

Abstract
Enterococci are major contributors of hospital-acquired infections and have emerged as important reservoirs for the dissemination of antibiotic resistance traits. The ability to form biofilms on medical devices is an important aspect of pathogenesis in the hospital environment. The Enterococcus faecalis Fsr quorum system has been shown to regulate biofilm formation through the production of gelatinase, but the mechanism has been hitherto unknown. Here we show that both gelatinase (GelE) and serine protease (SprE) contribute to biofilm formation by E. faecalis and provide clues to how the activity of these proteases governs this developmental process. Confocal imaging of biofilms suggested that GelE− mutants were significantly reduced in biofilm biomass compared to the parental strain, whereas the absence of SprE appeared to accelerate the progression of biofilm development. The phenotype observed in a SprE− mutant was linked to an observed increase in autolytic rate compared to the parental strain. Culture supernatant analysis and confocal microscopy confirmed the inability of mutants deficient in GelE to release extracellular DNA (eDNA) in planktonic and biofilm cultures, whereas cells deficient in SprE produced significantly more eDNA as a component of the biofilm matrix. DNase I treatment of E. faecalis biofilms reduced the accumulation of biofilm, implying a critical role for eDNA in biofilm development. In conclusion, our data suggest that the interplay of two secreted and coregulated proteases—GelE and SprE—is responsible for regulating autolysis and the release of high-molecular-weight eDNA, a critical component for the development of E. faecalis biofilms.
Bacteria are often found in nature as communities of sessile surface-adherent populations covered in a slimy matrix composed of exopolysaccharides, protein, and DNA (9, 19, 23). Bacteria present within these communities (also referred to as biofilms) exhibit social behavior analogous to that found in higher organisms in that they can communicate and rapidly adapt to changing growth environments (5, 23, 55).
The gram-positive opportunistic pathogen, Enterococcus faecalis develops persistent biofilm-like vegetations on implant devices, including orthopedic implants, urethral stents, catheters, and heart valves, making it a leading cause of nosocomial infection (29). Enterococci are becoming increasingly resistant to many conventional antibiotics (22). Compounding the drug resistance phenotypes displayed by clinical isolates is the observation that enterococci growing as biofilms are more resistant to vancomycin, ampicillin, and linezolid than their planktonic counterparts (44). Epidemiological data also suggest enterococci to be important reservoirs for the transmission of antibiotic resistance genes among different species of bacteria (7, 56).
Of the factors reported to be important for E. faecalis biofilm formation (29), the enterococcal surface protein (Esp) and the secreted metalloprotease, gelatinase (GelE), are known to be expressed as variable traits (33, 47). More recently, Tendolkar et al. (51a) identified a locus from a clinical E. faecalis urinary tract isolate that they termed biofilm enhancer in Enterococcus (bee locus). The genes from this locus resemble the pilin biosynthetic genes identified by Nallapareddy et al. (33a) and have been shown to contribute to biofilm formation, but were found to be present in less than 5% of clinical isolates. It is noteworthy that Arciola et al. (3) recently correlated the presence of the esp gene and high phenotypic expression of gelatinase with the ability of E. faecalis epidemic clones from orthopedic implant infections to form biofilms. The esp gene that encodes the surface-associated Esp is located on a 153-kb pathogenicity island, and its expression significantly increases the bacterial cell surface hydrophobicity and attachment on a substratum (51, 52). The expression of GelE is dependent on the fsr regulatory system (38, 39) and is known to vary among strains of E. faecalis due to a defined 23.9-kb deletion in the genome that encompasses the fsr genes (33). The fsr locus consists of four genes, designated fsrA, fsrB, fsrC, and fsrD (32). The fsrC and fsrA genes encode a two-component sensor kinase-response regulator pair (39). The fsrD codes for a peptide lactone that functions in a cell-density-dependent manner (31). FsrB is thought to be responsible for the proteolytic cleavage and cyclization of FsrD (32). It is likely that FsrC sensor histidine kinase senses the accumulation of the FsrD peptide in the extracellular space, leading to activation of the response regulator FsrA. The gene encoding GelE is located immediately adjacent to the 3′ end of fsrC and is cotranscribed with sprE, which encodes a secreted serine protease (38, 39). Mutations in the fsr locus and its downstream target gelE resulted in poor biofilm-forming capabilities, indicating that biofilm formation in Enterococcus is dependent on quorum sensing (20, 30, 36). Mutants defective in fsr quorum signaling were restored to wild-type biofilm levels by the addition of purified GelE, indicating that GelE alone is a major contributor to biofilm development (20).
The mechanism by which GelE positively regulates biofilm formation has hitherto been unknown. It was hypothesized that GelE, like Esp, may be able to modify the bacterial cell surface hydrophobicity by virtue of its ability to cleave substrates at hydrophobic residues (6, 27, 28). An alternate hypothesis involves the ability of GelE to activate cell wall autolysins (48, 54). SprE has also been shown to be an important virulence factor since an sprE gene disruption resulted in decreased virulence in a mouse peritonitis model (39, 50), a Caenorhabditis elegans model (15, 49), and a rabbit endophthalmitis model (14).
In the present study, we investigated the role of both extracellular secreted proteases in biofilm formation by comparing isogenic single ΔgelE and ΔsprE and double protease ΔgelE-sprE deletion mutants of E. faecalis V583. Further, the ability to regulate autolysis with the concomitant release of extracellular DNA (eDNA) was shown to be a key contributor to the overall development of E. faecalis biofilms.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The different strains and plasmids used in the present study are listed in Tables 1 and 2, respectively. Strains were cultured in Todd-Hewitt broth (THB) or M17 medium (Difco Laboratories) and grown at 37°C unless otherwise indicated. Escherichia coli EC1000 was used for plasmid constructions. The antibiotics used for selection in E. coli were chloramphenicol, kanamycin, and spectinomycin at concentrations of 10, 50, and 150 μg/ml, respectively, and those used for E. faecalis included chloramphenicol, tetracycline, and spectinomycin at concentrations of 15, 15, and 750 μg/ml, respectively.
TABLE 1.
E. faecalis strains used in this study
| Strain | Relevant genotype | Complementation in pAT28 | Relevant phenotypea | Origin |
|---|---|---|---|---|
| V583 | Parental | GelE+ SprE+ | Clinical isolate | |
| VT01 | ΔgelE | GelE− SprE+ | V583 | |
| VT02 | ΔsprE | GelE+ SprE− | V583 | |
| VT03 | ΔgelE-sprE | GelE- SprE− | V583 | |
| VT05 | gelE | gelE promoter gelE | GelE+ SprE+ Specr | pVT05→VT01 |
| VT07 | gelE sprE | gelE promoter gelE sprE | GelE+ SprE+ Specr | pVT07→VT03 |
| VT08 | sprE | gelE promoter sprE | GelE+ SprE+ Specr | pVT08→VT02 |
| VT09 | Parental | GelE+ SprE+ Gfp Tetr | pMV158gfp→V583 | |
| VT10 | ΔgelE | GelE− SprE+ Gfp Tetr | pMV158gfp→VT01 | |
| VT11 | ΔsprE | GelE+ SprE− Gfp Tetr | pMV158gfp→VT02 | |
| VT12 | ΔgelE-sprE | GelE- SprE− Gfp Tetr | pMV158gfp→VT03 |
Specr, spectinomycin resistance; Tetr, tetracycline resistance.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pLT06 | Integration vector, Cmr derivative of pCJK47 | L. Thurlow, unpublished data |
| pVT01 | pLT06 containing a 2.0-kb EcoRI/BamHI fragment containing engineered gelE deletion | This study |
| pVT02 | pLT06 containing a 2.0-kb EcoRI/BamHI fragment containing engineered sprE deletion | This study |
| pVT03 | pLT06 containing a 2.0-kb EcoRI/BamHI fragment containing engineered gelE sprE deletion | This study |
| pAT28 | Broad-host-range shuttle vector, spectinomycin resistance | 53 |
| pVT05 | pAT28 containing 1748-bp EcoRI/XhoI fragment containing the native gelE promoter along with full-length gelE | This study |
| pVT07 | pAT28 containing 2687-bp EcoRI/BamHI fragment containing the native gelE promoter and full-length gelE sprE | This study |
| pVT08 | pAT28 containing 2123-bp EcoRI/BamHI fragment containing the native gelE promoter, a truncated gelE, and full-length sprE | This study |
| pMV158gfp | Gram-positive replicative vector expressing a Gfp reporter | 1 |
Construction of E. faecalis V583 in-frame protease deletion mutants.
In-frame deletions of gelE, sprE, and gelE-sprE were constructed by using pLT06, an E. coli enterococcal temperature-sensitive cloning vector that had selectable and counterselectable markers that aided in the selection of mutants containing the targeted deletions (L. Thurlow and L. E. Hancock, unpublished results). The vector pLT06 is a derivative of pCJK47 (25), retaining the counterselection properties on dl-p-chlorophenylalanine containing agar due to the presence of the pheS dominant-negative allele (25). In addition, pLT06 contains a chloramphenicol resistance marker and origin of replication from pWV01 (26).
Flanking regions (∼1 kb) from both the 5′ and the 3′ ends of the targeted proteases were PCR amplified with the primers listed in Table 3. For the construction of pVT01 (gelE deletion), the primers GelEP1 and GelEP2 were used to amplify the region 5′ to gelE on the V583 genome. The primers GelEP3 and GelEP4 were used to amplify the region 3′ to gelE. GelEP1 and GelEP2 contained EcoRI and XhoI sites, respectively, and GelEP3 and GelEP4 contained SalI and BamHI restriction sites to facilitate cloning. Each PCR product was digested with the corresponding restriction enzymes, and both products were ligated into pLT06 cut with EcoRI and BamHI, prior to electroporation into E. coli EC1000. The correct constructs were identified by selection on LB agar plates containing chloramphenicol at 10 μg/ml, screened by restriction digest analysis, and further sequenced for verification. A similar approach was used in the construction of pVT02 (sprE deletion) using the primer pairs SprEP1 and SprEP2, as well as SprEP3 and SprEP4; and for pVT03 (gelE-sprE deletion) the primer pairs GelEP1 and GelEP2 were used, along with SprEP3 and SprEP4. The isolated plasmids were electroporated into electrocompetent E. faecalis V583 (10). E. faecalis V583 transformants were selected by growth at 28°C on THB agar containing chloramphenicol at 15 μg/ml and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) at 120 μg/ml. Blue colonies were inoculated into fresh THB containing chloramphenicol at 15 μg/ml. Cultures were grown overnight at 28°C, diluted 1:100 into fresh medium, and grown for an additional 2.5 h at 28°C and then shifted to 42°C for an additional 2.5 h to favor single-site integration of plasmids into the E. faecalis V583 genome. Serial dilutions of the integrants were plated onto THB agar plates supplemented with 15 μg of chloramphenicol/ml. Colony PCR was used to confirm single-site integration for each construct using vector-specific primers OriF or SeqR, along with primers targeted to regions 5′ or 3′ to the site of insertion (GelEUp, GelEDown, SprEUp, or SprEDown). A positive colony was then cultured in the absence of selection until the culture reached stationary phase (∼2 × 109 CFU/ml). Serial dilutions were prepared, and fresh medium (THB) was inoculated such that it contained 100 CFU/ml. Serial dilutions (1:500 and 1:1,000) were plated on MM9YEG agar supplemented with 10 mM dl-p-Cl-Phe and X-Gal at 120 μg/ml. Counterselection using dl-p-Cl-Phe has been shown to favor the selection of colonies that have lost the plasmid (25). Colony PCR using the primers GelEUp and GelEDown for VT01, SprEUp and SprEDown for VT02, and GelEUp and SprEDown for VT03 were used to confirm the gene deletion in the genome. Phenotypic confirmation of the protease deletions were also visualized on THB agar containing 1.5% skim milk.
TABLE 3.
Oligonucleotides used in this study
| Primer | Sequence (5′-3′) |
|---|---|
| GelEP1 | GAGAGAATTCGATTGGCTTAGTCATTGAAGC T |
| GelEP2 | CTCTCTCGAGAAAGATGCCTGTACCTAAAATG |
| GelEP3 | GAGAGTCGACCAGGTAAACCAACCAAGTGAAT |
| GelEP4 | CTCTGGATCCCCGTGATTCTGGAAATTCCGAG |
| SprEP1 | GAGAGAATTCGTGAACGCTACAGATGGAACAA |
| SprEP2 | CTCTCTCGAGTTCATTCATTGACCAGAACAGA |
| SprEP3 | GAGAGTCGACCTCGGAATTTCCAGAATCACGG |
| SprEP4 | CTCTGGATCCAGGTTCAGCGTTATCTACTAAG |
| GelEUp | CGCCAGAGATTTCACCTGACT |
| GelEDown | GGTACTTTTATCGTAACTTACAC |
| SprEUp | CAATCGGTTGATGCAATCGGTG |
| SprEDown | GTACGAGCATTCGCAGTAAATTC |
| GelEprom | GAGAGAATTCGCTATGGTATTGAGTTATGAGG |
| Ef1091-5′ | GCTATGTTGGCTACTCAAGTG |
| Ef1091-3′ | TGTCCTGCAGGTGCTTTTAC |
| Ef0887-5′ | GAGAGAATTCGAAGGAATTGTCATTGTGCG |
| Ef0887-3′ | CAAAACGGATCCGACTCGC |
| Ef2490-5′ | GAGAGTCGACAGCAAAAGGGTTGTTGAAATA |
| Ef2490-3′ | CTCTGCATGCCCTACTTTCTCTGTTACTTAAT |
| Ef2488-5′ | GAGAGGATCCTAAACAGGGGAGTGTGTGACATG |
| Ef2488-3′ | GAGAGCATGCCAAACACCAAATGCATTATTTA |
| Ef2194-5′ | GGAACAACGCTAAACTTTTAC |
| Ef2194-3′ | CGCTCCTATTCTGCTGCTAA |
| OriF | CAATAATCGCATCCGATTGCA |
| SeqR | CCTATTATACCATATTTTGGAC |
Complementation of E. faecalis V583 in-frame protease deletion mutants.
In-frame protease deletions of E. faecalis V583 were complemented with full-length gelE, sprE, and gelE-sprE, each with the native gelE promoter region in a pAT28 vector (53) and were denoted pVT05, pVT08, and pVT07, respectively. The gelE complement insert was cloned by PCR amplification of V583 genome with primers GelEprom and SprEP2 and subsequently inserted as an EcoRI/XhoI fragment into pAT28 cut with EcoRI/SalI. The gelE-sprE double protease complement construct was cloned by PCR amplification of V583 genome using primers GelEprom and GelEP4 and inserted as an EcoRI/BamHI fragment into EcoRI/BamHI-cut pAT28 vector. Plasmid pVT08 was constructed by digesting pVT07 with AflII and NcoI, followed by Klenow treatment to make it blunt ended, and then the molecule was circularized by self-ligation to obtain an in-frame deletion of gelE. These constructs were transformed into the corresponding protease deletion mutants, and phenotypic complementation was confirmed by zymography using skim milk at a final concentration of 0.02% as a substrate (24).
Biofilm assay on polystyrene microtiter plates.
Biofilm formation on polystyrene was quantified with crystal violet staining method as previously described (20). Each assay was performed in octuplicate and repeated five times. Statistical significance was calculated by using Dunnett's test (GraphPad Software, San Diego, CA).
Cell surface hydrophobicity assay.
The cell surface hydrophobicities of E. faecalis V583 and isogenic protease mutant strains were carried out as previously described (43). The percentage of bacterial adhesion to hydrocarbon was calculated as follows: [1 − (ODF/ODI)] × 100, where ODI and ODF are the optical densities of cells resuspended in PUM buffer (100 mM potassium phosphate [pH 7.1], 30 mM urea, 800 μM MgSO4·H2O) determined at the beginning and the end of the experiment, respectively. Statistical significance was computed by using the Dunnett's test (GraphPad Software, San Diego, CA).
Autolysis assay.
Autolysis assay was carried out as previously reported (11).
Isolation of eDNA from E. faecalis planktonic culture supernatants.
Supernatants from 24-h-old grown cultures were passed through a sterile syringe filter (0.2-μm pore size; Nalgene) and concentrated ∼20-fold using a 10-kDa cutoff membrane (YM-10 Centricon centrifugal filter devices; Millipore) according to the manufacturer's instructions. The concentrated samples were loaded on a 1% agarose gel and stained with ethidium bromide to visualize high-molecular-weight DNA. Densitometric spot comparisons were performed by using Alphaimager software (Alpha-Innotec, San Leandro, CA).
eDNA from culture supernatants was isolated by using the Wizard genomic DNA purification kit according to the manufacturer's instructions, and chromosomal DNA was isolated as previously described (37). For comparative PCR, primers listed in Table 3 were designed to amplify genes from regions of the E. faecalis V583 genome, including Ef0887, Ef1091, Ef2194, Ef2488, Ef2490, and Ef1818 (gelE).
Laser scanning confocal microscopy.
E. faecalis strains V583, VT01, VT02, and VT03 were transformed with pMV158GFP (34) to constitutively express Gfp for confocal imaging. The resulting strains were designated VT09, VT10, VT11, and VT12, respectively. Confocal microscopy was performed on E. faecalis biofilms grown on glass coverslips. Sterile glass coverslips were placed on the bottom of six-well tissue culture plates and submerged with 5 ml of M17 broth, seeded with a 1:100 dilution from an overnight culture (approximately 5 to 10 × 106 CFU), and grown for 24 h at 37°C. For 2-, 3-, and 4-day-old biofilms, the culture supernatants were replaced with fresh medium daily. Just prior to imaging, biofilms were gently rinsed three times with sterile phosphate-buffered saline, followed by 10 min of staining with 5 ml of propidium iodide (PI; 1 μM). The coverslips were mounted on a microscope slide and sealed with clear nail polish to prevent dehydration. Slides were visualized by using a Zeiss LSM 5 Pascal laser scanning confocal microscope. The LSM 5 system was equipped with a Zeiss Axioplan 2 MOT research microscope, a fully motorized stage, a Plan Apohromat objective (×63/1.4 oil) and differential contrast interference. Dual fluorescence emission imaging of green fluorescent protein (GFP) and PI was accomplished using a 488-nm line of 458/488/514 argon gas ion laser to excite GFP and a 543-nm line of HeNe laser to excite PI. A secondary HFT 545 dichroic was used to split the emission signals into two signals, the shorter wavelengths passed through a band-pass 505- to 530-nm filter to image GFP fluorescence, and the longer wavelength passed through a long-pass 560-nm filter to image PI fluorescence. For z-series, the Airy units of the longer and shorter wavelengths were adjusted to give an optical slice thickness of 0.7 μm, and this thickness was used as the slice interval. Biofilm quantification was carried out using the COMSTAT analysis package (21). Volumetric analysis (μm3) of representative confocal images portraying regions within the biofilm stained by PI were carried out using the 3D Object counter plug-in in the NIH Image J software. For determination of statistical significance, the data were natural log transformed, and an unpaired t test was performed using GraphPad (GraphPad Software, San Diego, CA).
DNase I treatment of biofilms.
To assess the significance of eDNA for E. faecalis biofilms, 6-, 12-, and 24-h-old biofilms were treated with 100 Kunitz units per ml of DNase I. The control contained denatured DNase I that was heated at 100°C for 15 min. The biofilms were imaged by using confocal laser scanning microscopy (CLSM).
RESULTS
Construction and complementation of E. faecalis V583 isogenic protease mutants.
Kristich et al. recently developed a pheS counterselectable vector system, pCJK47 to generate markerless in-frame isogenic deletion mutations in E. faecalis OG1RF (25). However, this vector system was unsuitable for studies with strain V583 due to the unavailability of selectable resistance markers, as well as difficulty associated with conjugal mating of strains possessing multiple plasmids. In the present study, we used the plasmid pLT06 (a derivative of pCJK47), which encodes resistance to chloramphenicol and contains a temperature-sensitive replication origin from pWV01 (26).
Extracellular protease deletion mutants VT01 (ΔgelE), VT02 (ΔsprE), and VT03 (ΔgelE-sprE) (Fig. 1A) were constructed by using the markerless exchange vectors pVT01, pVT02, and pVT03, respectively. The respective plasmids were integrated into the V583 genome by homologous recombination. Subsequent plasmid excision was counterselected by plating on medium containing dl-p-chlorophenylalanine as described previously (25). Roughly 50% of the isolates growing in the presence of dl-p-chlorophenylalanine yielded the expected gene deletion for each of the plasmid constructs. The proteolytic phenotypes of the mutants were compared to V583 and were consistent with previous reports (24). Strains VT01 and VT03 lacked a zone of proteolysis on skim milk agar, whereas strain VT02 showed a smaller zone compared to V583 (data not shown).
FIG. 1.

Extracellular protease deletion mutations affect E. faecalis V583 biofilm development. (A) Diagrammatic depiction of extracellular protease deletions. VT01, VT02, and VT03 correspond to E. faecalis V583 strains harboring ΔgelE, ΔsprE, and ΔgelE-sprE protease deletions, respectively. Solid lines indicate chromosome, boxed arrows indicate genes, and curved arrows indicate promoter regions. The schematic is not drawn to scale. (B) Biofilm formation of extracellular protease mutants on polystyrene microtiter plates. The biofilm density within microtiter plate wells was assayed as a function of crystal violet stain retained by the biofilm biomass. Mutant strains complemented with gelE, sprE, and gelE-sprE are designated VT04, VT05, and VT06, respectively. Assays were performed in triplicate, and error bars indicate the standard error of the mean.
E. faecalis V583 isogenic protease mutants (VT01, VT02, and VT03) were complemented with full-length genes of gelE, sprE, and gelE-sprE in trans under the control of the native gelE promoter. Complementation confirmed that the protease-negative phenotypes were a result of targeted protease deletions and not due to polar effects of gene mutations elsewhere on the chromosome (data not shown).
Biofilm formation of E. faecalis V583 isogenic protease mutants.
Quantitative analysis of biofilms formed by the protease deletion mutants on polystyrene confirmed previous findings (20). VT01 (ΔgelE) and the double protease deletion strain VT03 (ΔgelE-sprE) were significantly reduced in biofilm biomass compared to strain V583 (Dunnett's test, P < 0.05) (Fig. 1B). Interestingly, deletion of sprE (VT02) marginally increased the biofilm biomass, although this did not appear to be statistically significant (Dunnett's test, P = 0.30). Complementation of the protease-negative strains restored biofilm formation to near wild-type levels, suggesting no polar effects for the deletion mutations (Fig. 1B).
Given the differences in biofilm biomass on polystyrene, we sought to determine whether mutant cells exhibited any differences in primary biofilm mat formation on a glass substrate. CLSM analysis of the structural and spatial organization of 24-h-old biofilms (Fig. 2) showed a dense and compact parental V583 biofilm (VT09). Consistent with our earlier observations, VT10 (ΔgelE) and VT12 (ΔgelE-sprE) displayed poor biofilms (decreased by ca. 60 and 50%, respectively, compared to VT09; Table 4) and was composed mainly of isolated and sparse distributions of cells on the glass surface. In contrast, biofilms of VT11 (ΔsprE) were more dense than those formed by the parental strain (increased by ca. 55%; see Table 4) and appeared to have a rugged, mountainous surface terrain consistent with an early initiation of microcolony development.
FIG. 2.

Confocal analysis of 1-day-old biofilms of E. faecalis wild type and isogenic protease deletion mutants. All strains constitutively expressed Gfp from pMV158GFP (see Materials and Methods) and were grown on glass coverslips in M17 medium. Panels A, B, C, and D are representative biofilm projections of VT09, VT10, VT11, and VT12, respectively. Below each panel is the z-projection for the corresponding image, and the depth of the biofilm is indicated by the height of the z-stack (see Table 4). The inset scale bar represents 10 μm.
TABLE 4.
COMSTAT analysis of wild-type and isogenic protease mutant biofilm images
| Day | Biofilm | Mean ± SD
|
||
|---|---|---|---|---|
| Biomass (μm3/μm2) | Mean thickness (μm) | Maximum thickness (μm) | ||
| 1 | VT09 | 6.7 ± 0.93 | 6.3 ± 0.98 | 6.3 ± 0.98 |
| VT10 | 2.6 ± 0.37 | 2.4 ± 0.41 | 2.45 ± 0.49 | |
| VT11 | 10.5 ± 0.33 | 10.1 ± 0.48 | 10.15 ± 0.49 | |
| VT12 | 3.0 ± 0.48 | 2.4 ± 0.49 | 2.45 ± 0.5 | |
| 4 | VT09 | 5.6 ± 0.38 | 7.9 ± 1.04 | 11.2 ± 0.0 |
| VT10 | 0.035 ± 0.04 | 0.022 ± 0.027 | 8 ± 2.26 | |
| VT11 | 11.61 ± 0.46 | 19.9 ± 0.01 | 20.8 ± 0.0 | |
| VT12 | 0.194 ± 0.04 | 0.28 ± 0.03 | 7.2 ± 2.26 | |
Extracellular proteases do not affect the cell surface hydrophobicity of E. faecalis V583.
To determine whether the ability of GelE to enhance biofilm formation resulted from an increase in overall cell surface hydrophobicity, we tested whether a ΔgelE mutation would decrease the overall hydrophobicity of cells. The assay was carried out by quantifying the population of bacteria that were able to separate into an organic phase (n-hexadecane) depending on the degree of cell surface hydrophobicity displayed. The presence or absence of either protease in both single- and double-deletion protease mutants did not result in significant differences in partitioning into the n-hexadecane phase relative to the wild-type V583 strain (Fig. 3, Dunnett's test, P < 0.05).
FIG. 3.

Cell surface hydrophobicity of E. faecalis V583 and extracellular protease mutants. The overall measure of hydrophobicity of wild-type and mutant populations were calculated as the percent bacteria that adhered to hydrocarbon (BATH). Assays were performed in triplicate, and error bars represent the standard error of the mean.
Extracellular proteases modify the rate of E. faecalis V583 autolysis.
Given the ability of enterococcal proteases to modify autolysins (48) and based on the observations seen using confocal imaging of biofilms, we hypothesized that GelE and SprE may differentially regulate the autolysis rates of E. faecalis. We observed that VT01 (ΔgelE) and VT03 (ΔgelE-sprE) exhibited a decrease in the rate of autolysis compared to V583 (Fig. 4A), a finding consistent with observations reported by Waters et al. (54). In contrast, VT02 (ΔsprE) displayed a significant increase in the rate of autolysis compared to V583 (Fig. 4A, Student t test, P < 0.05).
FIG. 4.

Extracellular proteases influence autolysis rates and eDNA release. (A) Differences in autolysis rates of V583 (•) and extracellular protease mutants VT01 (▪), VT02 (▴), and VT03 (⧫) are exhibited as percent values of the initial optical density at 600 nm (OD600). Assays were performed in quadruplicate, and error bars denote the standard error of the mean calculated from three independent assays. (B) High-molecular-weight bacterial chromosomal DNA was detected by ethidium bromide staining, after 20-fold concentration of 24-h-old culture supernatants. Lanes: 1, V583; 2, VT01; 3, VT02; 4, VT03; and 4, 1-kb DNA ladder showing the 12-, 10-, and 8-kb bands (the 12-kb band is labeled in lane 5).
eDNA in E. faecalis V583 culture supernatants.
Based on the altered rates of autolysis, we hypothesized that eDNA resulting from cell lysis would be more abundant in culture supernatants of E. faecalis V583 than mutants deficient in GelE production. Concentrated (20-fold) supernatant fractions were assessed for the presence of eDNA by agarose gel electrophoresis. High-molecular-weight DNA was detected in V583 and VT02 (ΔsprE) fractions (Fig. 4B, lanes 1 and 3), but not in mutants VT01 and VT03 (Fig. 4B, lanes 2 and 4), a finding consistent with a decreased rate of autolysis in strains lacking GelE. Densitometric determination of band intensity between DNA present in V583 and VT02 culture supernatants indicated a ∼2-fold increase in the amount of eDNA from an SprE− mutant, a finding consistent with a role for SprE as a negative regulator of autolysis. Initiation of DNA release in V583 culture supernatants followed expression of GelE in the transition to stationary phase (data not shown), a finding consistent with the earlier observation that GelE initiates autolysis. Finally, comparative PCR using eDNA and chromosomal DNA as templates confirmed that eDNA was indeed chromosomal in nature since amplification with primer pairs targeted to randomly distributed regions of the V583 genome could be amplified from both templates (data not shown).
Tracking cell death in enterococcal biofilms.
Because autolysis and biofilm formation of E. faecalis was directly dependent on the presence of gelatinase, we questioned whether biofilms formed by the parental strain would contain foci of lysed cells compared to VT01 (ΔgelE). To test this, biofilms of V583 and VT01 expressing Gfp (VT09 and VT10, respectively) were grown over a period of 3 days and were stained for the presence of DNA and dead cells with PI. Regions within the biofilm of VT09 contained concentrated foci of DNA (as detected by PI staining) in contrast to the few random dead cells in VT10 biofilms (Fig. 5). ImageJ analysis software was used to quantify the amount of PI-stained volumes within the biofilm as a measure of eDNA present in the biofilm. From this analysis, it is apparent that a common feature shared by both GelE− mutant and wild-type cell populations is the presence of damaged cells capable of taking up PI, and this cell population is accounted for in our analysis. A property unique to the wild-type cells compared to the GelE− mutant is the presence of larger volumes of PI staining associated with lysed cells. The mean values for PI-stained volumes is ∼4.4-fold higher in the wild-type strain (113.5 ± 59.88) than in the GelE− mutant (25.80 ± 5.09), and this was shown to be statistically significant (P = 0.0004) by using an unpaired t test, after data transformation, to account for the fact that stained volumes present in the V583 biofilms were not normally distributed compared to VT01 biofilms. A graph of this analysis is shown in Fig. 5C, and the z-stack image comparing V583 and VT01 biofilms stained with PI is also shown in Fig. 5. Collectively, these results suggest that GelE enhances biofilm formation by inducing lysis in discrete pockets of cells that appear to initiate biofilm development.
FIG. 5.
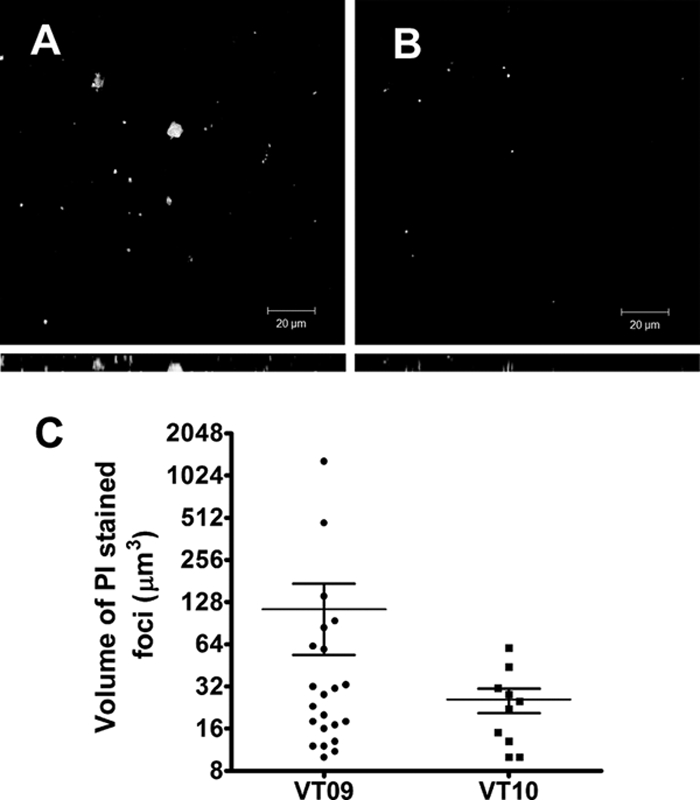
Bacterial cell death and eDNA release in 3-day-old biofilms. E. faecalis biofilms grown in M17 medium over a period of 3 days were stained with PI (1 μM) before being visualized by CLSM. (A) Top-down view of VT09 biofilm displaying discrete foci of lysed bacteria, along with dead bacterial cells. (B) View of isolated dead bacterial cells within the VT10 biofilm. Below each panel is the z-projection for the corresponding image, and the depth of the biofilm is indicated by the height of the z-stack. The inset scale bar represents 20 μm. (C) Volumetric analysis of PI-stained foci for VT09 and VT10 biofilms. Vertical scatter plots with each of the values of stained foci (cubic microns) are shown along with the mean and standard error of the mean.
Since the expression of GelE and SprE may only be optimally activated after the establishment of a quorum of bacteria on a surface, we hypothesized that we would see more prominent defects in the differentiation of the biofilm at later stages of development rather than the initial stages of attachment and proliferation. Consistent with this hypothesis, we observed that VT10 (ΔgelE) and VT12 (ΔgelE-sprE) were able to form a primary biofilm matt on a glass surface within 48 to 72 h of growth (data not shown). However, unlike the parental VT09 or VT11 (ΔsprE), even after 96 h of growth these two strains were not able to differentiate into microcolonies (Fig. 6). PI staining of the dead bacteria and eDNA in 4-day-old biofilms revealed clusters of dead bacteria around the base and stalk of a microcolony, whereas live bacteria interspersed with DNA frequently occupied the top of microcolonies within biofilms (Fig. 6). This suggested that pockets or clusters of dead cells that are dependent on the expression of GelE visualized at an earlier phase of biofilm development (Fig. 5) may actually be sites of initial microcolony development. Consistent with a role for SprE in negatively regulating GelE activity, we observed significantly more biofilm biomass (107% increase compared to the wild type) in an SprE− mutant after 96 h of growth than in the parental strain (Fig. 6 and Table 4).
FIG. 6.

Comparison of biofilm architectures and relative eDNA localization. Four-day-old Gfp-expressing strains of E. faecalis V583 and isogenic protease mutants were grown in M17 and stained for the presence of eDNA with PI (1 μM) as indicated in Materials and Methods. Live bacteria are green, and eDNA and dead cells are visualized in red. High concentrations of eDNA laced among live bacteria present on each raised microcolony and surroundings appear in shades of yellow. Panels A, B, C, and D are representative biofilm projections of VT09, VT10, VT11, and VT12, respectively. Below each panel is the z-projection for the corresponding image, and the depth of the biofilm is indicated by the height of the z-stack (see Table 4). The inset scale bar represents 20 μm.
Functional role of eDNA in enterococcal biofilms.
To determine whether eDNA of E. faecalis played a structural role in biofilm development, we analyzed the affect of DNase I on biofilm formation. Static biofilms grown on glass substrates were treated with DNase I after 6, 12, and 24 h of growth. Biofilm defects were most pronounced after early treatments of DNase I at 6 and 12 h of biofilm growth (Fig. 7). The affect of DNase I treatment at later stages of development was less significant, as exhibited by the 24-h treatment.
FIG. 7.

DNase I inhibits biofilm formation at early stages of development. V583 biofilms grown on glass coverslips were treated with DNase I after 6, 12, and 24 h of growth (represented in panels B, C, and D, respectively) and analyzed after 26 h by CLSM. The biofilm micrograph on the far left (panel A) shows a control experiment with heat-inactivated DNase I introduced after 6 h of biofilm development. Below each panel is the z-projection for the corresponding image, indicating the depth of the biofilm. The inset scale bar represents 20 μm.
DISCUSSION
The importance of extracellular proteases of E. faecalis in pathogenesis has been well demonstrated in a number of biological models (14, 15, 49, 50). Components of the host innate immune response are known to be cleaved by the proteolytic activity of gelatinase and include LL37 (45), α-defensin (46), and the complement components C3a and C3b (35), providing a mechanism for host immune evasion. GelE has also been shown to cleave fibrin, possibly enhancing efficient dissemination of the organism in vivo (54). Aside from its proteolytic affects on host factors, gelatinase has also been shown to have a positive role in E. faecalis biofilm development (20, 25). Hence, our present focus was to elucidate the mechanism behind GelE-dependent biofilm development and to further examine the role of SprE in that process.
A speculative role for GelE in biofilm development included its potential ability to increase cell surface hydrophobicity by cleaving surface polypeptides at hydrophobic residues (6, 27). Although cell surface hydrophobicity has previously been proposed to be a key factor in the initial attachment of bacteria to a substratum (12), our analysis of the different protease mutants does not support a role for GelE or SprE in altering cell surface hydrophobicity since the deletion of either protease singly or in tandem resulted in minimal changes. A second hypothesis centered on the ability of GelE to alter rates of autolysis, based on observations by Shockman and Cheney (48) and Waters et al. (54). Our data appear to confirm the importance of autolysis in driving the development of E. faecalis biofilms, since we observed altered rates of autolysis, changes in eDNA release, and differences in biofilm development in mutants defective in extracellular protease production. The contributions of both proteases to the process of biofilm development was readily observed only after confocal analysis. We did not initially observe a contribution for SprE in the microtiter plate biofilm assay. The apparent discrepancy between the two assays is consistent with observations reported by Tendolkar et al. (51) in which the plate assay significantly underestimated biofilm biomass compared to confocal imaging and COMSTAT analysis.
The major autolysin, AtlE of Staphylococcus epidermidis was recently shown to contribute to biofilm development through the generation of eDNA upon autolytic activation (40). A role for muramidase 2, a major autolysin of E. faecalis, in biofilm formation was reported by Mohamed et al. (30), and these authors concluded that it played a major role in the initial adherence phase of biofilm development. The findings reported by Qin et al. (40) that eDNA is an integral component of the biofilm matrix in S. epidermidis biofilms may warrant a reevaluation of the role of autolytic processes in biofilm development in E. faecalis. The observed alterations in eDNA release that are dependent on protease activity and appear to mediate the ability of E. faecalis to develop microcolonies within biofilms suggest that autolytic processes may govern not only initial attachment but also the subsequent development of the biofilm. Our findings have not only confirmed the role for GelE in activating autolysis since its deletion resulted in autolysis and biofilm defects but also provide direct evidence that SprE is involved in negatively regulating autolysis, eDNA release, and biofilm maturation.
Previous reports have identified and characterized SprE as a virulence factor whose activity is altered in the presence of GelE (24). This activity is similar in nature to that reported for the corresponding homologous extracellular proteases of S. aureus, where the metalloprotease aureolysin processes the cotranscribed SspA (V8 protease) (41). In S. aureus, SspA is known to alter the autolytic profile (41), which is consistent with our observations for the role of GelE and SprE in regulating autolysis. Because our data suggest that SprE prevents early maturation of biofilms by negatively regulating GelE activity, we postulated whether there would be a fitness cost associated with the bacterial cell in the absence of SprE. Our observations suggest that the quick biofilm maturation phenotype of VT02 is associated with cell surface perturbations that may be disadvantageous at a planktonic level of existence. For instance, the SprE− mutant is at least fourfold more sensitive to vancomycin compared to wild-type V583 (data not shown). Hence, it would seem that the trade-off for rapid biofilm development is costly and, in an evolutionary sense, unstable.
It has been observed in several model systems that eDNA serves as an important matrix component of microbial biofilms (2, 40, 42, 57). Consistent with a role for eDNA as a matrix component, we observed that treating a developing biofilm with DNase I at 6 and 12 h postinoculation resulted in diminished biofilm accumulation compared to a heat-inactivated DNase I control. In contrast, the addition of DNase I at 24 h showed only a marginal reduction in biofilm accumulation, suggesting that changes in the matrix composition may take place at later stages of development. Consistent with our findings, the observation that disrupting biofilms with DNase I treatment works better at earlier stages of development has been reported for Pseudomonas aeruginosa (57) and S. aureus (42) biofilms.
Although the factors regulating the spatial death of a subpopulation of bacterial cells in a biofilm are not clear, the extracellular nature of the proteases and their opposing phenotypes may play a role in this process. Our current model (Fig. 8) proposes two possible means by which these proteases may exert their regulatory affects on biofilm development. The first mechanism involves an autolytic pathway, wherein GelE localizes to the cell wall of the producing cell to activate autolysis. If insufficient levels of SprE are present to control the autolytic activation induced by GelE, then that cell will likely undergo autolysis. The second mechanism would involve an allolytic or fratricidal event, wherein GelE freely diffuses from the producer cell to a target sibling cell to activate autolysins present on the sibling cell wall. A delay in responding to the quorum signal by siblings would render them susceptible to the action of autolysins activated by GelE secreted from another cell. SprE would also likely be present in this extracellular environment, but differences in diffusion and affinity for the cell wall may likely give rise to regions in the biofilm where GelE could act independently of SprE activity. In the rare instances in which GelE would function independently of SprE, a sibling cell would lyse providing the necessary eDNA scaffold on which a developing biofilm could form. Consistent with the above model is the fact that only a few pockets within the observed biofilms give rise to cell lysis, which is indicative of the fact that the process is highly regulated.
FIG. 8.

Model of GelE-mediated lysis in E. faecalis biofilm development. The model presents two mechanisms by which GelE could mediate lytic activity. The first mechanism is referred to as autolysis (A), and gelatinase ( ) from the producer cell could activate a putative autolysin (▴) on the cell surface, resulting in autolysis. The presence of SprE (
) from the producer cell could activate a putative autolysin (▴) on the cell surface, resulting in autolysis. The presence of SprE ( ) is predicted to regulate the GelE-mediated autolysin activation. The second mechanism, referred to as fratricide (allolysis) (B), allows for the diffusion of GelE () from the producer cell (A) to a susceptible sibling (B), wherein the sibling cell undergoes lysis following autolysin (▴) activation by GelE. The extent of bystander or sibling lysis would potentially be regulated by the presence of SprE () in the environment. The mechanism of SprE-mediated regulation is unknown but may involve alteration of the putative autolysin, rendering it to an inactive form (▪).
) is predicted to regulate the GelE-mediated autolysin activation. The second mechanism, referred to as fratricide (allolysis) (B), allows for the diffusion of GelE () from the producer cell (A) to a susceptible sibling (B), wherein the sibling cell undergoes lysis following autolysin (▴) activation by GelE. The extent of bystander or sibling lysis would potentially be regulated by the presence of SprE () in the environment. The mechanism of SprE-mediated regulation is unknown but may involve alteration of the putative autolysin, rendering it to an inactive form (▪).
In recent times, bacterial death in biofilms has been compared to programmed cell death in eukaryotes (4). Often such comparisons propagate the idea that defective cells within a biofilm population are eliminated in response to environmental challenges due to their altruistic suicidal acts (4). Our model adds to this complexity by proposing that GelE-mediated lysis appears to be an important aspect of biofilm development by E. faecalis. The cytotoxic activity of GelE toward the producer cell (autolysis) or sibling cells (allolysis) by the activation of autolysins may result in the release of eDNA crucial for the early development of biofilms. Although in this case a subpopulation of cells may not be defective per se, their inability to produce the immunity factor (SprE) would result in their death. Allolysis has also recently been referred to as microbial fratricide (“sibling killing sibling”), and this term has been applied to the competence developmental program in Streptococcus pneumoniae (18), and a model was proposed on how this process might contribute to the development of biofilms (16). Allolysis (18) and cannibalism (17) regulate the differentiation of competent cells in S. pneumoniae and sporulation in Bacillus subtilis, respectively. Consistent with these fratricidal systems, a model for how fratricide in E. faecalis regulates the development of biofilms is also proposed (Fig. 8). Interestingly, all three processes of differentiation may be considered attributes of multicellularity resulting from cell-cell communication and involve quorum sensing, killing factors, and immunity proteins (8, 13). In E. faecalis biofilm development, quorum sensing is mediated through a peptide lactone (FsrD) originally characterized as the gelatinase biosynthesis-activating pheromone (31). The extracellular accumulation of the peptide triggers expression of both GelE (the effector) and SprE (the regulator). Both proteases are cotranscribed, suggesting an equal number of both molecules in the extracellular milieu. For this reason, we anticipate that most cells would be protected from autolysis or allolysis. However, within discrete foci, the balance of these two proteases may not be the same, giving rise to effector-mediated processes in the absence of regulatory control. Ongoing studies will better clarify which of the two mechanisms (autolysis versus fratricide) plays the dominant role in E. faecalis biofilm development.
Acknowledgments
We thank Gary Dunny and Chris Kristich (University of Minnesota) for plasmid pCJK47 and Arne Heydorn (Technical University of Denmark, Kongens Lyngby) for the COMSTAT software. We also thank Helmut Hirt for a critical review of the manuscript and helpful comments in the preparation of the manuscript.
This study was supported by a Heartland Affiliate Beginning Grant-in-Aid 0660072Z from the American Heart Association (to L.E.H.) and a grant-in-aid from the Terry C. Johnson Cancer Center at Kansas State University (V.C.T.).
Footnotes
Published ahead of print on 13 June 2008.
REFERENCES
- 1.Acebo, P., C. Nieto, M. A. Corrales, M. Espinosa, and P. Lopez. 2000. Quantitative detection of Streptococcus pneumoniae cells harbouring single or multiple copies of the gene encoding the green fluorescent protein. Microbiology 146(Pt. 6)1267-1273. [DOI] [PubMed] [Google Scholar]
- 2.Allesen-Holm, M., K. B. Barken, L. Yang, M. Klausen, J. S. Webb, S. Kjelleberg, S. Molin, M. Givskov, and T. Tolker-Nielsen. 2006. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 591114-1128. [DOI] [PubMed] [Google Scholar]
- 3.Arciola, C. R., L. Baldassarri, D. Campoccia, R. Creti, V. Pirini, J. Huebner, and L. Montanaro. 2008. Strong biofilm production, antibiotic multi-resistance, and high gelE expression in epidemic clones of Enterococcus faecalis from orthopaedic implant infections. Biomaterials 29580-586. [DOI] [PubMed] [Google Scholar]
- 4.Bayles, K. W. 2007. The biological role of death and lysis in biofilm development. Nat. Rev. Microbiol. 5721-726. [DOI] [PubMed] [Google Scholar]
- 5.Ben Jacob, E., I. Becker, Y. Shapira, and H. Levine. 2004. Bacterial linguistic communication and social intelligence. Trends Microbiol. 12366-372. [DOI] [PubMed] [Google Scholar]
- 6.Carniol, K., and M. S. Gilmore. 2004. Signal transduction, quorum-sensing, and extracellular protease activity in Enterococcus faecalis biofilm formation. J. Bacteriol. 1868161-8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cetinkaya, Y., P. Falk, and C. G. Mayhall. 2000. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 13686-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claverys, J. P., and L. S. Havarstein. 2007. Cannibalism and fratricide: mechanisms and raisons d'etre. Nat. Rev. Microbiol. 5219-229. [DOI] [PubMed] [Google Scholar]
- 9.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 2841318-1322. [DOI] [PubMed] [Google Scholar]
- 10.Cruz-Rodz, A. L., and M. S. Gilmore. 1990. High efficiency introduction of plasmid DNA into glycine treated Enterococcus faecalis by electroporation. Mol. Gen. Genet. 224152-154. [DOI] [PubMed] [Google Scholar]
- 11.Del Papa, M. F., L. E. Hancock, V. C. Thomas, and M. Perego. 2007. Full activation of Enterococcus faecalis gelatinase by a C-terminal proteolytic cleavage. J. Bacteriol. 1898835-8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donlan, R. M., and J. W. Costerton. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15167-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellermeier, C. D., E. C. Hobbs, J. E. Gonzalez-Pastor, and R. Losick. 2006. A three-protein signaling pathway governing immunity to a bacterial cannibalism toxin. Cell 124549-559. [DOI] [PubMed] [Google Scholar]
- 14.Engelbert, M., E. Mylonakis, F. M. Ausubel, S. B. Calderwood, and M. S. Gilmore. 2004. Contribution of gelatinase, serine protease, and fsr to the pathogenesis of Enterococcus faecalis endophthalmitis. Infect. Immun. 723628-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garsin, D. A., C. D. Sifri, E. Mylonakis, X. Qin, K. V. Singh, B. E. Murray, S. B. Calderwood, and F. M. Ausubel. 2001. A simple model host for identifying gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 9810892-10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilmore, M. S., and W. Haas. 2005. The selective advantage of microbial fratricide. Proc. Natl. Acad. Sci. USA 1028401-8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Pastor, J. E., E. C. Hobbs, and R. Losick. 2003. Cannibalism by sporulating bacteria. Science 301510-513. [DOI] [PubMed] [Google Scholar]
- 18.Guiral, S., T. J. Mitchell, B. Martin, and J. P. Claverys. 2005. Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: genetic requirements. Proc. Natl. Acad. Sci. USA 1028710-8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall-Stoodley, L., J. W. Costerton, and P. Stoodley. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 295-108. [DOI] [PubMed] [Google Scholar]
- 20.Hancock, L. E., and M. Perego. 2004. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 1865629-5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heydorn, A., A. T. Nielsen, M. Hentzer, C. Sternberg, M. Givskov, B. K. Ersboll, and S. Molin. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146(Pt. 10)2395-2407. [DOI] [PubMed] [Google Scholar]
- 22.Huycke, M. M., D. F. Sahm, and M. S. Gilmore. 1998. Multiple-drug resistant enterococci: the nature of the problem and an agenda for the future. Emerg. Infect. Dis. 4239-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jefferson, K. K. 2004. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 236163-173. [DOI] [PubMed] [Google Scholar]
- 24.Kawalec, M., J. Potempa, J. L. Moon, J. Travis, and B. E. Murray. 2005. Molecular diversity of a putative virulence factor: purification and characterization of isoforms of an extracellular serine glutamyl endopeptidase of Enterococcus faecalis with different enzymatic activities. J. Bacteriol. 187266-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristich, C. J., J. R. Chandler, and G. M. Dunny. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57131-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maguin, E., P. Duwat, T. Hege, D. Ehrlich, and A. Gruss. 1992. New thermosensitive plasmid for gram-positive bacteria. J. Bacteriol. 1745633-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makinen, P. L., D. B. Clewell, F. An, and K. K. Makinen. 1989. Purification and substrate specificity of a strongly hydrophobic extracellular metalloendopeptidase (“gelatinase”) from Streptococcus faecalis (strain 0G1-10). J. Biol. Chem. 2643325-3334. [PubMed] [Google Scholar]
- 28.Makinen, P. L., and K. K. Makinen. 1994. The Enterococcus faecalis extracellular metalloendopeptidase (EC 3.4.24.30; coccolysin) inactivates human endothelin at bonds involving hydrophobic amino acid residues. Biochem. Biophys. Res. Commun. 200981-985. [DOI] [PubMed] [Google Scholar]
- 29.Mohamed, J. A., and D. B. Huang. 2007. Biofilm formation by enterococci. J. Med. Microbiol. 561581-1588. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed, J. A., W. Huang, S. R. Nallapareddy, F. Teng, and B. E. Murray. 2004. Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect. Immun. 723658-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakayama, J., Y. Cao, T. Horii, S. Sakuda, A. D. Akkermans, W. M. de Vos, and H. Nagasawa. 2001. Gelatinase biosynthesis-activating pheromone: a peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol. Microbiol. 41145-154. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama, J., S. Chen, N. Oyama, K. Nishiguchi, E. A. Azab, E. Tanaka, R. Kariyama, and K. Sonomoto. 2006. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheromone propeptide corresponding to staphylococcal agrD. J. Bacteriol. 1888321-8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama, J., R. Kariyama, and H. Kumon. 2002. Description of a 23.9-kilobase chromosomal deletion containing a region encoding fsr genes which mainly determines the gelatinase-negative phenotype of clinical isolates of Enterococcus faecalis in urine. Appl. Environ. Microbiol. 683152-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33a.Nallapareddy, S. R., K. V. Singh, J. Sillanpää, D. A. Garsin, M. Höök, S. L. Erlandsen, and B. E. Murray. 2006. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Investig. 1162799-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nieto, C., and M. Espinosa. 2003. Construction of the mobilizable plasmid pMV158GFP, a derivative of pMV158 that carries the gene encoding the green fluorescent protein. Plasmid 49281-285. [DOI] [PubMed] [Google Scholar]
- 35.Park, S. Y., K. M. Kim, J. H. Lee, S. J. Seo, and I. H. Lee. 2007. Extracellular gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph and human serum. Infect. Immun. 751861-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pillai, S. K., G. Sakoulas, G. M. Eliopoulos, R. C. Moellering, Jr., B. E. Murray, and R. T. Inouye. 2004. Effects of glucose on fsr-mediated biofilm formation in Enterococcus faecalis. J. Infect. Dis. 190967-970. [DOI] [PubMed] [Google Scholar]
- 37.Pospiech, A., and B. Neumann. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11217-218. [DOI] [PubMed] [Google Scholar]
- 38.Qin, X., K. V. Singh, G. M. Weinstock, and B. E. Murray. 2001. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J. Bacteriol. 1833372-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin, X., K. V. Singh, G. M. Weinstock, and B. E. Murray. 2000. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect. Immun. 682579-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin, Z., Y. Ou, L. Yang, Y. Zhu, T. Tolker-Nielsen, S. Molin, and D. Qu. 2007. Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology 1532083-2092. [DOI] [PubMed] [Google Scholar]
- 41.Rice, K., R. Peralta, D. Bast, J. de Azavedo, and M. J. McGavin. 2001. Description of staphylococcus serine protease (ssp) operon in Staphylococcus aureus and nonpolar inactivation of sspA-encoded serine protease. Infect. Immun. 69159-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice, K. C., E. E. Mann, J. L. Endres, E. C. Weiss, J. E. Cassat, M. S. Smeltzer, and K. W. Bayles. 2007. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 1048113-8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenberg, M., A. Perry, E. A. Bayer, D. L. Gutnick, E. Rosenberg, and I. Ofek. 1981. Adherence of Acinetobacter calcoaceticus RAG-1 to human epithelial cells and to hexadecane. Infect. Immun. 3329-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandoe, J. A., J. Wysome, A. P. West, J. Heritage, and M. H. Wilcox. 2006. Measurement of ampicillin, vancomycin, linezolid, and gentamicin activity against enterococcal biofilms. J. Antimicrob. Chemother. 57767-770. [DOI] [PubMed] [Google Scholar]
- 45.Schmidtchen, A., I. M. Frick, E. Andersson, H. Tapper, and L. Bjorck. 2002. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46157-168. [DOI] [PubMed] [Google Scholar]
- 46.Schmidtchen, A., I. M. Frick, and L. Bjorck. 2001. Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivates antibacterial alpha-defensin. Mol. Microbiol. 39708-713. [DOI] [PubMed] [Google Scholar]
- 47.Shankar, N., A. S. Baghdayan, and M. S. Gilmore. 2002. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417746-750. [DOI] [PubMed] [Google Scholar]
- 48.Shockman, G. D., and M. C. Cheney. 1969. Autolytic enzyme system of Streptococcus faecalis. V. Nature of the autolysin-cell wall complex and its relationship to properties of the autolytic enzyme of Streptococcus faecalis. J. Bacteriol. 981199-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sifri, C. D., E. Mylonakis, K. V. Singh, X. Qin, D. A. Garsin, B. E. Murray, F. M. Ausubel, and S. B. Calderwood. 2002. Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect. Immun. 705647-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh, K. V., X. Qin, G. M. Weinstock, and B. E. Murray. 1998. Generation and testing of mutants of Enterococcus faecalis in a mouse peritonitis model. J. Infect. Dis. 1781416-1420. [DOI] [PubMed] [Google Scholar]
- 51.Tendolkar, P. M., A. S. Baghdayan, M. S. Gilmore, and N. Shankar. 2004. Enterococcal surface protein, Esp, enhances biofilm formation by Enterococcus faecalis. Infect. Immun. 726032-6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51a.Tendolkar, P. M., A. S. Baghdayan, and N. Shankar. 2006. Putative surface proteins encoded within a novel transferable locus confer a high-biofilm phenotype to Enterococcus faecalis. J. Bacteriol. 1882063-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toledo-Arana, A., J. Valle, C. Solano, M. J. Arrizubieta, C. Cucarella, M. Lamata, B. Amorena, J. Leiva, J. R. Penades, and I. Lasa. 2001. The enterococcal surface protein, Esp, is involved in Enterococcus faecalis biofilm formation. Appl. Environ. Microbiol. 674538-4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trieu-Cuot, P., C. Carlier, C. Poyart-Salmeron, and P. Courvalin. 1990. A pair of mobilizable shuttle vectors conferring resistance to spectinomycin for molecular cloning in Escherichia coli and in gram-positive bacteria. Nucleic Acids Res. 184296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waters, C. M., M. H. Antiporta, B. E. Murray, and G. M. Dunny. 2003. Role of the Enterococcus faecalis GelE protease in determination of cellular chain length, supernatant pheromone levels, and degradation of fibrin and misfolded surface proteins. J. Bacteriol. 1853613-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waters, C. M., and B. L. Bassler. 2005. Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21319-346. [DOI] [PubMed] [Google Scholar]
- 56.Weigel, L. M., D. B. Clewell, S. R. Gill, N. C. Clark, L. K. McDougal, S. E. Flannagan, J. F. Kolonay, J. Shetty, G. E. Killgore, and F. C. Tenover. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 3021569-1571. [DOI] [PubMed] [Google Scholar]
- 57.Whitchurch, C. B., T. Tolker-Nielsen, P. C. Ragas, and J. S. Mattick. 2002. Extracellular DNA required for bacterial biofilm formation. Science 2951487. [DOI] [PubMed] [Google Scholar]