

Abstract
Francisella tularensis, a small gram-negative intracellular bacterium responsible for causing tularemia, is highly pathogenic and classified as a category A agent of bioterrorism. As for other intracellular pathogens, successful protective immune responses to Francisella tularensis require rapid and efficient induction of gamma interferon (IFN-γ) production. Studies using intracellular bacteria such as Listeria monocytogenes as well as Francisella suggest that natural killer (NK) and T cells are important sources of IFN-γ. However, comprehensive characterization of specific sources of IFN-γ produced during Francisella infection in vivo remains incomplete, and depletion of NK cells before infection of mice with the F. tularensis live vaccine strain (LVS) has little impact on the course or outcome of infection. In this study, we determined the cell subpopulations that respond quickly to intradermal F. tularensis LVS infection of mice by producing IFN-γ within hours to a few days. Splenic and liver lymphocytes were obtained from LVS-infected mice and analyzed for IFN-γ mRNA by reverse transcription-PCR, for intracellular cytokine expression by multiparameter flow cytometry, and for ex vivo production of IFN-γ protein by enzyme-linked immunosorbent assay. Cells producing IFN-γ were readily detectable by day 3 after infection, and numbers progressively increased through days 5 to 7. Importantly, the cell types responsible for IFN-γ production were much more varied than expected: these included not only NK cells and T cells, which might be predicted, but also other cells, including dendritic cells (DCs), “NK DCs,” NK T cells, and neutrophils. Most importantly, since RAG-1 knockout mice appeared to exhibit a frequency of IFN-γ-producing cells comparable to that of intact wild-type mice, early IFN-γ production by innate immune cells does not depend on the presence of T or B cells.
Francisella tularensis is a small gram-negative bacterium responsible for causing tularemia, a zoonosis that occasionally affects humans (35). Human tularemia occurs in Eurasia and North America (39), and in the United States the incidence is about 200 cases per year (7). Contact, ingestion, or inhalation provides the route of infection, and when the disease is left untreated, the mortality rate of respiratory disease is up to 30% (16). The ability to cause severe disease by the airborne route, coupled with the low infectious dose (<10 CFU), makes F. tularensis a potential agent of bioterrorism (15), and it has been classified by the U.S. Centers for Disease Control as a category A agent (http://www.bt.cdc.gov/agent/agentlist-category.asp).
As an occasional disease, appropriately diagnosed tularemia is easily controlled by antibiotic therapy, but vaccine development is of interest to ensure protection against endemic disease or a possible biological attack. Currently, only live attenuated vaccines have been shown to provide substantial protection against infection with fully virulent F. tularensis in animals (20, 38) and probably humans (11, 38, 49). One such vaccine, the live vaccine strain (LVS) derived from type B F. tularensis, has shown some effectiveness in humans and in animal models (5, 20, 41, 42). In mice, LVS is attenuated only when it is given intradermally (i.d.), with a 50% lethal dose on the order of 106 CFU, but the 50% lethal dose is 10° to 101 CFU when the vaccine is given intraperitoneally or intravenously (22, 26). Murine survival of a sublethal vaccinating dose of LVS provides specific protective immunity that is T cell mediated (13, 55), similar to that observed in humans (49). Innate immune responses, however, provide initial control of F. tularensis LVS replication for 2 to 3 weeks, as illustrated by the observation that severe combined immunodeficiency (SCID) mice lacking all lymphocytes survive i.d. LVS infection for more than 20 days (23).
In infections with many intracellular pathogens, early production of gamma interferon (IFN-γ) is a key innate immune mechanism that limits initial bacterial replication, presumably permitting and directing development of adaptive immune responses (18, 29, 30). Similar to the case with other intracellular bacteria, IFN-γ knockout (KO) mice die within 5 to 7 days after a normally sublethal i.d. infection with LVS. Furthermore, if anti-IFN-γ is administered before or within 2 days after infection, C57BL/6J mice, athymic mice, T-cell KO mice, and SCID mice die 5 to 7 days after i.d. LVS infection (23). IFN-γ mRNA is detectable in spleen cells by 2 days after intraperitoneal or i.d. infection (9, 47), and IFN-γ protein is detectable by enzyme-linked immunosorbent assay (ELISA) in sera and in organ homogenates of i.d. LVS-infected mice within 2 to 3 days (21). In murine listeriosis, activated natural killer (NK) cells, NK T cells, and recently described “NK dendritic cells (NK DCs)” or “IK DCs” appear to be prominent sources of IFN-γ (8, 40, 50, 51), while under certain conditions in vitro macrophages (36) and traditional DCs (27) may produce IFN-γ. B cells (28) and neutrophils (3) have also been reported as a possible source of IFN-γ. The early activation of NK cells appears to be at least partially responsible for IFN-γ expression in lungs after respiratory infection with LVS (33), and liver NK cells stimulated with LVS ex vivo produce IFN-γ (54). Surprisingly, however, depletion of NK cells prior to sublethal infection with LVS i.d. (31) or lethal infection with LVS intranasally (33) has little impact on the course of infection. Furthermore, in vitro studies on the mechanisms by which CpG oligonucleotides provide short-term protection against lethal LVS challenge indicate a clear role for NK cells in controlling intramacrophage LVS growth, but in vivo NK cell depletion failed to ablate CpG-stimulated protection (K. Elkins et al., submitted for publication). Overall, studies of the cellular sources of IFN-γ during infection with intracellular bacteria have often focused on individual cell types and relied heavily on in vitro approaches. These considerations, coupled with interest in the overall role of NK cells in host responses to Francisella infection, prompted a detailed and comprehensive study of all the in vivo cellular sources of IFN-γ following Francisella LVS infection. Thus, reverse transcription-PCR (RT-PCR), multiparameter flow cytometry combined with intracellular cytokine staining, and protein assessment by ELISA were used to directly characterize in vivo activated splenic cells and liver leukocytes that produce IFN-γ within a few days in response to i.d. LVS vaccination. Our data indicate that the cell types responsible for innate, lymphocyte-independent IFN-γ production are highly diversified and include not only traditional NK cells and T cells but also neutrophils, DCs, NK T cells, and hybrid “NK DCs.”
MATERIALS AND METHODS
Experimental animals.
Six- to 12-week-old specific-pathogen-free male C57BL/6J and B6.129S7-Rag1<tm1Mom>/J mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice were housed in sterile microisolator cages in a barrier environment at CBER/FDA, fed autoclaved food and water ad libitum, and routinely tested for common murine pathogens by a diagnostic service provided by the Division of Veterinary Services, CBER. Within an experiment, all mice were age matched. All experiments were performed under protocols approved by the Animal Care and Use Committee of CBER.
Bacteria and growth conditions.
F. tularensis LVS (ATCC 29684) was grown to mid-log phase in modified Mueller-Hinton (MH) broth (Difco Laboratories, Detroit, MI), as previously described (2, 26), harvested, and frozen in 1-ml aliquots in broth alone at −70°C. Bacteria were periodically thawed for use, and viability was quantified by plating serial dilutions on MH agar plates. There was <10% variation from the original CFU.
Bacterial infections.
Groups of mice were immunized by i.d. injection with 1 × 105 CFU LVS diluted in 0.1 ml phosphate-buffered saline (PBS; BioWhittaker, Walkersville, MD) containing <0.01 ng of endotoxin/ml and were sacrificed for analysis on the indicated days. Actual doses of inoculated bacteria were simultaneously determined by plate counts; control groups received 0.1 ml PBS i.d.
Determination of bacterial organ burdens.
Determinations of numbers of CFU in organs of various mice were performed as previously described (26). Briefly, spleens and livers were removed aseptically. Spleens were disrupted with a 3-ml syringe plunger in 10 ml of sterile PBS-2% fetal bovine serum, while livers were emulsified in a stomacher (Tekmar, Cincinnati, OH) in 10 ml of sterile PBS. Appropriate dilutions were plated on MH plates. After 2 days of incubation at 37°C and 5% CO2, bacterial CFU were counted and the results adjusted according to the dilution factors used.
Preparation of splenocytes and liver leukocytes.
Disrupted spleens were used to prepare single-cell suspensions, and erythrocytes were lysed with ammonium chloride (ACK lysing buffer; BioWhittaker). Cells were washed, and viability was assessed by exclusion of trypan blue. For liver leukocyte preparations, emulsified livers were washed three times with room temperature PBS. Cells were collected and suspended in 2.5 ml/liver of 40% Percoll (Sigma, St. Louis, MO). After centrifugation at 1,400 rpm for 20 min at room temperature, the top layer was removed, the interface was collected, and the cells were resuspended in PBS-2% fetal bovine serum. Erythrocytes were lysed with ACK lysing buffer, cells were washed, and viability was assessed by exclusion of trypan blue.
Real-time PCR.
Total RNA was extracted from tissue samples and from single-cell suspensions. Fragments of harvested spleens and livers were obtained, submerged in RNA stabilization solution (RNAlater; Ambion, Austin, TX), and stored at −20°C. Similarly, 1 × 106 to 5 × 107 cells from spleen and liver cell preparations were suspended in RNAlater and stored at −20°C until further characterization. RNA was purified using commercial kits for tissue samples (RNeasy Midi kit; Qiagen, Valencia, CA) and for cell suspensions (RNeasy Mini kit; Qiagen), performed according to the manufacturer's directions. One to two micrograms of RNA was used to synthesize cDNA, using the commercially available SuperScript first-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Semiquantitative real-time PCR amplification was completed with an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). One microliter of each cDNA was diluted to a volume of 25 μl of PCR mix (Applied Biosystems) containing 0.1 μM and 0.2 μM of each primer and probe, respectively. IFN-γ-specific primer and probe sequences were as follows: forward primer, 5′-AGCAACAGCAAGGCGAAAA; reverse primer, 5′-CTGGACTGTGGGTTGTTGA; and probe, 5′-6-carboxyfluorescein-CCTCAAACTTGGCAATACTCATGAATGCATCC-6-carboxytetramethylrhodamine. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Taqman reagents (Applied Biosystems) were used to measure GAPDH mRNA levels as an internal standard. For amplification, the activation of uracil-DNA glycosylase at 50°C for 2 min and the initial denaturation at 95°C for 10 min were followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 min. The level of IFN-γ mRNA relative to the GAPDH mRNA concentration was calculated by using the following formula: relative mRNA expression = 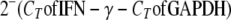 , where CT represents the crossing point (the beginning of the PCR exponential phase).
, where CT represents the crossing point (the beginning of the PCR exponential phase).
In vitro cell cultures and ELISA.
The assay for monitoring IFN-γ production in vitro was carried out by a previously described procedure (10). Briefly, 5 × 106 splenocytes and liver leukocytes were cultured in 24-well plates for 3 days in complete Dulbecco's modified Eagle's medium (DMEM; BioWhittaker), defined as DMEM containing 10% fetal calf serum (HyClone, Logan, UT), 1% glutamine (Lonza, Allendale, NJ), 1% HEPES (BioWhittaker), 1% sodium bicarbonate (BioWhittaker), 1% sodium pyruvate (BioWhittaker), and 1% nonessential amino acids (BioWhittaker). Culture supernatants were assayed using standard sandwich ELISAs according to the manufacturer's instructions (BD Pharmingen, San Diego, CA). The absorbance was read at 405 nm on a VersaMax tunable microplate reader with a reference wavelength of 630 nm (Molecular Devices, Sunnyvale, CA). Cytokines were quantitated by comparison to recombinant standard proteins (BD Pharmingen), using four-parameter fit regression in SOFTmax Pro ELISA analysis software (Molecular Devices). The limit of detection for IFN-γ was approximately 50 pg/ml.
Flow cytometry.
Single-cell suspensions were prepared from splenocytes and liver leukocytes as described above. Essentially all cells obtained by these methods were CD45+ leukocytes. Cells were resuspended in complete DMEM supplemented with 10 μg/ml brefeldin A (Sigma) and incubated for 4 h. Because bacteria were present in cell preparations at all time points studied (see Fig. 1A), no additional stimuli were added exogenously during the incubation period. Cells were harvested from the dish, washed, and resuspended in flow cytometry buffer (PBS-2% serum [FB]). Nonspecific binding of antibodies was inhibited by blocking Fc receptors with anti-CD16 (Fc block; BD Pharmingen) for 10 min on ice. To discriminate live from dead cells, a staining step for dead cells was performed using a commercially available kit following the manufacturer's instructions (Live/Dead staining kit; Invitrogen). The cells were then washed in FB and stained for cell surface markers, as previously described (4, 55). Antibody concentrations were previously optimized for use in seven- to nine-color staining protocols, as required, using appropriate fluorochrome-labeled isotype-matched control antibodies. The following antibodies were used: anti-CD45/B220 (clone RA3-6B2), anti-CD19 (clone 1D3), anti-T-cell receptor β (anti-TCRβ; clone H57-597), anti-CD45 (clone 30-F11), anti-CD4 (clone RM4-5), anti-CD8β (H35-17.2), anti-NK1.1 (clone PK136), anti-CD11b (clone M1/70), anti-Thy1.2 (clone 30-H12), anti-Gr1 (clone RB6-8C5), anti-CD11c (cloneHL3), anti-CD80 (clone 16-10A1), anti-CD86 (clone H1.2F3), and anti-CD40 (clone 3/23) (all above antibodies were purchased from BD Pharmingen). Anti-F4/80 (clone 704; Serotec, Raleigh, NC) and anti-I-A/I-E (clone M5/114.15.2; Biolegend, San Diego, CA) were also used for surface staining. For intracellular staining (ICS) of IFN-γ, cells were fixed for 20 min with 2% paraformaldehyde (EMS, Hatfield, PA), washed, permeabilized with saponin, and stained with anti-IFN-γ (clone XMG1.2; BD Pharmingen). After 30 min of incubation, cells were washed in FB and fixed in 0.5% paraformaldehyde. Ten to thirty thousand total events were counted using an analytical LSR II flow cytometer (Becton Dickinson). Data analyses were performed using FlowJo (Tree Star, Inc.) software, v7.1.3. The starting point for all analyses was live (Pacific Blue negative) CD45+ cells, with aggregates excluded by comparison of forward scatter A (FSC-A) versus FSC-H.
FIG. 1.

Increases in numbers of splenic and liver leukocytes follow the bacterial organ burdens after LVS infection of mice. C57BL/6J mice were infected with 105 CFU of LVS i.d. (A) CFU were estimated in homogenized spleens (black bars) and livers (hatched bars) by conventional plate counts. (B) Pooled single-cell suspensions of spleen (black bars) and liver (hatched bars) cells were prepared from five mice at the indicated time points. Viability was assessed by exclusion of trypan blue; only viable cell numbers are shown. Results shown were derived from a single representative experiment of three total experiments of similar design. Between experiments, numbers of CFU varied 30% or less, and numbers of viable murine cells varied <20%.
In vitro cell purification.
A MACS Midi system and the appropriate magnetic beads (Miltenyi Biotec, Auburn, CA) were used to enrich cell subpopulations according to standard protocols. Anti-CD19 beads were used to enrich B cells, anti-CD11c beads were used to enrich DCs, anti-DX5 beads were used to enrich NK cells, anti-Thy1.2 beads were used to enrich T cells, and anti-CD11b beads were used to enrich neutrophils, macrophages, and subpopulations of DCs and NK cells. Single-cell suspensions of splenocytes obtained from either LVS-infected or control mice were treated with the appropriate amount of magnetic beads according to the manufacturer's instructions. The composition and relative purity of the resulting enriched cells were assessed by multiparameter flow cytometry.
RESULTS
Characterization of total IFN-γ production profiles in spleen and liver following vaccinating LVS infection.
To characterize the time course and extent of development of the early innate IFN-γ response, wild-type (WT) C57BL/6J mice were infected i.d. with F. tularensis LVS and sacrificed on days 1, 3, 5, and 7. Spleens and livers were harvested to analyze total leukocyte numbers, bacterial burdens, and ex vivo IFN-γ production. In both spleens and livers, bacterial CFU peaked at 1 × 106 to 2 × 106 CFU/organ after 3 days of LVS infection and declined thereafter; bacteria were cleared from spleens somewhat faster than from livers (Fig. 1A). In parallel with the quantitation of infection as reflected by the CFU, numbers of eukaryotic leukocytes in organs were determined. Spleens from naive mice averaged 4 × 107 to 8 × 107 cells per spleen, while numbers of recovered liver leukocytes were 2 × 106 to 7 × 106, on average (Fig. 1B). The average cell number in spleen or liver did not change appreciably during the first 3 days of LVS infection. By 7 days after LVS infection, however, total cell numbers in spleens increased twofold. In livers, even more dramatic increases were found: after 5 days of infection, cell numbers were about fourfold higher than those for naive mice, and after 7 days, they were about ninefold higher. These increases were accompanied by changes in the cellular composition of spleens and livers. In particular, the relative proportions of NK cells, DCs, and T cells in livers increased (data not shown).
To evaluate the expression of IFN-γ mRNA in response to LVS infection, RNA was purified from liver and spleen sections and from liver and spleen leukocyte cell suspensions at the indicated time points after LVS infection and then analyzed by RT-PCR. Similar to bacterial organ burdens, IFN-γ mRNA obtained from cell preparations peaked by 3 days of LVS infection in both the spleen and liver (Fig. 2A). A comparison of RT-PCR results obtained for cell suspensions (Fig. 2A) with those obtained for organs demonstrated similar time courses; however, the amount of IFN-γ mRNA detected in total liver was much lower than that obtained from liver leukocyte suspensions, indicating that the majority of the IFN-γ mRNA was likely produced by liver leukocytes, not by other cells of the liver parenchyma (data not shown).
FIG. 2.

IFN-γ mRNA and IFN-γ ex vivo secretion are detected in both spleen and liver quickly after LVS infection. C57BL/6J mice were infected with 105 CFU of LVS i.d., and pooled single-cell suspensions of spleen (▪) and liver (□) cells were prepared from five mice at the indicated time points. (A) IFN-γ-specific mRNA prepared from splenocytes (▪) and liver leukocytes (□) at the indicated time points after LVS infection was quantified by real-time PCR and normalized in relationship to GAPDH expression. (B) To assess secretion of IFN-γ protein, splenocytes and liver leukocytes were cultured for 3 days, and the harvested supernatants were analyzed for secreted IFN-γ by ELISA. Results are means ± standard deviations for three individual cultures established using pooled cells as described above. Results shown were derived from a single representative experiment of two (panel A) or three (panel B) total experiments of similar design.
To assess the ex vivo secretion of IFN-γ protein, splenocytes and liver leukocytes were harvested at different time points after LVS infection and cultured for 3 days, and the harvested supernatants were then analyzed by ELISA for secreted IFN-γ. As expected, production of IFN-γ protein (Fig. 2B) lagged behind expression of IFN-γ mRNA (Fig. 2A). Secreted protein amounts from both spleen and liver cells peaked after 5 days of LVS infection (Fig. 2B). Notably, ex vivo IFN-γ production by liver cells was higher than that by spleen cells on a per-cell basis at all time points examined.
Direct determination of cellular subpopulations responsible for IFN-γ production in spleen and liver following vaccinating LVS infection.
To directly examine the cellular sources of early IFN-γ production after LVS infection, ICS and analyses by flow cytometry were applied. Mice were infected i.d. with LVS and sacrificed to obtain splenic and liver leukocytes at various time points after LVS infection, and the resulting cells were subjected to staining for a panel of cell surface markers as well as for intracellular ex vivo accumulation of IFN-γ. Consistent with the lack of detectable IFN-γ mRNA or ex vivo secreted IFN-γ (Fig. 2A), few IFN-γ-expressing (IFN-γ+) cells were detected in naive mice or on day 1 after LVS infection (Fig. 3A). CD45+ IFN-γ-producing cells were readily detectable by day 3 after LVS infection, reaching a peak by days 5 to 7. On day 3, an average of 1.4% of spleen cells exhibited detectable intracellular IFN-γ; by day 5 (Fig. 3B) and day 7, 2.5% and 2.0%, respectively, of splenocytes were IFN-γ+. In the liver, 2.5%, 4.4%, and 6.2% IFN-γ+ leukocytes were found after 3, 5, and 7 days of LVS infection, respectively (Fig. 3A).
FIG. 3.

IFN-γ+ cells are detected by ICS in both spleen and liver quickly after LVS infection. C57BL/6J mice were infected with 105 CFU of LVS i.d., and pooled single-cell suspensions of splenocytes from five naive and LVS-infected mice were prepared on the indicated days. (A) The percentages of IFN-γ+ cells in splenocytes and liver leukocytes were assessed by ICS and flow cytometry. (B) Representative dot plots after exclusion of aggregated cells (using forward light scatter patterns) for cells obtained from naive mice treated only with isotype control antibodies (left), cells obtained from naive mice stained with anti-CD45 and anti-IFN-γ (middle), and cells obtained from mice 5 days after LVS infection and stained with anti-CD45 and anti-IFN-γ (right), illustrating the gating strategy and patterns of IFN-γ+ cells. Results shown were derived from a single representative experiment of three total experiments of similar design.
More detailed analysis suggested that different cell types were involved in IFN-γ production at different time points after LVS infection (Table 1). Three distinct patterns were discerned. Between day 3 and day 5 after LVS infection, conventional NK cells (CD45+ NK1.1+ TCRβ−) and NK T cells (CD45+ NK1.1+ TCRβ+) produced IFN-γ, but production by these cells declined thereafter. In contrast, substantial numbers of macrophages (CD45+ F4/80+ CD11b+ Gr1− CD11c−), neutrophils (CD45+ CD11b+ Gr1+ CD11c−), and DCs (CD45+ CD11c+ B220+/−) produced IFN-γ for a longer period, spanning between day 3 and day 7. Finally, conventional T cells (CD45+ TCRβ+ NK1.1−) contributed minimally to IFN-γ production until about day 7, the last time point assessed in these studies.
TABLE 1.
Evolution of cell types producing IFN-γ after i.d. LVS infectiona
| Cell type | % IFN-γ+ cells after LVS infection on day:
|
||||
|---|---|---|---|---|---|
| 0 | 1 | 3 | 5 | 7 | |
| Spleen cells | |||||
| NK cells | 1.0 | 2.4 | 21.0 | 15.0 | 2.5 |
| NK T cells | 2.6 | 2.0 | 8.0 | 8.0 | 3.3 |
| Macrophages | 0 | 0 | 1.9 | 4.3 | 1.1 |
| Polymorphonuclear leukocytes | 0 | 0 | 3.8 | 0.9 | 1.2 |
| DCs | 2.1 | 1.2 | 7.5 | 11.9 | 10.1 |
| T cells | 0.1 | 0.4 | 1.0 | 2.0 | 2.6 |
| Liver cells | |||||
| NK cells | 0.9 | 0.8 | 8.2 | 14.8 | 7.4 |
| NK T cells | 1.3 | 3.9 | 11.6 | 6.1 | 4.7 |
| Macrophages | 1.1 | 0.2 | 2.3 | 3.1 | 3.0 |
| Polymorphonuclear leukocytes | 0 | 1.9 | 2.1 | 3.5 | 3.2 |
| DCs | 1.3 | 2.2 | 9.7 | 14.9 | 9.6 |
| T cells | 0.2 | 0.3 | 2.9 | 3.9 | 8.5 |
Mice were infected with 105 CFU of LVS i.d. At the indicated time points after infection, mice were sacrificed, and splenic and liver leukocytes were prepared as pools from five individual mice and processed for determination of IFN-γ+ cells by ICS as described in Materials and Methods. The percentage of IFN-γ+ cells of each subtype, defined using multiparameter flow cytometry and a panel of markers as described in the text, is shown.
Approximately 40 to 45% of splenocytes are B lymphocytes, but initial studies yielded ambivalent results regarding IFN-γ production by this large population in LVS-infected mice. To examine the contribution of B cells directly, B cells were enriched from LVS-infected mice by use of anti-CD19 beads; such B-cell preparations were 92 to 95% CD19+ B220+. Although unseparated splenocytes from LVS-infected mice contained readily detectable IFN-γ+ cells by ICS (Fig. 4A), enriched B cells did not (Fig. 4B). Furthermore, enriched B cells did not secrete detectable IFN-γ upon ex vivo culture (Fig. 4C). Other reports indicate that murine B cells can produce IFN-γ under some circumstances following in vitro stimulation with specific antigen (28). Here, however, B cells did not appear to contribute significantly, if at all, to IFN-γ production after a vaccinating LVS infection, despite the presence of LVS bacteria during the culture period (Fig. 1A).
FIG. 4.

B cells do not contribute to IFN-γ production after LVS infection. C57BL/6J mice were infected with 105 CFU of LVS i.d. Single-cell suspensions of pooled splenocytes from five naive and LVS-infected mice were prepared on day 4 and enriched for B cells by use of anti-CD19+ MACS beads. The percentage of IFN-γ+ enriched B cells was assessed by ICS and flow cytometry (B), and the percentage of IFN-γ expression in total splenocytes, obtained and analyzed prior to B-cell enrichment, is shown as a positive control (A). (C) The amount of IFN-γ in supernatants after 3 days of ex vivo culture was assessed by ELISA. Data indicate means ± standard deviations for three individual cultures established using pooled cells as described above. Results shown were derived from a single experiment of this design and are consistent with similar results obtained using unseparated cells.
To further facilitate evaluation of the contributions of non-B-cell subpopulations to IFN-γ production in LVS-infected mice, splenocytes were obtained from LVS-infected mice on day 4, enriched for DX5+ NK cells, CD11c+ DCs, CD11b+ myeloid cells, and Thy1.2+ T cells, and evaluated by flow cytometry for expression of intracellular IFN-γ and by ex vivo culture for IFN-γ secretion. Each enriched cell preparation exhibited substantial numbers of IFN-γ+ cells, and supernatants contained large quantities of secreted IFN-γ protein (data not shown). However, characterization of the enriched cells by flow cytometry demonstrated that each enriched fraction remained heterogeneous. In subsequent studies, we therefore relied on multiparameter flow cytometry without separation of cells to precisely define IFN-γ+ cells on the basis of large panels of cell surface markers.
To define all subtypes of IFN-γ+ cells as well as to assess the total number of each cell subpopulation in spleens and livers producing IFN-γ, mice were infected with LVS for 4 days, the percentage of defined IFN-γ-positive cells from each organ was determined by ICS, and numbers were adjusted based on the total number of the starting single-cell preparations. In this case, cell surface markers to distinguish newly defined “NK DCs” (6, 40, 46, 53) from NK cells, NK T cells, and traditional DCs were available and included in the analyses. The strategy for defining these subpopulations is illustrated by representative dot plots shown in Fig. 5. Splenocytes were prepared 4 days after LVS infection and analyzed by flow cytometry by gating on single live cells and CD45 and then displaying expression of CD11c in relationship to expression of NK1.1. Thus, 1.9% of splenocytes were CD45+ CD11c+ NK1.1− DCs, and 26.5% of these contained detectable intracellular IFN-γ (Fig. 5). Similar stepwise gating approaches were used to define all cells from LVS-infected spleens that contained intracellular IFN-γ 4 days after infection. The absolute number and type of cells producing IFN-γ varied between organs after 4 days of LVS infection (Fig. 6). In LVS-infected spleens, DCs (defined in these analyses as CD45+ CD11c+ NK1.1− cells) and T cells contributed the most to producing IFN-γ, followed by NK DCs (CD45+ CD11c+ NK1.1+), NK T cells (CD45+ NK1.1+ TCRβ+), and traditional NK cells (CD45+ CD11c− NK1.1+ TCRβ−). In this study, the number of NK cells producing IFN-γ was derived by subtracting the number of CD45+ NK1.1+ TCRβ+ NK T cells from the total number of CD45+ CD11c− NK1.1+ cells. The overall number of liver leukocytes producing IFN-γ was about one-fifth that of splenocytes, reflecting the difference in the total number of lymphoid cells present in each organ. In LVS-infected livers, IFN-γ production was dominated by NK DCs, DCs, and conventional NK cells, with T cells and NK T cells providing only 10% of the total at this time point. In both organs, numbers of neutrophils and macrophages producing IFN-γ were small but, nonetheless, readily and reproducibly detectable.
FIG. 5.

NK cells, DCs, and “NK DCs” in LVS-infected spleens produce IFN-γ. C57BL/6J mice were infected with 105 CFU of LVS i.d. Single-cell suspensions of pooled splenocytes from LVS-infected mice were prepared on day 4 and stained with a panel of antibodies to cell surface markers as well as with a dye to reveal viability. Total splenocytes were gated to eliminate aggregates, using the relationship between FSC-H and FSC-A, then to eliminate dead cells, then to show positive expression of CD45, and then to show expression of CD11c in relationship to expression of NK1.1, as shown by representative dot plots. Gates for NK1.1+ CD11c−, NK1.1+ CD11c+, and NK1.1− CD11c+ subpopulations were established by comparison to appropriate isotype control-treated cells. The proportion of IFN-γ+ splenocytes within each subpopulation is shown. Results shown were derived from a single representative experiment of three total experiments of similar design.
FIG. 6.

Quantification of IFN-γ-producing cell subpopulations after LVS infection. C57BL/6J mice were infected with 105 CFU of LVS i.d. Single-cell suspensions of pooled splenocytes from five naive and LVS-infected mice were prepared on day 4, and IFN-γ+ splenocytes (A) and liver leukocytes (B) were analyzed by ICS and flow cytometry. The total number of IFN-γ+ cells of each subpopulation, as defined by a panel of cell surface markers (see text), was derived by multiplying the percentage of IFN-γ+ cells by the number of total viable cells obtained. Results shown were derived from a single representative experiment of three total experiments of similar design.
Contribution of lymphocytes to regulation of early IFN-γ production by myeloid cells.
To evaluate whether the absence of T and B cells affected the total production of IFN-γ, RAG-1 KO mice, which lack both B and T cells, were infected i.d. with LVS for 4 days. Total cell numbers, IFN-γ message, ex vivo IFN-γ secretion, and the cellular source of IFN-γ production were then evaluated as described above and compared to results for intact WT mice. The total number of viable cells in spleens of LVS-infected RAG-1 KO mice doubled to about 1 × 107 per mouse, compared to 5 × 106 to 6 × 106 in naive RAG-1 KO mice; this is about 10% of the numbers found for normal C57BL/6J mice. Numbers of liver leukocytes from naive and infected RAG-1 KO mice were similar to the numbers found in WT mice (1 × 106 and 6.8 × 106, respectively). Four days after LVS infection, RAG-1 KO mice exhibited somewhat larger bacterial organ burdens in spleens than WT mice did, averaging about 4 × 106 bacteria per organ, while CFU numbers in livers from each type of mouse were comparable (about 5 × 106). In evaluations by RT-PCR and ex vivo ELISA, the relative production of IFN-γ in RAG-1 KO mice was comparable to or higher than that of WT mice (data not shown), likely reflecting the lack of B cells in RAG-1 KO mice to contribute to IFN-γ production. Indeed, ICS analyses demonstrated that approximately 14% of RAG-1 KO splenocytes expressed IFN-γ 4 days after LVS infection, compared to about 2% of cells from WT mice. However, there were no obvious differences in IFN-γ production by liver leukocytes between the two mouse strains: approximately 4% of cells from both groups produced IFN-γ. Most importantly, detailed evaluation by ICS of the types of RAG-1 KO cells from LVS-infected mice demonstrated that numbers of splenic (Fig. 7A) and liver (Fig. 7B) IFN-γ+ cells, as well as the distribution of IFN-γ+ cells among the various subpopulations identified, were comparable to the numbers found in LVS-infected WT mice. Taken together, therefore, these data indicate that a wide variety of myeloid and lymphoid cells of the innate immune system, including unexpected cell types such as DCs, produce IFN-γ very quickly after LVS vaccination. This production is not dependent on the activity of T or B cells, but T cells contribute to production at later times, when adaptive immune responses develop. The patterns of different cell types producing IFN-γ evolved over time, possibly reflecting migration of responding cells as well as evolution of control in different microenvironments within infected tissues.
FIG. 7.

Development of IFN-γ-producing myeloid cells after LVS vaccination does not depend on T or B lymphocytes. C57BL/6J (WT) or C57-RAG-1 mice were infected with 105 CFU of LVS i.d. Single-cell suspensions of pooled splenocytes from five WT and three RAG-1 knockout naive and LVS-infected mice were prepared on day 4, and IFN-γ+ splenocytes (A) and liver leukocytes (B) were analyzed by ICS and flow cytometry. The total number of IFN-γ+ cells of each subpopulation, as defined by a panel of cell surface markers (see text), was derived by multiplying the percentage of IFN-γ+ cells by the number of total viable cells obtained. The absolute number of cells for each cell subpopulation, illustrated by sections in the stacked bars, is also provided in the legend on the right. Results shown were derived from a single representative experiment of three total experiments of similar design.
DISCUSSION
Understanding both innate and adaptive immune responses against a vaccine strain of F. tularensis is of interest for basic immunology as well as for vaccine development for intracellular pathogens. Studies using LVS illustrate the potential of a live attenuated vaccine, and at the same time, LVS is an excellent tool for studying immune responses to intracellular pathogens in animal models. In this report, we focused our attention on the characterization of IFN-γ-producing cells during the early stage of F. tularensis LVS vaccination, particularly those expressing NK cell markers, given previous unexpected results regarding the minimal impact of NK cell depletion on LVS vaccination (31, 33). In order to obtain a dynamic evaluation of IFN-γ responses, we determined IFN-γ production over time, using several complementary approaches. By 3 days after sublethal i.d. LVS infection, bacteria in the spleen and liver reached peak levels (Fig. 1A); concurrently, IFN-γ mRNA, IFN-γ secreted protein, and a substantial number of IFN-γ-producing cells, as determined by ICS, were detectable (Fig. 2 and 3). By day 5, a surge of secreted IFN-γ was accompanied by increased total cell numbers (Fig. 1B) and IFN-γ+ cells (Fig. 3A) in both organs. Finally, by day 7, innate immune responses appeared to be effective, since bacterial organ burdens started to decline, while IFN-γ-producing cells as well as IFN-γ secretion were still present at substantial levels and T cells were activated.
Following initial qualitative characterization of the time course of IFN-γ production in spleens and livers from LVS-infected mice (Table 1), we selected 4 days of infection for further detailed studies. At this time point, all of the different IFN-γ+ cell types detected throughout the first week were in some stage of IFN-γ production. Studies with separated splenocyte subpopulations clearly established that B cells (CD19+ enriched cells) did not contribute (Fig. 4). Detailed analysis at day 4, using unmanipulated total numbers of splenic and liver lymphocytes for multicolor flow cytometry, permitted calculation of the total number of IFN-γ+ cells of each cell type in each organ (Fig. 5 to 7). This analysis revealed that conventional NK cells, NK T cells, and NK DCs as well as different myeloid cells, including numerous DCs, were involved in IFN-γ production during innate immune responses (Table 1; Fig. 4 to 6), in a T-cell-independent manner (Fig. 7). The results were remarkable for illustrating functional redundancy that ensures many sources of this critical cytokine, with a large variety of cells involved, as well as the evolution of their engagement over time. Not surprisingly, production by different cell types peaked at different time points. Although neutrophils and macrophages producing IFN-γ in vivo following LVS infection were reproducibly detectable, their numerical contribution to the total production was minimal throughout the week studied.
One protein clearly important in regulating IFN-γ production is MyD88, part of the signal transduction cascade engaged upon the activation of Toll-like receptors (TLRs) secondary to the recognition of pathogens. The absence of MyD88 renders mice susceptible to many intracellular pathogens (19, 25, 43), including LVS (10), at least in part because of poor production of IFN-γ. Thus, it will be important to next assess the role of MyD88 and associated TLRs in initiating IFN-γ production by individual cell types. In addition to being critical for survival of initial infection with virtually any intracellular pathogen, the sources of innate, early IFN-γ production are of interest because IFN-γ regulates the expression of such a large variety of genes. At least 200 genes, many involved in antigen presentation, antimicrobial functions, and leukocyte trafficking, are regulated by IFN-γ (44). Given the global importance of this mediator, it is perhaps not surprising that so many innate immune cell types are endowed with the ability to respond and to produce it following infection; this redundancy in function likely ensures a sufficient IFN-γ response that leads to gene activation, the associated functional cascade, and ultimately, survival.
NK cells and T lymphocytes have commonly been considered the principal sources of IFN-γ during infections with intracellular bacteria. In classic studies, NK cells were identified as those responding to Listeria exposure in vitro (52), and in vivo or in vitro depletion of NK cells followed by Listeria exposure reduced IFN-γ production at day 1 but not day 7 or 14 after infection (50). Similarly, using Listeria monocytogenes-infected mice, NK cells were described as the main source of IFN-γ in spleens and livers of RAG mice about 18 h after infection; major histocompatibility complex class II-positive IFN-γ+ cells were not detected (51). Most recently, murine cells bearing both NK markers and CD11c, which may more closely resemble NK cells than DCs in function (6) and which produce abundant IFN-γ upon Listeria infection, have been described (40). Indeed, these NK DCs may be the most important cell type contributing to control of Listeria infection. The relationship between NK DCs and another unusual population, dubbed “IK DCs,” for IFN-producing killer DCs, remains to be fully clarified (8, 53). In this study, we were readily able to recognize LVS-stimulated IFN-γ production by several NK-related subpopulations, including traditional NK cells (defined as CD45+ NK1.1+ CD11c− TCRβ− cells), NK T cells (defined as CD45+ NK1.1+ CD11c− TCRβ+ cells), and NK DCs (defined as CD45+ NK1.1+ CD11c+ cells).
Other studies showed clearly that CD8α+ DCs can be a major source of IFN-γ production upon in vitro culture of splenocytes with Listeria (37). Furthermore, mice lacking the cytokine receptor common gamma subunit (γC− mice), which are severely deficient in NK cells, exhibited normal early IFN-γ production and control of Listeria infection (1), and transfer of DCs into Listeria-infected γC− mice greatly increased IFN-γ production and control of infection (48). Similarly, in the present study, CD11c+ cells which clearly lacked NK markers were the dominant IFN-γ+ cell type found in LVS-infected spleens as well as in livers. Given the importance and complexity of the many DC subpopulations now being defined, future studies will concentrate on delineating the DC subpopulations responding to Francisella in greater detail.
Another interesting alternative source of IFN-γ production quickly after infection is “bystander” CD8+ T cells, which are apparently non-antigen specific. Thus, when murine spleen cells were incubated in vitro with Burkholderia pseudomallei, both NK cells and CD8+ cells produced IFN-γ within 24 to 48 h (32), and CD8+ T cells in mice infected with Burkholderia pseudomallei produced IFN-γ within 16 h of infection (32). However, such cells were not readily detectable in either spleens or livers of LVS-infected mice.
Indeed, for LVS infection, thus far only NK cells have been implicated in innate IFN-γ production. Mouse liver lymphocytes produced IFN-γ in vitro when they were stimulated with LVS or type A Francisella, and at least half of those lymphocytes were NK cells (54). Similarly, 72 h after intranasal LVS infection, CD11b+ DX5+ NK1.1+ lung cells harvested and then cultured in vitro with phorbol myristate acetate and ionomycin appeared to be the major producers of IFN-γ (33). Furthermore, NK cell depletion followed by intranasal LVS infection reduced serum IFN-γ levels compared to those of control mice. It is important, however, that strong polyclonal stimulation, while useful for increasing IFN-γ detection, may facilitate the expansion and functions of one cell type over another during in vitro culture, thereby altering the observed pattern. In this study, we instead elected to study cells obtained from LVS-infected mice by ICS without further deliberate stimulation during the in vitro culture period required for brefeldin addition beyond that provided by the numbers of bacteria present in vivo and carried over into the in vitro cultures. Strikingly, this approach revealed a much greater variety of cell types involved in innate IFN-γ production following LVS infection in vivo than that obtained by in vitro studies or using ex vivo phorbol myristate acetate stimulation. Indeed, by 4 days after i.d. LVS infection, the numbers of DCs, traditional NK cells, and NK DCs producing IFN-γ were roughly equivalent. If the principal function of NK cells during intracellular infections is the production of IFN-γ, the identification of large numbers of non-NK-related cells producing IFN-γ provides an explanation as to why NK cell depletion has apparently little impact during LVS vaccination.
Another understudied aspect of infections that disseminate to organs of the reticuloendothelial system is the role of liver leukocytes. The liver, as a peripheral site of infection, is an important site of cell-mediated immune surveillance and effector functions (34, 45) and is rich in NK cells and unconventional T cells, such as NK T cells (24, 45). LVS infects livers of mice within 3 days of infection by any route and replicates in both Kupffer cells and hepatocytes (12). As noted above, mouse liver lymphocytes cultured with LVS in vitro produced IFN-γ. At least half of the production was attributed to NK1.1+ NK cells, but the balance of the cells were not characterized (54). Here we demonstrated qualitatively similar results between liver leukocytes and splenocytes (Fig. 7), although some quantitative differences were evident. The numbers of leukocytes, particularly T cells, in LVS-infected livers increased at an even higher rate than did the numbers of splenocytes (Fig. 1B); it remains to be determined whether this resulted from local proliferation and/or migration from professional immune system organs. Bacterial organ burden data for livers were always slightly higher than those for spleens at all time points, and bacterial clearance was somewhat slower in livers (Fig. 1A). Most notably, the proportion of leukocytes producing IFN-γ consistently appeared to be higher in livers than in spleens (Fig. 2B and 3A; Table 1).
The production of IFN-γ from some cells may depend on signals released by other cell types, and the lack of those cells therefore alters IFN-γ production. To address this possibility, we compared the response of LVS-infected, T- and B-cell-deficient RAG mice to that of WT mice. In the absence of B and T cells, the numbers of IFN-γ-producing cells were comparable between the two strains of mice (Fig. 7), indicating that myeloid cells did not require the presence of lymphocytes to respond properly to LVS infection. This is consistent with the observation that IFN-replete but lymphocyte-deficient SCID mice infected with LVS i.d. survive for up to 3 weeks after infection (23). The lack of lymphocytes that might otherwise confound flow cytometry analyses or separation studies also served to further confirm the predominance of the numbers of IFN-γ+ DCs in LVS-infected mice (Fig. 7).
These analyses are, of course, subject to the limitations of the techniques applied. Only cells from infected organs that were amenable to preparation in single-cell suspensions were included in the flow cytometry analyses. Furthermore, detection of intracellular cytokine does not necessarily result in secreted protein. Nonetheless, the concordance between the presence of IFN-γ mRNA, IFN-γ+ cells by ICS, and IFN-γ protein secretion in vitro following in vivo LVS infection lends confidence to the interpretation that the flow analyses accurately detected at least a large proportion of the cells involved. Obviously, the limited sensitivity of ICS may still fail to detect some cells, even in single-cell preparations. Our intent here was to evaluate IFN-γ production in vivo by using an approach that resembles clinical vaccination and infection as closely as possible. Despite such acknowledged limitations, this approach has nonetheless served to reveal a complex innate immune response and, in particular, has emphasized the unexpected contribution of DCs as a major IFN-γ-producing cell type in both spleens and livers from LVS-infected mice. Other studies have shown that subtypes of DCs, particularly plasmacytoid DCs, produce IFN-α in response to viral infection or stimulation (14). The present study firmly establishes DCs as a major source of IFN-γ production after intracellular bacterial infection. The specific relationship between subsets of cytokine-producing DCs, infection of DCs, and antigen presentation is currently unknown (17) and is a subject of intense current study. Further studies to completely characterize the subtypes and functions of DCs that respond to LVS infection, as well as the infection status of DCs that produce cytokines, are under way. We expect such studies to have significant implications for immunotherapeutic strategies and for vaccine development.
Acknowledgments
We thank our colleagues Siobhán Cowley and Ronald Rabin for critical reviews of the manuscript.
This work was supported in part by an interagency agreement with DMID/NIAID/NIH.
We declare no conflict of interest or financial interests in this study.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 23 June 2008.
REFERENCES
- 1.Andersson, A., W. J. Dai, J. P. Di Santo, and F. Brombacher. 1998. Early IFN-gamma production and innate immunity during Listeria monocytogenes infection in the absence of NK cells. J. Immunol. 1615600-5606. [PubMed] [Google Scholar]
- 2.Baker, C. N., D. G. Hollis, and C. Thornsberry. 1985. Antimicrobial susceptibility testing of Francisella tularensis with a modified Mueller-Hinton broth. J. Clin. Microbiol. 22212-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogdan, C., and U. Schleicher. 2006. Production of interferon-gamma by myeloid cells—fact or fancy? Trends Immunol. 27282-290. [DOI] [PubMed] [Google Scholar]
- 4.Bosio, C. M., and K. L. Elkins. 2001. Susceptibility to secondary Francisella tularensis LVS infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect. Immun. 69194-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke, D. S. 1977. Immunization against tularemia: analysis of the effectiveness of live Francisella tularensis vaccine in prevention of laboratory-acquired tularemia. J. Infect. Dis. 13555-60. [DOI] [PubMed] [Google Scholar]
- 6.Caminschi, I., F. Ahmet, K. Heger, J. Brady, S. L. Nutt, D. Vremec, S. Pietersz, M. H. Lahoud, L. Schofield, D. S. Hansen, M. O'Keeffe, M. J. Smyth, S. Bedoui, G. M. Davey, J. A. Villadangos, W. R. Heath, and K. Shortman. 2007. Putative IKDCs are functionally and developmentally similar to natural killer cells, but not to dendritic cells. J. Exp. Med. 2042579-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. 2002. Tularemia—United States, 1990-2000. MMWR Morb. Mortal. Wkly. Rep. 51181-184. [PubMed] [Google Scholar]
- 8.Chan, C. W., E. Crafton, H. N. Fan, J. Flook, K. Yoshimura, M. Skarica, D. Brockstedt, T. W. Dubensky, M. F. Stins, L. L. Lanier, D. M. Pardoll, and F. Housseau. 2006. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat. Med. 12207-213. [DOI] [PubMed] [Google Scholar]
- 9.Cole, L. E., K. L. Elkins, S. M. Michalek, N. Qureshi, L. J. Eaton, P. Rallabhandi, N. Cuesta, and S. N. Vogel. 2006. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 1766888-6899. [DOI] [PubMed] [Google Scholar]
- 10.Collazo, C. M., A. Sher, A. I. Meierovics, and K. L. Elkins. 2006. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8779-790. [DOI] [PubMed] [Google Scholar]
- 11.Conlan, J. W. 2004. Vaccines against Francisella tularensis—past, present and future. Expert Rev. Vaccines 3307-314. [DOI] [PubMed] [Google Scholar]
- 12.Conlan, J. W., and R. J. North. 1992. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect. Immun. 605164-5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conlan, J. W., A. Sjöstedt, and R. J. North. 1994. CD4+ and CD8+ T-cell-dependent and -independent host defense mechanisms can operate to control and resolve primary and secondary Francisella tularensis LVS infection in mice. Infect. Immun. 625603-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalod, M., T. P. Salazar-Mather, L. Malmgaard, C. Lewis, C. Asselin-Paturel, F. Briere, G. Trinchieri, and C. A. Biron. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195517-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dennis, D. T., T. V. Inglesby, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, A. M. Friedlander, J. Hauer, M. Layton, S. R. Lillibridge, J. E. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, and K. Tonat. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 2852763-2773. [DOI] [PubMed] [Google Scholar]
- 16.Dienst, F. T., Jr. 1963. Tularemia: a perusal of three hundred thirty-nine cases. J. La. State Med. Soc. 115114-127. [PubMed] [Google Scholar]
- 17.Dudziak, D., A. O. Kamphorst, G. F. Heidkamp, V. R. Buchholz, C. Trumpfheller, S. Yamazaki, C. Cheong, K. Liu, H. W. Lee, C. G. Park, R. M. Steinman, and M. C. Nussenzweig. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science 315107-111. [DOI] [PubMed] [Google Scholar]
- 18.Dunn, P. L., and R. J. North. 1991. Early gamma interferon production by natural killer cells is important in defense against murine listeriosis. Infect. Immun. 592892-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelson, B. T., and E. R. Unanue. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J. Immunol. 1693869-3875. [DOI] [PubMed] [Google Scholar]
- 20.Eigelsbach, H. T., and C. M. Downs. 1961. Prophylactic effectiveness of live and killed tularemia vaccines. J. Immunol. 87415-425. [PubMed] [Google Scholar]
- 21.Elkins, K. L., A. Cooper, S. M. Colombini, S. C. Cowley, and T. L. Kieffer. 2002. In vivo clearance of an intracellular bacterium, Francisella tularensis LVS, is dependent on the p40 subunit of interleukin-12 (IL-12) but not on IL-12 p70. Infect. Immun. 701936-1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elkins, K. L., S. C. Cowley, and C. M. Bosio. 2003. Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect. 5132-142. [DOI] [PubMed] [Google Scholar]
- 23.Elkins, K. L., T. R. Rhinehart-Jones, S. J. Culkin, D. Yee, and R. K. Winegar. 1996. Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect. Immun. 643288-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emoto, M., and S. H. Kaufmann. 2003. Liver NKT cells: an account of heterogeneity. Trends Immunol. 24364-369. [DOI] [PubMed] [Google Scholar]
- 25.Feng, C. G., C. A. Scanga, C. M. Collazo-Custodio, A. W. Cheever, S. Hieny, P. Caspar, and A. Sher. 2003. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 1714758-4764. [DOI] [PubMed] [Google Scholar]
- 26.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 592922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frucht, D. M., T. Fukao, C. Bogdan, H. Schindler, J. J. O'Shea, and S. Koyasu. 2001. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol. 22556-560. [DOI] [PubMed] [Google Scholar]
- 28.Ganapamo, F., V. A. Dennis, and M. T. Philipp. 2001. CD19+ cells produce IFN-gamma in mice infected with Borrelia burgdorferi. Eur. J. Immunol. 313460-3468. [DOI] [PubMed] [Google Scholar]
- 29.Gazzinelli, R. T., M. Wysocka, S. Hayashi, E. Y. Denkers, S. Hieny, P. Caspar, G. Trinchieri, and A. Sher. 1994. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 1532533-2543. [PubMed] [Google Scholar]
- 30.Geng, Y., K. Berencsi, A. Gyulai, T. Valy-Nagy, E. Gonczol, and G. Trinchieri. 2000. Roles of interleukin-12 and gamma interferon in murine Chlamydia pneumoniae infection. Infect. Immun. 682245-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leiby, D. A., A. H. Fortier, R. M. Crawford, R. D. Schreiber, and C. A. Nacy. 1992. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect. Immun. 6084-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lertmemongkolchai, G., G. Cai, C. A. Hunter, and G. J. Bancroft. 2001. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J. Immunol. 1661097-1105. [DOI] [PubMed] [Google Scholar]
- 33.Lopez, M. C., N. S. Duckett, S. D. Baron, and D. W. Metzger. 2004. Early activation of NK cells after lung infection with the intracellular bacterium, Francisella tularensis LVS. Cell. Immunol. 23275-85. [DOI] [PubMed] [Google Scholar]
- 34.Masopust, D., and L. Lefrancois. 2003. CD8 T-cell memory: the other half of the story. Microbes Infect. 5221-226. [DOI] [PubMed] [Google Scholar]
- 35.Morner, T. 1992. The ecology of tularaemia. Rev. Sci. Technol. 111123-1130. [PubMed] [Google Scholar]
- 36.Munder, M., M. Mallo, K. Eichmann, and M. Modolell. 1998. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J. Exp. Med. 1872103-2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohteki, T., T. Fukao, K. Suzue, C. Maki, M. Ito, M. Nakamura, and S. Koyasu. 1999. Interleukin 12-dependent interferon γ production by CD8α+ lymphoid dendritic cells. J. Exp. Med. 1891981-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oyston, P. C., and J. E. Quarry. 2005. Tularemia vaccine: past, present and future. Antonie van Leeuwenhoek 87277-281. [DOI] [PubMed] [Google Scholar]
- 39.Oyston, P. C., A. Sjostedt, and R. W. Titball. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2967-978. [DOI] [PubMed] [Google Scholar]
- 40.Plitas, G., U. I. Chaudhry, T. P. Kingham, J. R. Raab, and R. P. DeMatteo. 2007. NK dendritic cells are innate immune responders to Listeria monocytogenes infection. J. Immunol. 1784411-4416. [DOI] [PubMed] [Google Scholar]
- 41.Saslaw, S., H. T. Eigelsbach, J. A. Prior, H. E. Wilson, and S. Carhart. 1961. Tularemia vaccine study. II. Respiratory challenge. Arch. Intern. Med. 107134-146. [DOI] [PubMed] [Google Scholar]
- 42.Saslaw, S., H. T. Eigelsbach, H. E. Wilson, J. Prior, and S. Carhart. 1961. Tularemia vaccine study. I. Intracutaneous challenge. Arch. Intern. Med. 107121-133. [DOI] [PubMed] [Google Scholar]
- 43.Scanga, C. A., A. Bafica, C. G. Feng, A. W. Cheever, S. Hieny, and A. Sher. 2004. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect. Immun. 722400-2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroder, K., P. J. Hertzog, T. Ravasi, and D. A. Hume. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75163-189. [DOI] [PubMed] [Google Scholar]
- 45.Seki, S., Y. Habu, T. Kawamura, K. Takeda, H. Dobashi, T. Ohkawa, and H. Hiraide. 2000. The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol. Rev. 17435-46. [DOI] [PubMed] [Google Scholar]
- 46.Shortman, K., and J. A. Villadangos. 2006. Is it a DC, is it an NK? No, it's an IKDC. Nat. Med. 12167-168. [DOI] [PubMed] [Google Scholar]
- 47.Stenmark, S., D. Sunnemark, A. Bucht, and A. Sjöstedt. 1999. Rapid local expression of interleukin-12, tumor necrosis factor alpha, and gamma interferon after cutaneous Francisella tularensis infection in tularemia-immune mice. Infect. Immun. 671789-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzue, K., T. Asai, T. Takeuchi, and S. Koyasu. 2003. In vivo role of IFN-gamma produced by antigen-presenting cells in early host defense against intracellular pathogens. Eur. J. Immunol. 332666-2675. [DOI] [PubMed] [Google Scholar]
- 49.Tärnvik, A. 1989. Nature of protective immunity to Francisella tularensis. Rev. Infect. Dis. 11440-451. [PubMed] [Google Scholar]
- 50.Teixeira, H. C., and S. H. E. Kaufmann. 1994. Role of NK1.1+ cells in experimental listeriosis. NK1+ cells are early IFN-γ producers but impair resistance to Listeria monocytogenes infection. J. Immunol. 1521873-1882. [PubMed] [Google Scholar]
- 51.Thale, C., and A. F. Kiderlen. 2005. Sources of interferon-gamma (IFN-gamma) in early immune response to Listeria monocytogenes. Immunobiology 210673-683. [DOI] [PubMed] [Google Scholar]
- 52.Tripp, C. S., S. F. Wolf, and E. R. Unanue. 1993. Interleukin 12 and tumor necrosis factor α are costimulators of interferon γ production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiological antagonist. Proc. Natl. Acad. Sci. USA 903725-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vremec, D., M. O'Keeffe, H. Hochrein, M. Fuchsberger, I. Caminschi, M. Lahoud, and K. Shortman. 2007. Production of interferons by dendritic cells, plasmacytoid cells, natural killer cells, and interferon-producing killer dendritic cells. Blood 1091165-1173. [DOI] [PubMed] [Google Scholar]
- 54.Wickstrum, J. R., K. J. Hong, S. Bokhari, N. Reed, N. McWilliams, R. T. Horvat, and M. J. Parmely. 2007. Coactivating signals for the hepatic lymphocyte gamma interferon response to Francisella tularensis. Infect. Immun. 751335-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yee, D., T. R. Rhinehart-Jones, and K. L. Elkins. 1996. Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 1575042-5048. [PubMed] [Google Scholar]