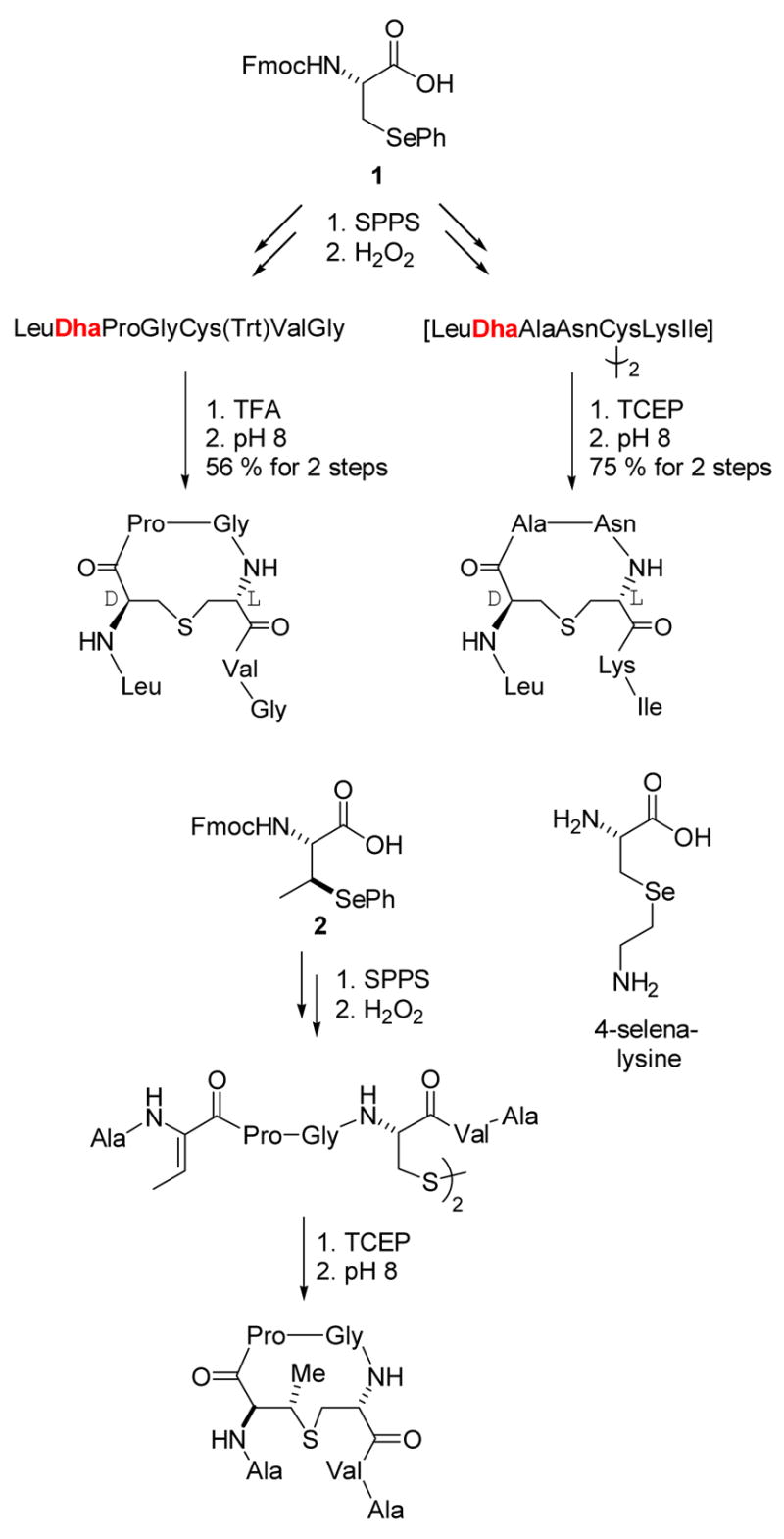
Preparation of dehydropeptides by oxidative elimination.
The oxidative elimination of selenoxides is a stereospecific process, and therefore use of monomer 2 provided convenient and stereospecific entry to Z-Dhb-containing peptides.24,25 With these tools in hand we chemically prepared dehydropeptides as substrates for heterologously expressed and purified nisin and subtilin cyclases (NisC and SpaC). However, our attempts to distinguish enzymatic cyclization from spontaneous non-enzymatic cyclization of these peptides remained inconclusive.26 Although disappointing at the time of these experiments, several years later we would show that the idea of using dehydrated peptides as substrates for NisC expressed in E. coli was sound, but that the full length dehydropeptide is needed for a sensitive assay (see below).16
Although we did not succeed in applying our methodology of dehydropeptide synthesis to investigation of the biosynthetic enzymes, the difficulties we encountered in our efforts to distinguish enzymatic from non-enzymatic cyclization led us to study the latter process in more detail. Thus, Dha and Dhb containing peptides were prepared that also incorporated disulfide-protected Cys residues (Scheme 1). Upon release of the free Cys residues by using the reducing agent tris(carboxyethyl)phosphine (TCEP), cyclization indeed occurred efficiently, and interestingly resulted in the stereoselective production of the naturally occurring diastereomers as the major and often only products (Scheme 1).22,24 This selectivity obtained in aqueous solution was quite unexpected as the protonation of the enolate intermediate was anticipated to be very fast and thus rather non-selective. Mechanistic studies on these non-enzymatic cyclizations subsequently showed the selectivity to be due to kinetic and not thermodynamic control.25 At the time, there was no conclusive evidence that the LanC proteins indeed were cyclases, which was assumed because disruption of LanC genes resulted in accumulation of dehydrated peptides.8,27,28 We therefore were curious as to whether the apparent inherent propensity of these short dehydropeptides to provide the same stereoselectivity as observed in the lantibiotics could be extended to the regioselectivity and chemoselectivity of cyclization. As a first test of this question, the dehydropeptide 3 was prepared using the methodology of Scheme 1. This peptide corresponds to the precursor to the A and B rings of nisin, the segment that interacts with lipid II.29 When the dehydropeptide 3 was allowed to cyclize non-enzymatically, the inherently more reactive Dha residues were preferentially attacked by the Cys residues in the peptide; whereas the A ring was still formed, the B-ring (dashed arrow) was not (Scheme 2).25 These results provided convincing additional evidence that a cyclase enzyme is needed to overcome the chemoselectivity of the non-enzymatic reaction and assure the correct connectivity of the lanthionine and methyllanthionine rings.
Scheme 2.
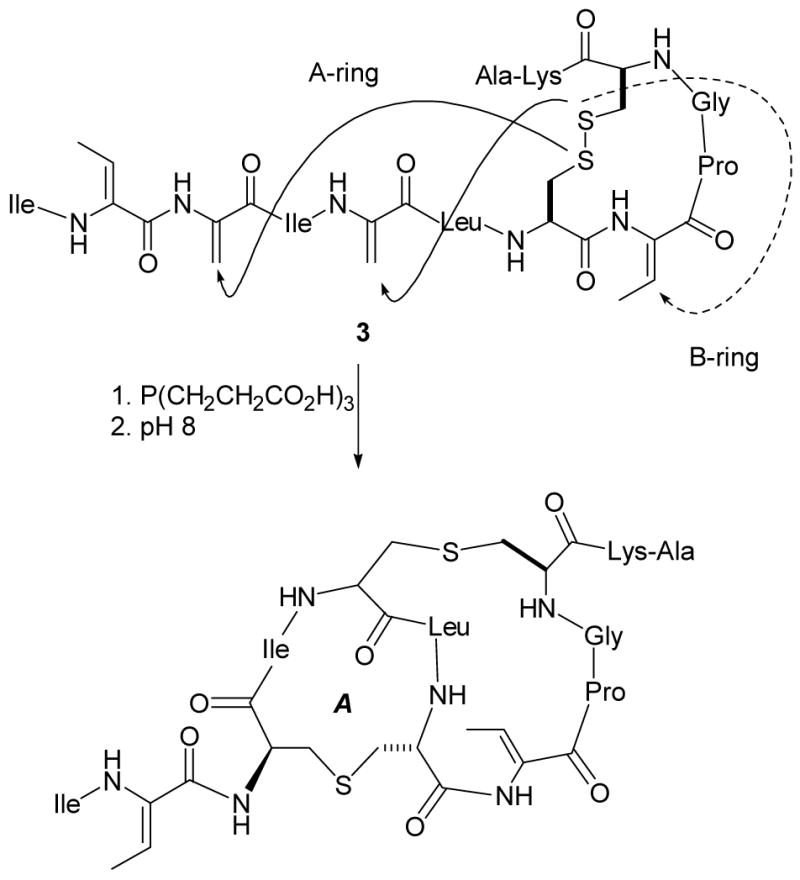
Attempted biomimetic synthesis of the A and B-rings of nisin.
A second project based on this methodology was the preparation of dehydropeptides for chemoselective ligations. The past decade has seen a resurgence in methods to achieve conjugations of two biomolecules or of a biomolecule with a biophysical probe or small molecule.30 By introducing a dehydroalanine into a peptide, a site-specific conjugation point is created for selective intermolecular reaction with nucleophiles. Indeed we showed that prenyl thiols and complex carbohydrates containing a thiol functionality could be efficiently ligated to dehydropeptides by this approach (eg Scheme 3).31,32 A drawback to the methodology was that the diastereoselectivities of the protonation at the α-carbon were modest at best, in contrast to the intramolecular cyclizations in Scheme 1. To overcome this problem, a collaborative project with David Gin’s laboratory was initiated based on development of an efficient solid phase synthesis of aziridine containing peptides in which the stereocenter at the α-carbon would be set. Subsequent ring opening of these aziridines with external nucleophiles allowed efficient ligation with a series of nucleophiles producing the desired bioconjugates with defined stereochemistry.33,34 Of particular interest are the ligations of thiol containing oligosaccharides in which both the stereochemistry at the anomeric center and at the α-carbon of the former aziridine are controlled (Scheme 4).
Scheme 3.
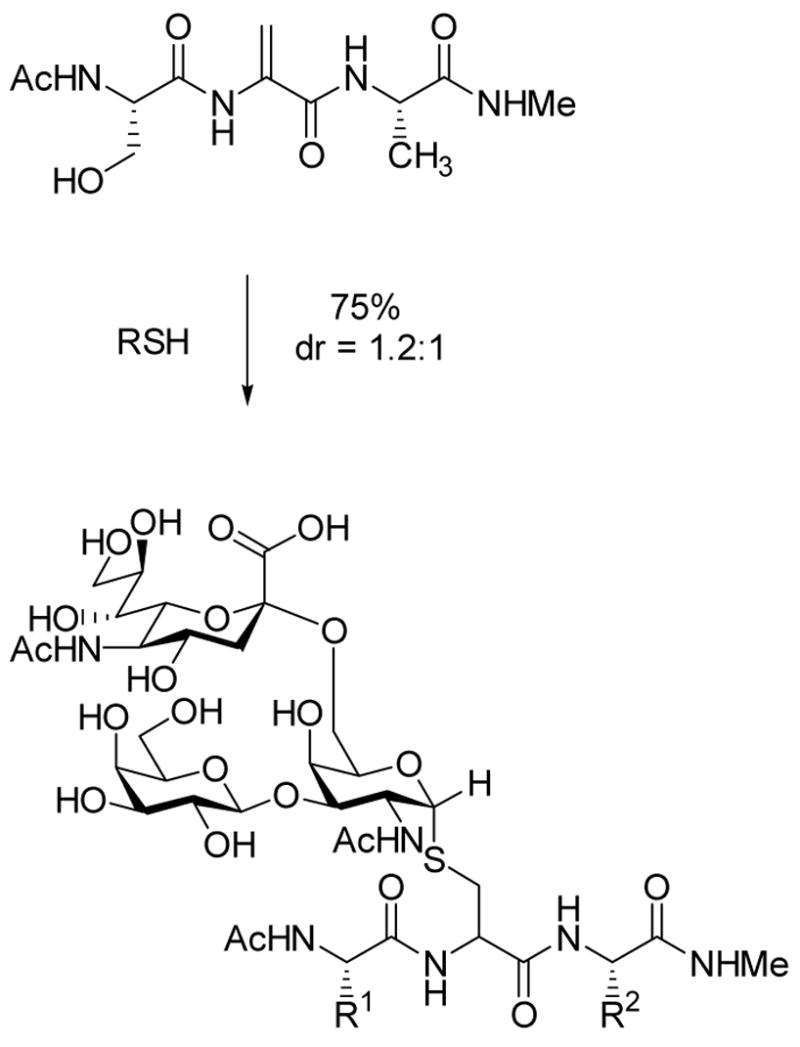
Conjugate addition of external thiol nucleophiles to Dha.32
Scheme 4.
By the end of 2002, our lantibiotic biosynthesis project appeared destined for disappointing termination. Although our biomimetic and chemoselective ligation chemistry was fruitful, our initial goals of preparing lantibiotics in vitro using the biosynthetic machinery appeared unattainable. Focus within the group, which from 1997 to 2000 had been almost entirely on lantibiotics, was now diverted to two new projects on the mechanisms of phosphite dehydrogenase and prostaglandin synthase (also called cyclooxygenase or COX).35 Phosphite dehydrogenase, discovered by Bill Metcalf in the Department of Microbiology at UIUC,36 catalyzes the oxidation of phosphite to phosphate concomitant with the reduction of NAD+ to NADH (Scheme 5).
Scheme 5.
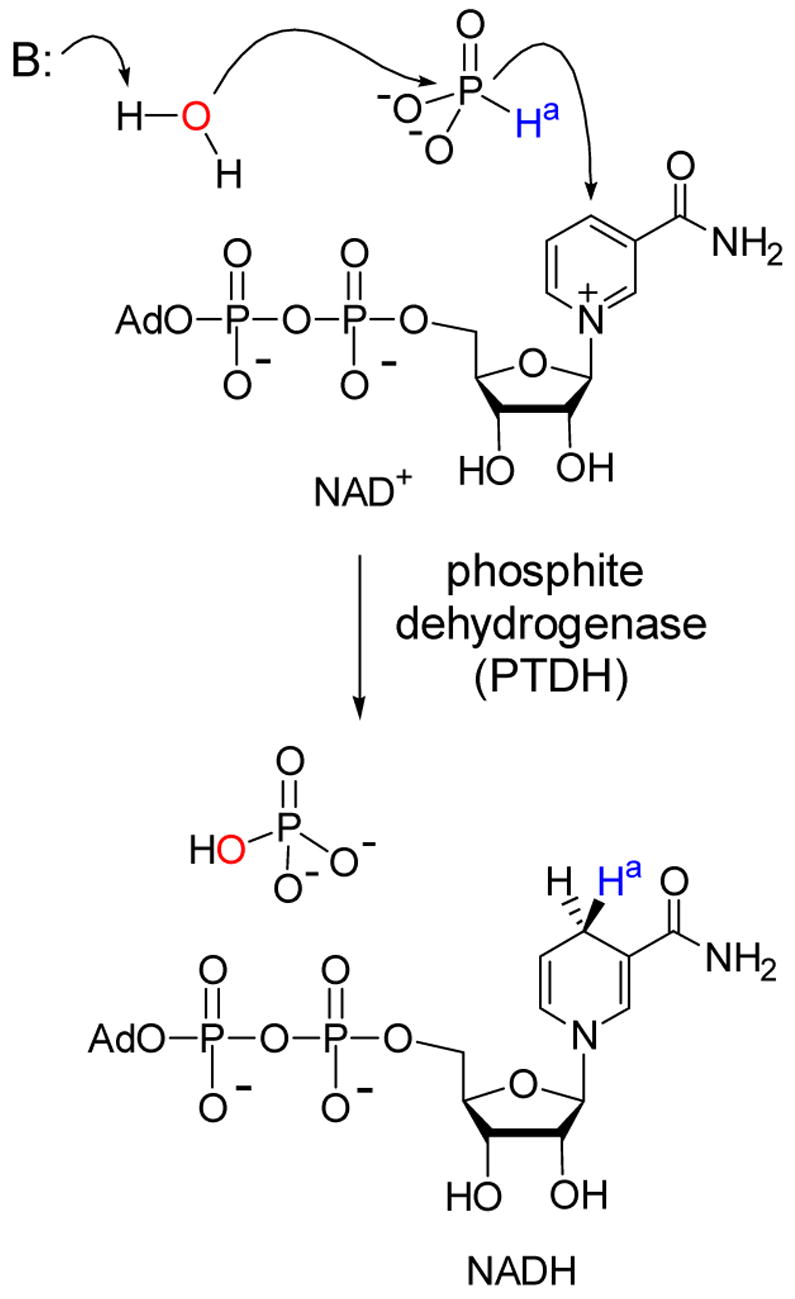
NAD+-dependent oxidation of phosphite by PTDH.
We became initially interested in the enzyme because it promotes a highly unusual hydride transfer reaction.37 Although catalyzed by a dehydrogenase, the reaction is formally a phosphoryl transfer from hydride to hydroxide. From a mechanistic perspective associative, dissociative (SN1) and concerted (SN2) mechanisms all appear to be improbable for this transformation. However, analysis of the solution thermodynamics of the transformation shows it to be 15 kcal/mol exergonic.37 At present, we do not know specifics of the mechanism of this hydride transfer and discussion of our mechanistic studies are beyond the scope of this perspective,38 but one practical application will be highlighted. It occurred to us that if this reaction was indeed exergonic, then phosphite dehydrogenase should be a good NADH regeneration enzyme that might be useful in biocatalysis. We have been able to turn this idea into practice (Scheme 6),39 and a subsequent joint investigation with Huimin Zhao’s laboratory employing rational design resulted in an engineered variant that uses both NAD+ and NADP+ effectively.40 We then utilized directed evolution techniques to improve on its expression and activity,41 and this latter mutant was further engineered in the Zhao laboratory to obtain a thermostable variant42 that rivals or outperforms other enzymatic NAD(P)H regeneration systems, at least on a laboratory scale. Hence, what attracted us initially to this enzyme was purely mechanistic curiosity, but detailed analysis of the reaction ultimately resulted in an engineered analog with great practical potential.
Scheme 6.
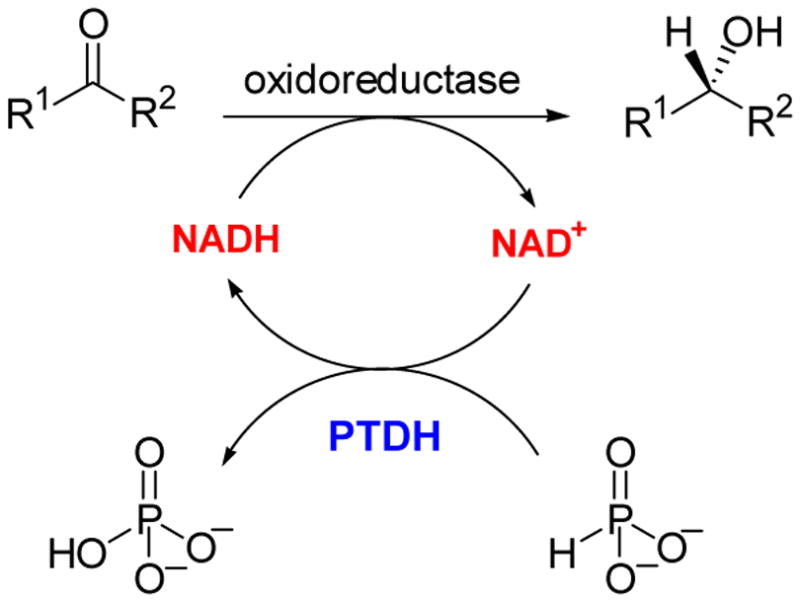
Use of PTDH for cofactor regeneration.39
In the Spring of 2003, one student was still working on the lantibiotic biosynthesis project with other students having switched to the phosphite dehydrogenase and ligation projects discussed in the previous section. In a last attempt to achieve in vitro lantibiotic biosynthesis, we had turned to the LanM proteins that combine the dehydratase and cyclase activities in one bifunctional enzyme in the biosynthesis of class II lantibiotics.8,43 One such protein, LctM involved in the biosynthesis of lacticin 481, was heterologously overexpressed in E. coli and purified. The enzyme and its substrate peptide LctA were incubated with DTT, Zn2+, ATP, and Mg2+ and a MALDI-MS was recorded. For almost 5 years we had always seen unmodified peptide no matter the ingredients of the assays. But this time, the assay product corresponded to a fourfold dehydrated peptide! (Figure 4).15 The subsequent determination of the location of those dehydrations and whether the cyclizations had also occurred was our long sought, but challenging task. Proteolytic removal of the leader peptide showed that the four dehydrations had taken place in the propeptide region of the substrate peptide and not in the leader peptide. Moreover, individual mutagenesis of the Ser/Thr residues as well as Fourier Transform MS/MS analysis in the laboratory of my colleague Neil Kelleher demonstrated that the Ser/Thr that were dehydrated corresponded with the anticipated sites of post-translational modification for lacticin 481 biosynthesis. Furthermore, the product after removal of the leader peptide showed the expected antimicrobial activity.15 Thus, after more than 5 years of unsuccessful attempts, in vitro biosynthesis of a lantibiotic was finally achieved.44
Figure 4.
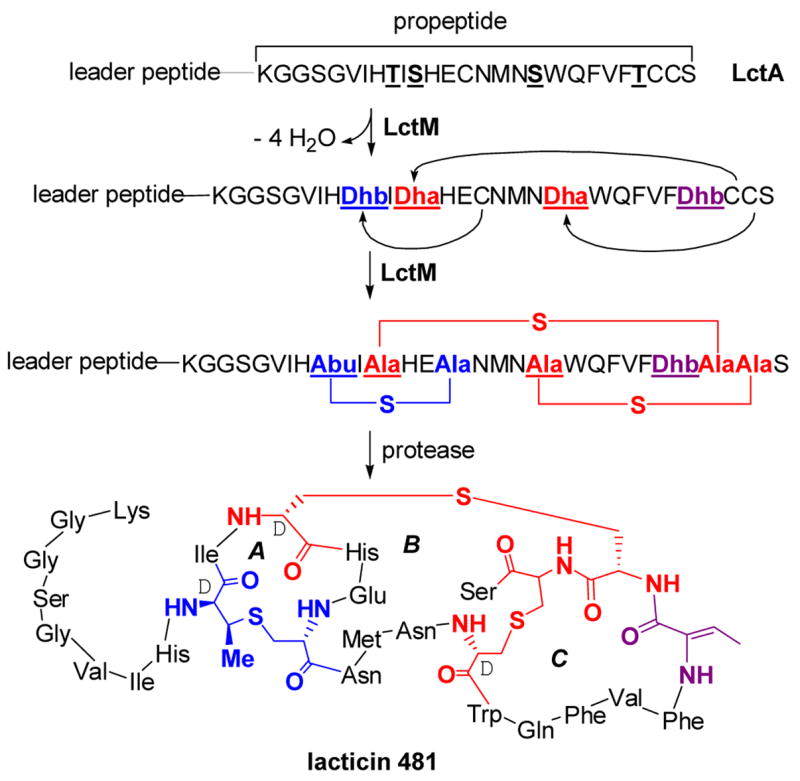
The post-translational maturation process of lacticin 481, a class II lantibiotic. LctM catalyzes the anti elimination of water from the underlined Ser and Thr residues in the propeptide region of LctA to generate Dha and Dhb residues. LctM also catalyzes the conjugate addition of three Cys residues in a regioselective manner to three of the dehydrated residues to generate three cyclic thioethers, one methyllanthionine (blue) and two lanthionines (red). The leader peptide is proteolytically removed by the N-terminal protease domain of LctT that also excretes the final product.45 The sequence of the leader peptide is MKEQNSFNLLQEVTESELDLILGA. Abu, S-2-aminobutyric acid.
With an active system in hand, the many questions that had initially attracted us to the lantibiotics could finally be investigated.46 LctM does not have homology with proteins of known function except for enzymes involved in the biosynthesis of other lantibiotics. Its C-terminal domain has homology with the LanC proteins suggesting that this domain is responsible for the cyclization. However, the N-terminal domain of LctM does not have homology with the LanB dehydratases utilized in class I lantibiotic biosynthesis but is nevertheless still expected to contain the dehydratase active site. Recent kinetic studies including one-turnover experiments demonstrated that each dehydration event is not kinetically coupled to a cyclization event.47 Furthermore, it appears that LctM does not release the substrate between dehydrations but rather completes all four dehydrations, in other words the enzyme is processive. ATP and Mg2+ are required for the dehydration but not for the cyclization reaction. In the dehydration process, ATP is used to first phosphorylate targeted Ser/Thr residues followed by elimination of phosphate to produce the Dha and Dhb structures.48 In addition to mechanistic studies, LctM has also been interrogated with respect to its substrate specificity. As had been anticipated when our program on lantibiotic biosynthesis was initiated in 1997, the enzyme is indeed quite promiscuous. It is capable of dehydrating Ser and Thr residues that are introduced at non-native positions in its substrate and even Ser and Thr residues in non-lantibiotic peptides.15,49
More recently, the class I lantibiotic nisin also succumbed to in vitro biosynthesis in our laboratory.16 An engineered Lactococcus lactis strain that does not produce nisin but carries a plasmid encoding NisA, NisB, and NisT was reported to secrete the dehydrated nisin prepeptide into the growth medium.50a As mentioned previously, we had synthetically prepared dehydrated peptides but because of the size of dehydrated NisA (51 amino acids, box Figure 3), these synthetic peptides were truncated analogs and we were unable to distinguish between enzymatic and non-enzymatic cyclization. With the engineered strain kindly provided by Gert Moll of BiOMaDe, we were able to purify the full length dehydrated substrate for NisC. Indeed, incubation of this dehydropeptide with the enzyme and subsequent proteolytic removal of the leader sequence produced nisin, identified by a sensitive bioassay, whereas non-enzymatic cyclization did not.16 This latter result was anticipated based on the results shown in Scheme 2. In collaboration with my colleague Satish Nair, the crystal structure of the cyclase NisC was also solved.16 The protein has a very similar fold as farnesyl transferase including the presence of an active site zinc at the top of an α,α-barrel. In farnesyl transferase, this zinc activates cysteines in its protein substrates for nucleophilic attack on farnesyl pyrophosphate,51 and a similar role is envisioned for the zinc in NisC,16,26 albeit that attack in this case is intramolecular onto a Michael acceptor (Scheme 7). An active site acid then stereoselectively protonates the enolate for a net anti addition providing the D-stereochemistry at the α-carbon.
Scheme 7.
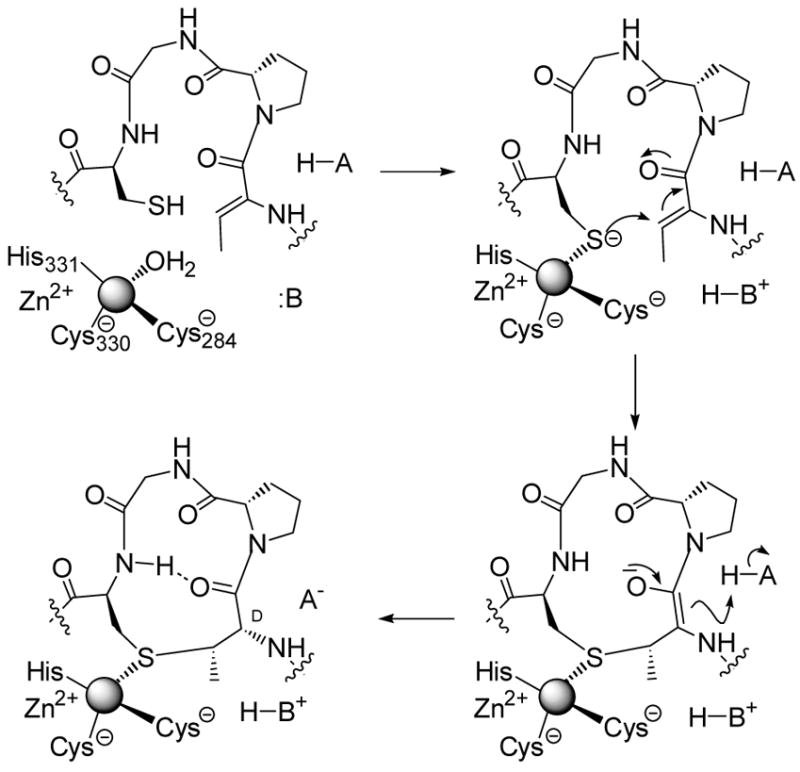
Proposed mechanism of enzymatic Lan and MeLan formation.
With the successful in vitro biosynthesis of the two lantibiotics described here as well as intriguing substrate specificity studies reported for several individual lantibiotic biosynthetic enzymes,50b,c,52,53 the future for further engineering of these structures is bright. Nisin has high affinity for lipid II, which it recognizes through a novel pyrophosphate binding cage that may form a blueprint for new antibiotics.29 Currently, therapeutic use of nisin is hampered due to problems of bioavailability and stability. Analogs that are smaller in size and perhaps have replaced the protease-sensitive amide linkages may help overcome these hurdles. Given the difficulty of installing lanthionine synthetically,54 the use of their biosynthetic machinery provides an attractive alternative route to such analogs for structure-activity studies. Other lantibiotics with entirely different primary and three-dimensional structures such as mersacidin and cinnamycin also have high affinity for their targets, lipid II and phosphatidyl ethanolamine, respectively.8 These impressive examples of molecular recognition suggest that the lanthionine motif is a naturally privileged architecture for constraining peptides into a bioactive conformation. In fact, lanthionines have very recently been found in morphogenetic peptides involved in sporulation in the streptomycetes,55 demonstrating that their use extends beyond antimicrobial peptides. Furthermore, synthetic non-natural lanthionine containing peptides have found use as mimics of disulfides in natural products or as structures that limit the conformational flexibility of bioactive compounds.54 The lanthionine moiety has been shown to confer higher chemical, proteolytic, and metabolic stability to such analogs.56 These biologically advantageous properties may therefore provide a framework for the design of a range of therapeutic peptides and lantibiotics. Certainly, the high tolerance of various lantibiotic modification enzymes suggests they are a class with much promise for biocatalysis. Coupled with the many other unique post-translational modifications found in lantibiotics (Figure 5), the prospects for diversity oriented biosynthesis are bright.
Figure 5.
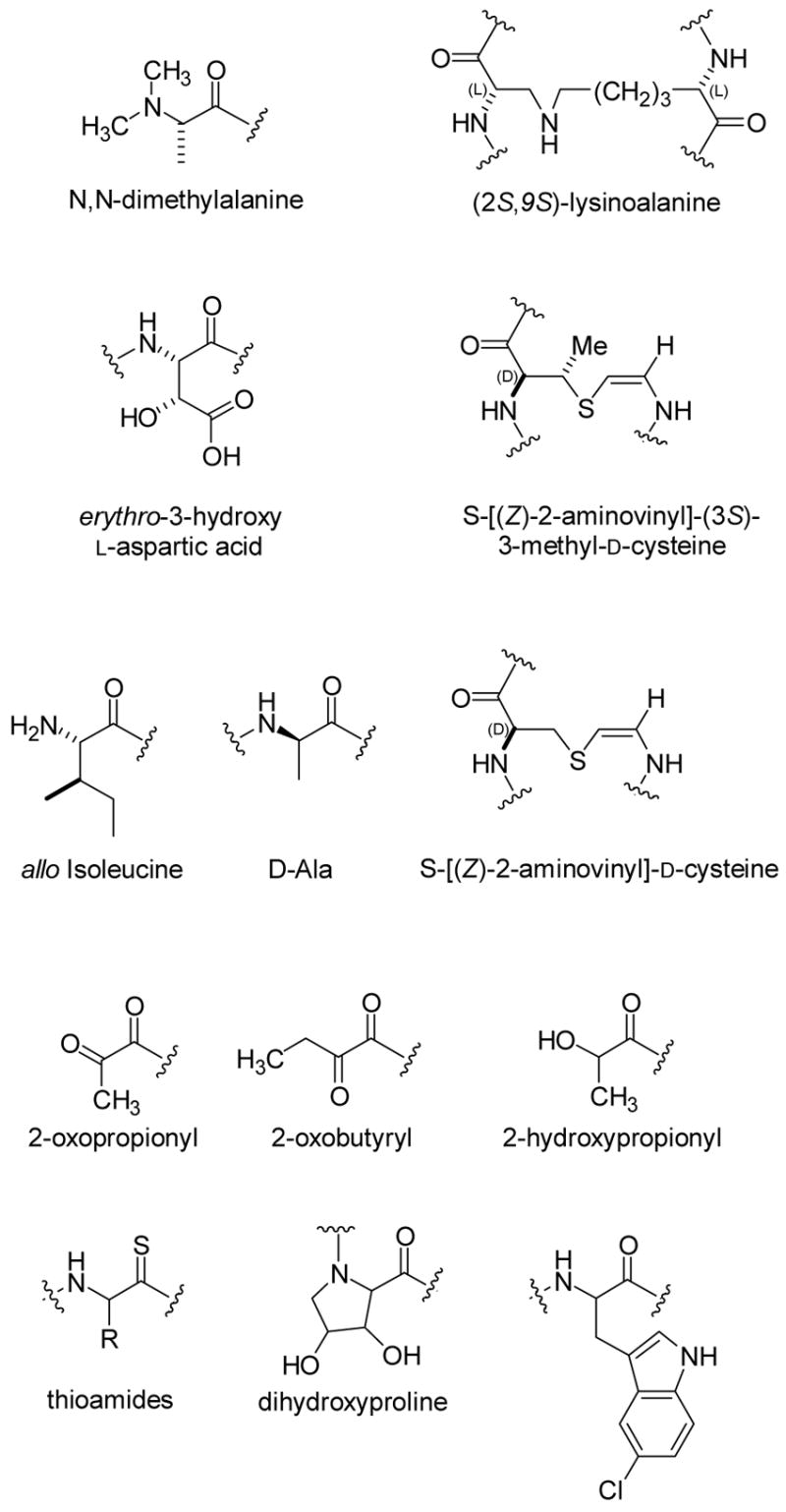
Various post-translational modifications of proteinogenic amino acids found in lantibiotic family members.8 The stereochemistry of the dihydroxyproline is currently unknown.57 Thioviramide contains an aminovinylcysteine suggesting it may be a lantibiotic, but it has not yet been shown to be gene encoded. It also contains the thioamides shown in the figure.58
Phosphonate natural products biosynthesis
Despite their ubiquitous use in antibacterial, antiviral, and antiparasitic therapies, and in agriculture as herbicides and pesticides, biosynthetic pathways for P-C bond containing natural products are relatively unexplored. A few of these compounds are shown in Scheme 8. Phosphonates and phosphinates are effective antibacterial and antifungal agents because the P-C bond is resistant to acid and base catalyzed hydrolysis, as well as phosphotransferase catalyzed hydrolysis. They exert their biological action as mimics of either carboxylic acids or phosphate esters. Fosfomycin is currently clinically used under the name Monurol® for the treatment of urinary tract infections.59 It is produced by Streptomyces wedmorensis, S. fradiae, and several Pseudomonas species,3e,60 and inactivates UDP-N-acetyl-glucosamine-3-O-enolpyruvyltransferase (MurA), an essential enzyme in cell wall biosynthesis.61 Phosphinothricin (PT) is an intermediate in the biogenesis of bialaphos, which is commercially used as a herbicide under the name BASTA®. Various formulations of phosphinothricin are widely used in agriculture, with annual sales in excess of $200 million per year.
Scheme 8.
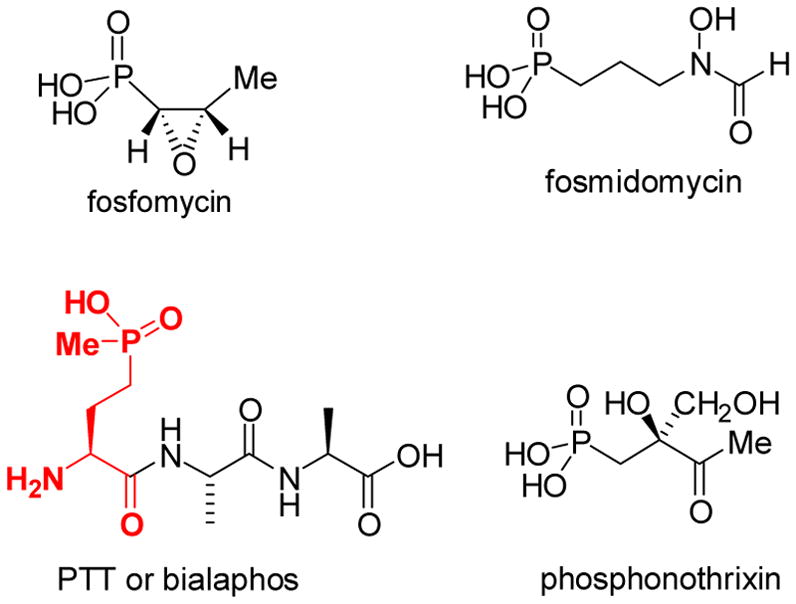
Several members of the phosphonate/phosphinate class of natural products. The phosphinothricin moiety in bialaphos is highlighted in red.
Seminal pioneering studies by Seto and coworkers using genetic techniques complemented by feeding studies with isotopically labeled precursors suggested the biosynthetic pathway for fosfomycin shown in Scheme 9.3e Using stereoselectively labeled precursors, Hammerschmidt and coworkers worked out the stereochemistry of the overall process.4 The methylation step that both groups proposed drew our initial interest. The methyl group in 2-hydroxypropyl phosphonic acid (HPP) was shown by labeling studies to be derived from methylcobalamin (MeCbl, Figure 2).3a What makes this methyl transfer very unusual is that it was proposed to take place as an anion (CH3-) from MeCbl to an electrophile, the aldehyde of PnAA (Scheme 9).3e,4a This proposal is fundamentally different from the accepted mechanisms for all MeCbl dependent methyltransferases in which methylation takes place via nucleophilic attack by the substrate at the methyl group of MeCbl (Figure 6).5 This then produces the highly nucleophilic Co(I) form of the cofactor, which can accept a methyl cation equivalent from a cellular methyl donor like S-adenosylmethionine (AdoMet) or methyl tetrahydrofolate (CH3-H4folate).
Scheme 9.
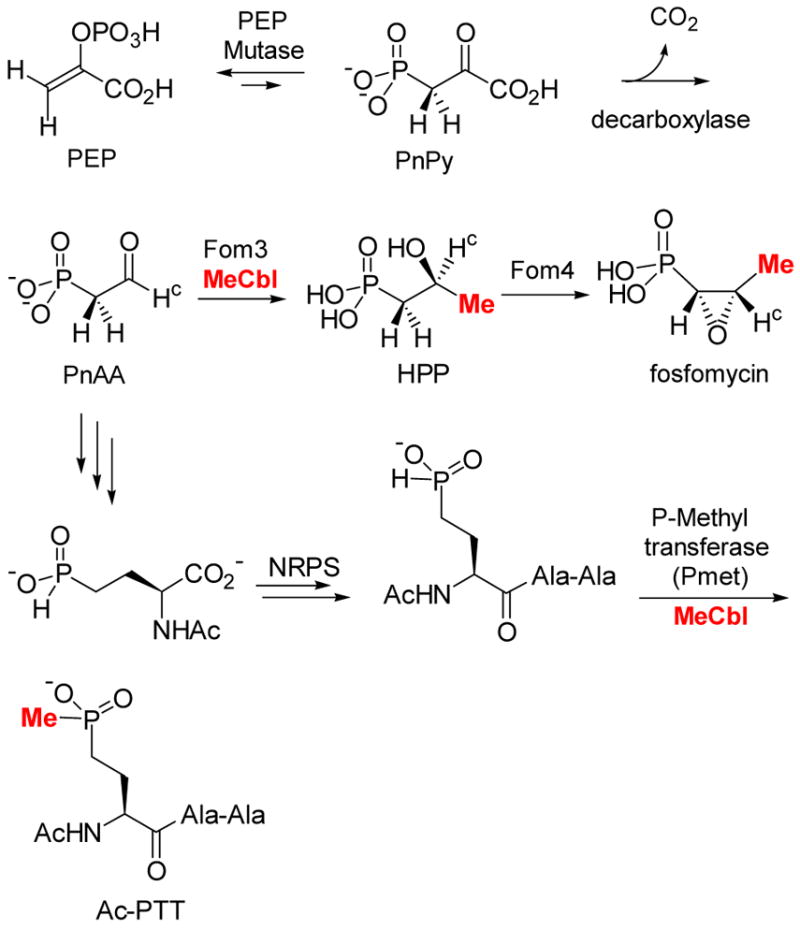
Proposed biosynthetic pathways of fosfomycin and bialaphos.3e
Figure 6.
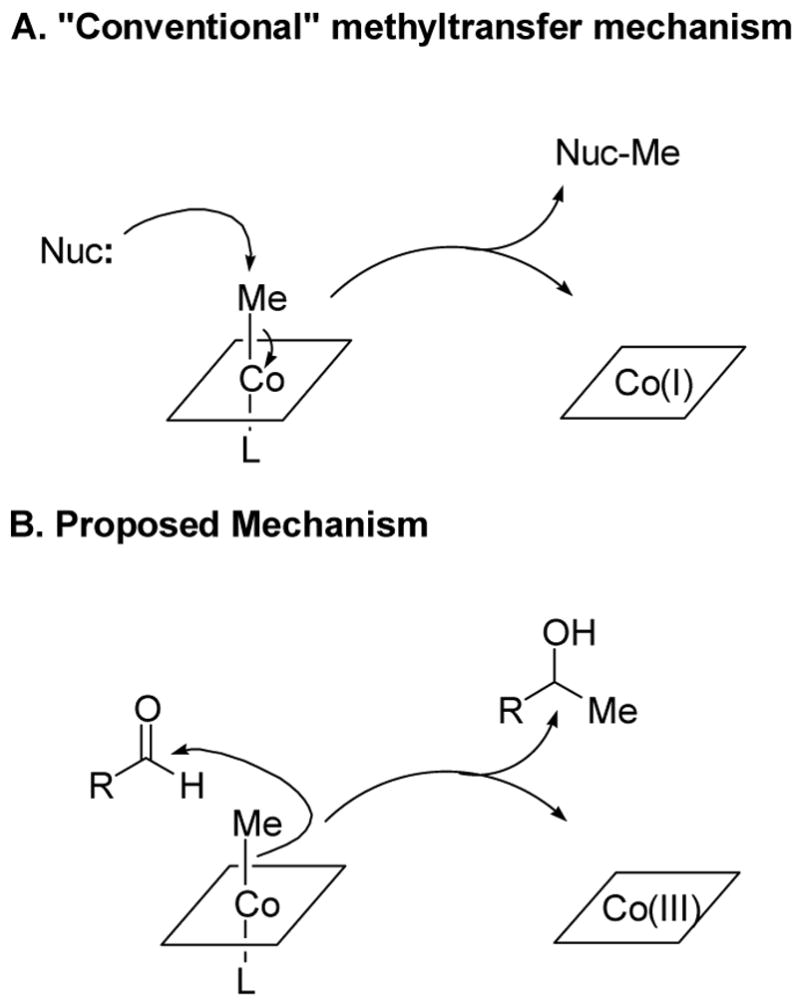
Conventional5 and unusual mechanisms3e,4a of B12-dependent methyl transfer. The corrin ligand ring system (Figure 2) is schematically represented by a square. L may be the benzimidazole ligand of B12 or an amino acid ligand from the protein.
Seto and coworkers also studied the biosynthesis of the phosphinate bialaphos (Figure 6) and showed that its methyl group attached to phosphorus is likely also derived from MeCbl.3b,d The high degree of homology between the two proteins catalyzing the methyl transfer steps, Pmet and Fom3, suggests they catalyze their reactions using similar mechanisms. The substrate for Fom3 has a very different chemical reactivity, however, from the substrate for the P-methyltransferase. The latter, through its thermodynamically unfavored tricoordinate tautomer 4, could act as a nucleophile according to the conventional mechanism of methyl transfer from MeCbl (Scheme 10). However, umpolung with thiamin pyrophosphate of the electrophilic aldehyde group of the PnAA substrate has been ruled out for Fom3 by the labeling studies of Hammerschmidt.4b
Scheme 10.
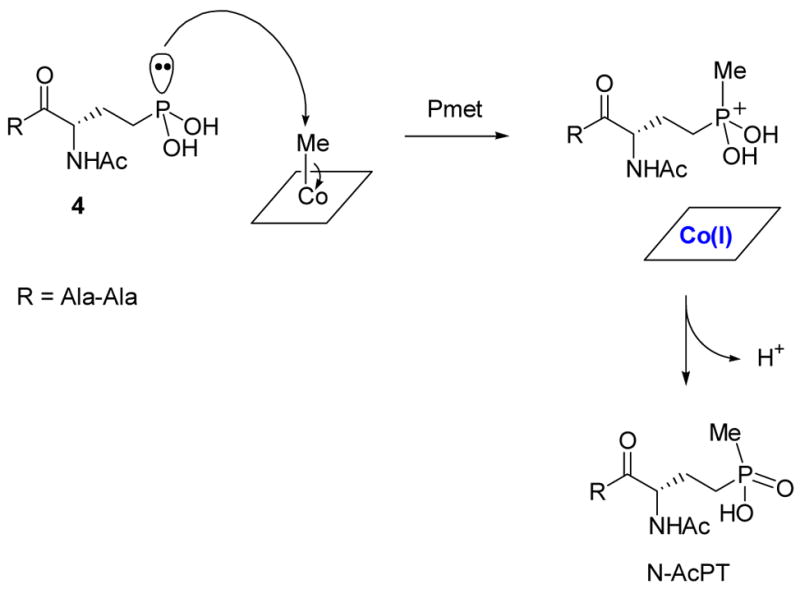
Possible heterolytic mechanism of P-methyltransferase (Pmet).
Intrigued by the apparent mechanistic paradox in how these enzymes would utilize B12 for their methyl transfer reactions, the biosynthesis of fosfomycin and bialaphos was the focus of one of my initial research proposals in 1996. The genes for both enzymes contain the sequence CysX3CysX2Cys. In postdoctoral work, I had encountered this sequence before, which at the time was known in two enzymes, the anaerobic ribonucleotide reductase and pyruvate formate lyase.62 In both enzymes, the cysteines are involved in an iron-sulfur cluster that was proposed to homolytically cleave the carbon sulfur bond of S-adenosyl methionine (SAM) to generate an adenosyl radical. This radical then abstracts a hydrogen atom from the substrate. From this information, we attempted to write a unifying mechanism for the two methyl transfer reactions. For the Pmet transformation, the 5′-deoxyadenosyl radical 5 could abstract a hydrogen atom from the phosphinate breaking the relatively weak P-H bond (Scheme 11).
Scheme 11.
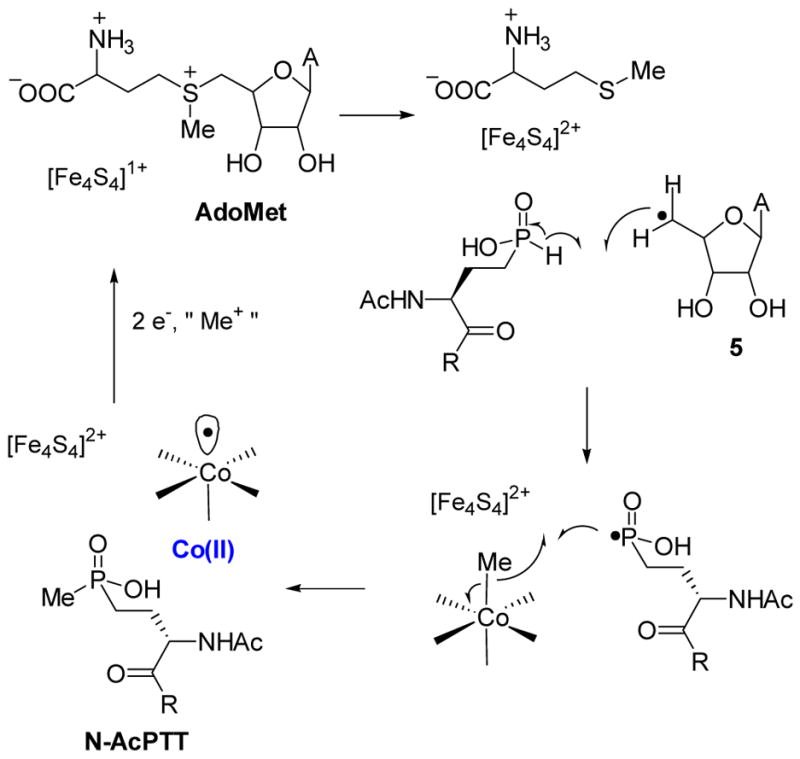
Possible homolytic mechanism of P-methyltransferase (Pmet).
This step is supported by radical chain reactions observed during addition of phosphinate radicals to alkenes in which the propagation step consists of hydrogen abstraction from the phosphinic acid by a carbon-based radical.63 The resulting phosphorus based radical could then potentially attack MeCbl to cleave the Co-C bond homolytically. Admittedly, this mechanism was (and is) highly speculative, but since we first formulated it, a number of reports in the literature provide support. In 1999, Sofia et al published a highly influential paper in which the authors showed that the CysX3CysX2Cys motif is widespread in nature and they termed this family the radical-SAM superfamily.64 Indeed, many radical-SAM proteins have now been studied and they all use an iron-sulfur cluster for the formation of the 5′-deoxyadenosyl radical that initiates catalysis of a wide range of transformations.65 P-methyltransferase and PnAA methyltransferase promote very different reactions than other confirmed radical-SAM proteins, but modern bioinformatics tools66 predict both a B12 and a SAM binding domain such that their inclusion in the family, although not proven, is likely. Indeed, Sofia et al proposed a number of putative methyl transferases in the superfamily with the role of CloN6 involved in the biosynthesis of clorobiocin recently confirmed by gene inactivation.67 In other developments supporting the model in Scheme 11, Kräutler and coworkers showed that carbon based free radicals react with MeCbl to give one-carbon homologated products (equation 1).68
 |
(1) |
However, the mechanism in Scheme 11 still poses problems for the reaction catalyzed by Fom3, because reaction of an acyl radical, if formed by abstraction of the hydrogen labeled Hc in PnAA (Scheme 9), with MeCbl would produce a ketone, not an alcohol, and would be inconsistent with Hammerschmidt’s labeling studies.4b Would Fom3 then indeed catalyze a methyl anion transfer or was there perhaps something missing in the proposed pathway?
Like our project on lantibiotics, our studies of the biosynthesis of fosfomycin and bialaphos initially met with many problems. Heterologously expressed proteins from S. wedmorensis were insoluble and fusion proteins that were soluble were not active. Having learned from our experience with the lantibiotics that it is often advantageous to move to a different but related system rather than spend too much time on an initially intractable system, we decided to clone the fosfomycin biosynthetic cluster of S. fradiae.69 Expression of the cluster in S. lividans resulted in the production of fosfomycin, the first successful example of heterologous expression. Gene inactivation analysis demonstrated that the gene fomC is absolutely essential to support fosfomycin production in S. lividans70 but this gene has no role in the previously proposed pathway in Scheme 9. FomC is predicted to be an iron-dependent dehydrogenase and we wondered whether perhaps PnAA must first be reduced to the corresponding alcohol before Fom3 carries out the methyl transfer reaction (Scheme 12). Consistent with this hypothesis, fosfomycin production could be restored in the inactivated fomC mutant by adding exogenous hydroxyethyl phosphonate (HEP) to the growth medium. Collectively, these results strongly argue that HEP is a biosynthetic intermediate and suggests that it may be the true substrate for Fom3 (Scheme 12). Our revised pathway for fosfomycin biosynthesis is fully consistent with the studies of the Seto and Hammerschmidt groups. Both laboratories had previously shown that HEP can be converted to fosfomycin by wild type producing organisms.3f,4 However, without compelling evidence HEP was considered to be an off-pathway intermediate. Recent cloning of the bialaphos biosynthetic cluster71 revealed an iron-dependent dehydrogenase with high sequence identity with FomC. Since PnAA is the last confirmed common intermediate in the biosynthetic pathways of these two compounds, it is likely that both dehydrogenases act on PnAA to give HEP, after which the two pathways diverge.
Scheme 12.
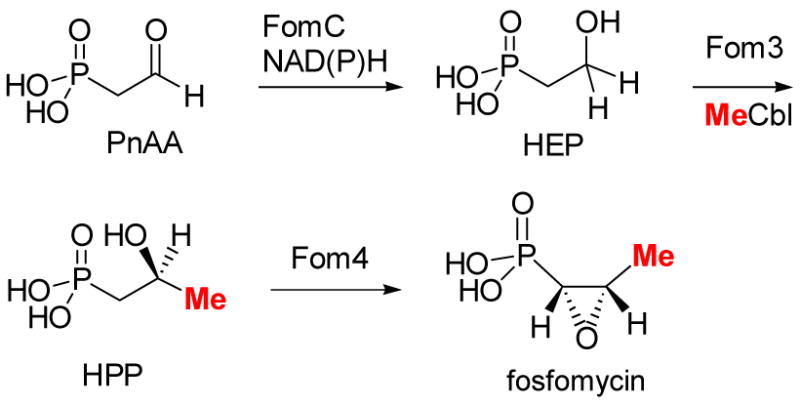
Revised biosynthetic pathway of fosfomycin
Hammerschmidt’s labeling studies are also consistent with Scheme 12. HEP labeled at C2 with deuterium resulted in label incorporation in the fosfomycin product when fed to S. fradiae. In the context of the original pathway (Scheme 8), these findings were interpreted to result from oxidation of HEP to PnAA, subsequent methyl transfer, and finally epoxidation (Scheme 13). However, the data can be equally well accounted for by the revised pathway of Scheme 12. Furthermore, oxygen labeling studies are more readily explained with the pathway in Scheme 12 than that of Scheme 13. S. fradiae converted HEP labeled with 18O in the hydroxyl group into fosfomycin in which 50 % of the label was retained in the epoxide oxygen.4a These elegant studies revealed for the first time the unusual nature of the epoxidation step,72 but as pointed out by Hammerschmidt, the retention of the label requires remarkably fast utilization of labeled PnAA since the half-life for exchange of oxygen in acetaldehyde is about 1 min at pH 7. The pathway in Scheme 12, however, does not require any kinetic limitations since the label would never be in an exchangeable position. The revised pathway also allows formulation of a unifying mechanism for the reactions catalyzed by Fom 3 and Pmet (Scheme 14). As proposed for Pmet (Scheme 11), Fom3 would generate a 5′-deoxyadenosyl radical, which then abstracts a hydrogen atom from C2 of HEP. Such hydrogen atom abstraction has ample precedent in adenosylcobalamin dependent enzymes.5 The ensuing carbon based radical could then react with the methyl group of MeCbl in similar fashion as the model studies of Kräutler (eq. 1) to produce HPP. Studies to verify this new model are currently underway.
Scheme 13.
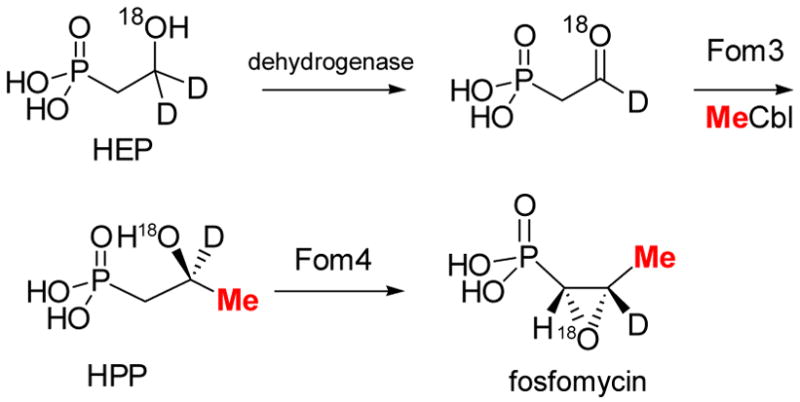
Labeling studies on fosfomycin biosynthesis.4
Scheme 14.
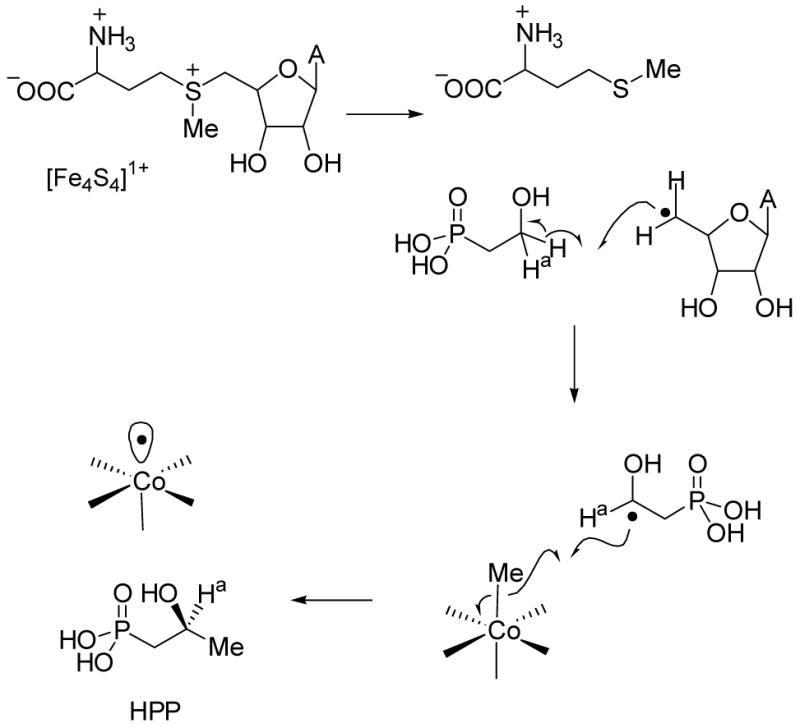
Proposed mechanism for Fom3.
Retrospective
Looking back at the relatively short history of our laboratory, two of the projects we initiated in the first year did not come to fruition until many years later and well after the tenure decision. The lantibiotics program ultimately delivered on the potential it contained and the biosynthesis of phosphonate natural products now forms a theme at the new Institute for Genomic Biology at UIUC. Both projects initially met typical obstacles inherent with starting a new program that hinges on multiple objectives that need to be met simultaneously. One of the early comments from an NIH reviewer still rings true today, the lantibiotic biosynthesis was a high risk, high pay-off project. In 1998, the project was fundable despite only limited preliminary results but in the current funding climate it might be difficult to support. Hopefully the scientific community and funding agencies will not fall prey to relying on only supporting ongoing research or sure-bet proposals and will continue to give young faculty the chance to take on a high risk, high impact problem.
A separate but equally important challenge that I suspect regularly faces assistant professors is to decide when to abandon a project when it is not looking promising. In our specific case, new projects on prostaglandin synthase and phosphite dehydrogenase were started in year 3 and along with a third initial project on B12-catalyzed dechlorination, 7 they carried the laboratory for the early years. Would the lantibiotics program have yielded results in a sufficiently timely manner if we had continued to put all our resources into it? Of course, one will never know. One frequently heard piece of advice for new faculty is to focus and not dilute their efforts, and I certainly endorse this recommendation. Our specific case allowed us to divert mid-flight because of the type of new projects that emerged with relatively short timelines and yet a high level of significance, and because of the availability and capability of new students in our graduate program. Without either of these conditions, temporarily redirecting our research program would not have been advisable. Furthermore, over the years seminal contributions have been made by many outstanding new investigators that stayed focus with smaller groups.
Acknowledgments
I am grateful to all my coworkers that have contributed to the success of our research, and to many colleagues for their advice and support. Our work on lantibiotic biosynthesis was supported by NIH (GM58822), the Burroughs Welcome Fund, and the Beckman Foundation. Research on phosphite dehydrogenase was supported by NIH (GM63003) and the Biotechnology Research and Development Consortium (Project 2-4-121), and our work on fosfomycin and bialaphos was supported by the University of Illinois.
References
- 1.(a) McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Science. 1993;262:1546–50. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]; (b) Kao CM, Katz L, Khosla C. Science. 1994;265:509–12. doi: 10.1126/science.8036492. [DOI] [PubMed] [Google Scholar]
- 2.Li YM, Milne JC, Madison LL, Kolter R, Walsh CT. Science. 1996;274:1188–93. doi: 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kuzuyama T, Hidaka T, Kamigiri K, Imai S, Seto H. J Antibiot. 1992;45:1812–4. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]; (b) Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. J Antibiot. 1992;45:781–7. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]; (c) Hidaka T, Goda M, Kuzuyama T, Takei N, Kidaka M, Seto H. Mol Gen Genet. 1995;249:274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]; (d) Hidaka T, Hidaka M, Kuzuyama T, Seto H. Gene. 1995;158:149–150. doi: 10.1016/0378-1119(95)00101-b. [DOI] [PubMed] [Google Scholar]; (e) Seto H, Kuzuyama T. Nat Prod Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]; (f) Seto H, Hidaka T, Kuzuyama T, Shibahara S, Usui T, Sakanaka O, Imai S. J Antibiot. 1991;44:1286–8. doi: 10.7164/antibiotics.44.1286. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hammerschmidt F. Angew Chem Int Ed Engl. 1994;33:341–342. [Google Scholar]; (b) Hammerschmidt F, Kählig H. J Org Chem. 1991;56:2364–2370. [Google Scholar]
- 5.Banerjee R, editor. The Chemistry and Biochemistry of B12. Wiley; New York: 1999. [Google Scholar]
- 6.(a) Burgess K, van der Donk WA, Jarstfer MB, Ohlmeyer MJ. J Am Chem Soc. 1991;113:6139–6144. [Google Scholar]; (b) Burgess K, van der Donk WA, Westcott SA. J Am Chem Soc. 1992;114:9350–9. [Google Scholar]; (c) Burgess K, van der Donk WA. J Am Chem Soc. 1994;116:6561–9. [Google Scholar]
- 7.Completing the spectrum from antibiotic biosynthesis to vitamin B12 chemistry, a third initial proposal focused on the B12-catalyzed dehalogenation of the priority pollutants perchloroethylene and trichloroethylene. Shey J, van der Donk WA. J Am Chem Soc. 2000;122:12403–12404.McCauley KM, Wilson SR, van der Donk WA. Inorg Chem. 2002;41:393–404. doi: 10.1021/ic011023x.McCauley KM, Wilson SR, van der Donk WA. Inorg Chem. 2002;41:5844–5848. doi: 10.1021/ic025714k.Shey J, McGinley CM, McCauley KM, Dearth A, Young B, van der Donk WA. J Org Chem. 2002;67:837–846. doi: 10.1021/jo0160470.McCauley KM, Wilson SR, van der Donk WA. J Am Chem Soc. 2003;125:4410–4411. doi: 10.1021/ja029692c.McCauley KM, Pratt DA, Wilson SR, Shey J, Burkey TJ, van der Donk WA. J Am Chem Soc. 2005;127:1126–36. doi: 10.1021/ja048573p.Pratt DA, van der Donk WA. J Am Chem Soc. 2005;127:384–96. doi: 10.1021/ja047915o.Pratt DA, van der Donk WA. Chem Commun. 2006:558–60. doi: 10.1039/b513624e.McGinley CM, Releya HA, van der Donk WA. Synlett. 2006:211–214.
- 8.Chatterjee C, Paul M, Xie L, van der Donk WA. Chem Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 9.Schnell N, Entian KD, Schneider U, Götz F, Zahner H, Kellner R, Jung G. Nature. 1988;333:276–278. doi: 10.1038/333276a0. [DOI] [PubMed] [Google Scholar]
- 10.Rogers LA. J Bacteriol. 1928;16:321–5. doi: 10.1128/jb.16.5.321-325.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross E, Morell JL. J Am Chem Soc. 1971;93:4634–4635. doi: 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]
- 12.Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Antonie van Leeuwenhoek. 1996;69:193–202. doi: 10.1007/BF00399424. [DOI] [PubMed] [Google Scholar]
- 13.Cotter PD, Hill C, Ross RP. Nat Rev Microbiol. 2005;3:777–88. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- 14.For a review, see: Kuipers OP, Bierbaum G, Ottenwälder B, Dodd HM, Horn N, Metzger J, Kupke T, Gnau V, Bongers R, van den Bogaard P, Kosters H, Rollema HS, de Vos WM, Siezen RJ, Jung G, Götz F, Sahl HG, Gasson MJ. Antonie van Leeuwenhoek. 1996;69:161–169. doi: 10.1007/BF00399421.
- 15.Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Science. 2004;303:679–81. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- 16.Li B, Yu JPJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 17.Xie L, Chatterjee C, Balsara R, Okeley NM, van der Donk WA. Biochem Biophys Res Commun. 2002;295:952–957. doi: 10.1016/s0006-291x(02)00783-0. [DOI] [PubMed] [Google Scholar]
- 18.Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl H, de Kruijff B. Science. 1999;286:2361–2364. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- 19.Breukink E, de Kruijff B. Nat Rev Drug Discov. 2006;5:321–32. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 20.Walker S, Chen L, Hu Y, Rew Y, Shin D, Boger DL. Chem Rev. 2005;105:449–76. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 21.Hasper HE, de Kruijff B, Breukink E. Biochemistry. 2004;43:11567–75. doi: 10.1021/bi049476b. [DOI] [PubMed] [Google Scholar]
- 22.Okeley NM, Zhu Y, van der Donk WA. Org Lett. 2000;2:3603–3606. doi: 10.1021/ol006485d. [DOI] [PubMed] [Google Scholar]
- 23.Seebeck FP, Szostak JW. J Am Chem Soc. 2006;128:7150–1. doi: 10.1021/ja060966w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H, van der Donk WA. Org Lett. 2002;4:1335–1338. doi: 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Y, Gieselman M, Zhou H, Averin O, van der Donk WA. Org Biomol Chem. 2003;1:3304–3315. doi: 10.1039/b304945k. [DOI] [PubMed] [Google Scholar]
- 26.Okeley NM, Paul M, Stasser JP, Blackburn N, van der Donk WA. Biochemistry. 2003;42:13613–13624. doi: 10.1021/bi0354942. [DOI] [PubMed] [Google Scholar]
- 27.Meyer C, Bierbaum G, Heidrich C, Reis M, Süling J, Iglesias-Wind MI, Kempter C, Molitor E, Sahl HG. Eur J Biochem. 1995;232:478–489. doi: 10.1111/j.1432-1033.1995.tb20834.x. [DOI] [PubMed] [Google Scholar]
- 28.Koponen O, Tolonen M, Qiao M, Wahlstrom G, Helin J, Saris Per EJ. Microbiology. 2002;148:3561–8. doi: 10.1099/00221287-148-11-3561. [DOI] [PubMed] [Google Scholar]
- 29.Hsu ST, Breukink E, Tischenko E, Lutters MA, De Kruijff B, Kaptein R, Bonvin AM, Van Nuland NA. Nat Struct Mol Biol. 2004;11:963–7. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 30.For some important examples and reviews, see Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–779. doi: 10.1126/science.7973629.Wallace CJ. Curr Opin Biotechnol. 1995;6:403–10. doi: 10.1016/0958-1669(95)80069-7.Lemieux GA, Bertozzi CR. TIBTECH. 1998;16:506–513. doi: 10.1016/s0167-7799(98)01230-x.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci USA. 1998;95:6705–10. doi: 10.1073/pnas.95.12.6705.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192–3. doi: 10.1021/ja021381e.Kimmerlin T, Seebach D. J Pept Res. 2005;65:229–60. doi: 10.1111/j.1399-3011.2005.00214.x.
- 31.Zhu Y, van der Donk WA. Org Lett. 2001;3:1189–1192. doi: 10.1021/ol015648a. [DOI] [PubMed] [Google Scholar]
- 32.Galoni D, van der Donk WA, Gin DY. Chem- Eur J. 2003;24:5997–6006. doi: 10.1002/chem.200305290. [DOI] [PubMed] [Google Scholar]
- 33.Galoni D, van der Donk WA, Gin DY. J Am Chem Soc. 2004:12712–3. doi: 10.1021/ja046793x. [DOI] [PubMed] [Google Scholar]
- 34.Galoni DP, Ide ND, Donk WAvd, Gin DY. J Am Chem Soc. 2005;127:7359–69. doi: 10.1021/ja050304r. [DOI] [PubMed] [Google Scholar]
- 35.For our work on PGHS and other systems involving amino acid radicals, see Peng S, Okeley NM, Tsai AL, Wu G, Kulmacz RJ, van der Donk WA. J Am Chem Soc. 2001;123:3609–3610. doi: 10.1021/ja015599x.Pesavento RP, van der Donk WA. Adv Protein Chem. 2001;58:317–385. doi: 10.1016/s0065-3233(01)58008-0.Peng S, Okeley NM, Tsai AL, Wu G, Kulmacz RJ, van der Donk WA. J Am Chem Soc. 2002;124:10785–10796. doi: 10.1021/ja026880u.van der Donk WA, Tsai AL, Kulmacz RJ. Biochemistry. 2002;41:15451–8. doi: 10.1021/bi026938h.Peng S, McGinley CM, van der Donk WA. Org Lett. 2004;6:349–352. doi: 10.1021/ol0361711.McGinley CM, van der Donk WA. J Label Compd Radiopharm. 2006;49:545–558. doi: 10.1002/jlcr.1073.McCauley KM, Vrtis JM, Dupont J, van der Donk WA. J Am Chem Soc. 2000;122:2403–2404.Pratt DA, Pesavento RP, van der Donk WA. Org Lett. 2005;7:2735–8. doi: 10.1021/ol050916g.Pesavento RP, Pratt DA, Jeffers J, van der Donk WA. Dalton Trans. 2006:3326 – 3337. doi: 10.1039/b516090a.
- 36.Costas AM, White AK, Metcalf WW. J Biol Chem. 2001;276:17429–36. doi: 10.1074/jbc.M011764200. [DOI] [PubMed] [Google Scholar]
- 37.Vrtis JM, White A, Metcalf WW, van der Donk WA. J Am Chem Soc. 2001;123:2672–2673. doi: 10.1021/ja004301k. [DOI] [PubMed] [Google Scholar]
- 38.(a) Relyea HA, van der Donk WA. Bioorg Chem. 2005;33:171–189. doi: 10.1016/j.bioorg.2005.01.003. [DOI] [PubMed] [Google Scholar]; (b) Relyea HA, Vrtis JM, Woodyer R, Rimkus SA, van der Donk WA. Biochemistry. 2005;44:6640–6649. doi: 10.1021/bi047640p. [DOI] [PubMed] [Google Scholar]; (c) Woodyer R, Wheatley J, Relyea H, Rimkus S, van der Donk WA. Biochemistry. 2005;44:4765–4774. doi: 10.1021/bi047868c. [DOI] [PubMed] [Google Scholar]; (d) Woodyer R, Zhao H, van der Donk WA. FEBS J. 2005;272:3816–27. doi: 10.1111/j.1742-4658.2005.04788.x. [DOI] [PubMed] [Google Scholar]
- 39.Vrtis JM, White A, Metcalf WW, van der Donk WA. Angew Chem Int Ed Engl. 2002;41:3257–3259. doi: 10.1002/1521-3773(20020902)41:17<3257::AID-ANIE3257>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 40.Woodyer R, van der Donk WA, Zhao H. Biochemistry. 2003;42:11604–11614. doi: 10.1021/bi035018b. [DOI] [PubMed] [Google Scholar]
- 41.Woodyer R, van der Donk WA, Zhao H. Comb Chem High Throughput Screen. 2006;9:237–245. doi: 10.2174/138620706776843246. [DOI] [PubMed] [Google Scholar]
- 42.Johannes TW, Woodyer RW, Zhao H. Appl Environ Microbiol. 2005;71:5728–5734. doi: 10.1128/AEM.71.10.5728-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siezen RJ, Kuipers OP, de Vos WM. Antonie van Leeuwenhoek. 1996;69:171–84. doi: 10.1007/BF00399422. [DOI] [PubMed] [Google Scholar]
- 44.Xie L, van der Donk WA. Curr Opin Chem Biol. 2004;8:498–507. doi: 10.1016/j.cbpa.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Uguen P, Hindré T, Didelot S, Marty C, Haras D, Le Pennec JP, Vallee-Rehel K, Dufour A. Appl Environ Microbiol. 2005;71:562–5. doi: 10.1128/AEM.71.1.562-565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patton GC, van der Donk WA. Curr Opin Microbiol. 2005;8:543–51. doi: 10.1016/j.mib.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Miller LM, Chatterjee C, van der Donk WA, Kelleher NL. J Am Chem Soc. 2006;128:1420–21. doi: 10.1021/ja057203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA. J Am Chem Soc. 2005;127:15332–3. doi: 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
- 49.Chatterjee C, Patton GC, Cooper L, Paul M, van der Donk WA. Chem Biol. 2006 doi: 10.1016/j.chembiol.2006.08.015. in press. [DOI] [PubMed] [Google Scholar]
- 50.(a) Kuipers A, De Boef E, Rink R, Fekken S, Kluskens LD, Driessen AJ, Leenhouts K, Kuipers OP, Moll GN. J Biol Chem. 2004;279:22176–82. doi: 10.1074/jbc.M312789200. [DOI] [PubMed] [Google Scholar]; (b) Kluskens LD, Kuipers A, Rink R, de Boef E, Fekken S, Driessen AJ, Kuipers OP, Moll GN. Biochemistry. 2005;44:12827–12834. doi: 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]; (c) Rink R, Kuipers A, de Boef E, Leenhouts KJ, Driessen AJ, Moll GN, Kuipers OP. Biochemistry. 2005;44:8873–82. doi: 10.1021/bi050081h. [DOI] [PubMed] [Google Scholar]; (d) Dawson MJ, Cortes Bargallo J, Rudd BAM, Boakes S, Bierbaum G, Hoffmann A, Schmitz S. 2005 p WO 2005/093069 A2. [Google Scholar]
- 51.Hightower KE, Fierke CA. Curr Opin Chem Biol. 1999;3:176–181. doi: 10.1016/s1367-5931(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 52.Cotter PD, O’Connor PM, Draper LA, Lawton EM, Deegan LH, Hill C, Ross RP. Proc Natl Acad Sci USA. 2005;102:18584–9. doi: 10.1073/pnas.0509371102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kupke T, Kempter C, Jung G, Götz F. J Biol Chem. 1995;270:11282–9. doi: 10.1074/jbc.270.19.11282. [DOI] [PubMed] [Google Scholar]
- 54.Paul M, van der Donk WA. Minirev Org Chem. 2005;2:23–37. [Google Scholar]
- 55.(a) Kodani S, Hudson ME, Durrant MC, Buttner MJ, Nodwell JR, Willey JM. Proc Natl Acad Sci USA. 2004;101:11448–11453. doi: 10.1073/pnas.0404220101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kodani S, Lodato MA, Durrant MC, Picart F, Willey JM. Mol Microbiol. 2005;58:1368–80. doi: 10.1111/j.1365-2958.2005.04921.x. [DOI] [PubMed] [Google Scholar]
- 56.(a) van der Meer JR, Polman J, Beerthuyzen MM, Siezen RJ, Kuipers OP, de Vos WM. J Bacteriol. 1993;175:2578–8. doi: 10.1128/jb.175.9.2578-2588.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bierbaum G, Szekat C, Josten M, Heidrich C, Kempter C, Jung G, Sahl HG. Appl Environ Microbiol. 1996;62:385–92. doi: 10.1128/aem.62.2.385-392.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Svensson CI, Rew Y, Malkmus S, Schiller PW, Taulane JP, Goodman M, Yaksh TL. J Pharmacol Exp Ther. 2003;304:827–32. doi: 10.1124/jpet.102.039750. [DOI] [PubMed] [Google Scholar]
- 57.Lazzarini A, Gastaldo L, Candiani G, Ciciliato I, Losi D, Marinelli F, Selva E, Parenti F. 2005 doi: 10.1016/j.chembiol.2007.11.009. p WO2005/014628 A1. [DOI] [PubMed] [Google Scholar]
- 58.Hayakawa Y, Sasaki K, Nagai K, Shin-ya K, Furihata K. J Antibiot (Tokyo) 2006;59:6–10. [Google Scholar]
- 59.Reeves DS. J Antimicrob Chemother. 1994;34:853–8. doi: 10.1093/jac/34.6.853. [DOI] [PubMed] [Google Scholar]
- 60.(a) Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. Science. 1969;166:122–3. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]; (b) Christensen BG, Leanza WJ, Beattie TR, Patchett AA, Arison BH, Ormond RE, Kuehl FA, Jr, Albers-Schonberg G, Jardetzky O. Science. 1969;166:123–5. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- 61.Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, Walsh CT. Biochemistry. 1994;33:10646–51. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- 62.Stubbe J, van der Donk WA. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 63.(a) Benschop HP, Platenburg DHJM. J Chem Soc Chem Commun. 1970:1098–1099. [Google Scholar]; (b) Farnham WB, Murray RK, Jr, Mislow K. J Chem Soc, Chem Commun. 1971:146–147. [Google Scholar]
- 64.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frey PA, Magnusson OT. Chem Rev. 2003;103:2129–48. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 66.Marchler-Bauer A, Bryant SH. Nucleic Acids Res. 2004;32:W327–31. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Westrich L, Heide L, Li SM. Chembiochem. 2003;4:768–73. doi: 10.1002/cbic.200300609. [DOI] [PubMed] [Google Scholar]
- 68.Mosimann H, Kräutler B. Angew Chem Int Ed Engl. 2000;39:393–395. doi: 10.1002/(sici)1521-3773(20000117)39:2<393::aid-anie393>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 69.Woodyer RD, Shao Z, Thomas P, Kelleher NL, Metcalf WM, van der Donk WA, Zhao H. 2006 doi: 10.1016/j.chembiol.2006.09.007. submitted for publication. [DOI] [PubMed] [Google Scholar]
- 70.Woodyer RD, Li G, Zhao H, van der Donk WA. 2006 submitted for publication. [Google Scholar]
- 71.(a) Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Appl Environ Microbiol. 2004;70:7093–102. doi: 10.1128/AEM.70.12.7093-7102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blodgett JA, Zhang JK, Metcalf WW. Antimicrob Agents Chemother. 2005;49:230–40. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.For more recent mechanistic and structural studies of this fascinating process, see Liu P, Murakami K, Seki T, He X, Yeung SM, Kuzuyama T, Seto H, Liu HW. J Am Chem Soc. 2001;123:4619–20. doi: 10.1021/ja004153y.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu HW. Angew Chem Int Ed Engl. 2002;41:4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2.Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu HW. Biochemistry. 2003;42:11577–86. doi: 10.1021/bi030140w.Liu P, Mehn MP, Yan F, Zhao Z, Que L, Jr, Liu HW. J Am Chem Soc. 2004;126:10306–12. doi: 10.1021/ja0475050.Higgins LJ, Yan F, Liu P, Liu HW, Drennan CL. Nature. 2005;437:838–44. doi: 10.1038/nature03924.McLuskey K, Cameron S, Hammerschmidt F, Hunter WN. Proc Natl Acad Sci USA. 2005;102:14221–6. doi: 10.1073/pnas.0504314102.