

Abstract
The obligate intracellular pathogen Chlamydia trachomatis secretes effector proteins across the membrane of the pathogen-containing vacuole (inclusion) to modulate host cellular functions. In an immunological screen for secreted chlamydial proteins, we identified CT049 and CT050 as potential inclusion membrane-associated proteins. These acidic, nonglobular proteins are paralogously related to the passenger domain of the polymorphic membrane protein PmpC and, like other Pmp proteins, are highly polymorphic among C. trachomatis ocular and urogenital strains. We generated antibodies to these Pmp-like secreted (Pls) proteins and determined by immunofluorescence microscopy that Pls1 (CT049) and Pls2 (CT050) localized to globular structures within the inclusion lumen and at the inclusion membrane. Fractionation of membranes and cytoplasmic components from infected cells by differential and density gradient centrifugation further indicated that Pls1 and Pls2 associated with membranes distinct from the bulk of bacterial and inclusion membranes. The accumulation of Pls1 and, to a lesser extent, Pls2 in the inclusion lumen was insensitive to the type III secretion inhibitor C1, suggesting that this translocation system is not essential for Pls protein secretion. In contrast, Pls secretion and stability were sensitive to low levels of β-lactam antibiotics, suggesting that a functional cell wall is required for Pls secretion from the bacterial cell. Finally, we tested the requirement for these proteins in Chlamydia infection by microinjecting anti-Pls1 and anti-Pls2 antibodies into infected cells. Coinjection of anti-Pls1 and -Pls2 antibodies partially inhibited expansion of the inclusion. Because Pls proteins lack classical sec-dependent secretion signals, we propose that Pls proteins are secreted into the inclusion lumen by a novel mechanism to regulate events important for chlamydial replication and inclusion expansion.
The obligate intracellular pathogen Chlamydia trachomatis infects epithelial cells of the conjunctiva and the genital tract to cause conjunctivitis and sexually transmitted diseases. Inflammatory damage from chronic infections can lead to severe sequelae, including infectious blindness (trachoma), pelvic inflammatory disease, and infertility (36). Epithelial cells are infected by elementary bodies (EB), the invasive, metabolically inactive form of C. trachomatis. After entry, EBs differentiate into metabolically active reticulate bodies (RBs) and replicate within a membrane-bound vacuole (inclusion) (28). By 20 to 24 h postinfection, depending on the serovar, RB replication becomes asynchronous, generating both RBs and EBs. At the end of the infectious cycle, EBs are released by lysis of the host cell or extrusion of the inclusion (19) and then infect neighboring cells.
At all stages of infection, C. trachomatis communicates with its host by secreting proteins (effectors) across the inclusion membrane to modulate host cellular pathways. Because C. trachomatis carries a functional type III secretion (TTS) system, it is commonly accepted that Chlamydia, like other gram-negative bacterial pathogens, uses this secretion apparatus to translocate most effector proteins into host cells (33). Indeed, a large number of chlamydial proteins have been identified as potential targets of TTS because their amino termini can impart the ability to be secreted by heterologous TTS systems on artificial substrates (12, 18, 41). These putative TTS substrates include cytoplasmic proteins, such as the actin nucleating protein Tarp (7), the putative cyclin modulator CT847 (6), and a diverse family of integral inclusion membrane proteins (Incs) (1, 37). Small-molecule inhibitors of the TTS apparatus decrease the transport of Inc proteins to the inclusion membrane surface (29, 45) and block chlamydial replication, indicating that TTS-dependent protein transport is central to chlamydial pathogenesis.
Although TTS plays a prominent role in delivering effector proteins, there is mounting evidence that this is not the only mechanism by which chlamydial proteins can access the host cell cytoplasm. For example, the protease CPAF, which degrades host proteins important for innate and adaptive immune responses (34, 46), is first secreted into the lumen of the inclusion before accessing the cytoplasm of the infected cell (38). Similarly, the protease Tsp/CT441 accesses the cytoplasm of infected cells, where it degrades RelA to inhibit NF-κβ-mediated signaling (23). CPAF and Tsp have classical signal peptides, suggesting that these proteins engage the general secretory (sec-dependent) pathway to exit the bacterial cell. Consistent with this model, the secretion of CPAF is insensitive to TTS inhibitors (45). A similar route may be used by the passenger domain of some chlamydial autotransporters. For instance, the passenger domain of the Chlamydia pneumoniae protein Cpn0796 is proteolytically processed during infection, and the resulting fragments access the cytoplasm of the infected cell (43). How Tsp, CPAF, and cleaved autotransporters are translocated across the inclusion membrane or the extent to which chlamydial proteins exported via the general secretory pathway access the host cytoplasm is unknown.
One potential mechanism for the delivery of sec-dependent proteins to the inclusion membrane and the host cytoplasm could involve outer membrane vesicles, which many gram-negative pathogens use to deliver membrane-bound toxins and periplasmic contents to host cells (21). This mechanism of protein delivery may occur in C. trachomatis, where extensive vesiculation of RB outer membranes has been observed in response to antibiotic treatment (13). Indeed, these outer membrane vesicles have been proposed to act as conduits for the delivery of bacterial antigens beyond the confines of the inclusion (13).
In this study, we characterized two novel chlamydial proteins with homology to the passenger domain of PmpC. Even though these proteins do not contain classical signal peptides, they are secreted into the inclusion lumen, where they localize to globular structures that closely associate with the inclusion membrane and perform functions important for efficient inclusion expansion. Interestingly, Pmp-like secreted protein 1 (Pls1) and Pls2 appear to follow a secretion route that parallels that of autotransporter proteins. Overall, these results highlight the varied and unusual strategies used by chlamydiae to deliver proteins to the host cell.
MATERIALS AND METHODS
Sequence analysis.
PSI BLAST searches (www.ncbi.nlm.nih.gov/blast/Blast.cgi) of the C. trachomatis genome (serovar D) (40) were performed with the predicted amino acid sequences of open reading frames (ORFs) CT049, CT050, and CT051. Alignments were assembled based on homology and similarities of the query sequences to the closest chlamydial homologue, PmpC. Searches for secretion signals and features of secreted proteins were performed with the web-based prediction programs SignalP 3.0 and Secretome 2.0 at the Technical University of Denmark website (www.cbs.dtu.dk).
Cell lines and C. trachomatis infections.
HeLa cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. C. trachomatis lymphogranuloma venereum serovar L2 was propagated in HeLa cells at 37°C in the presence of 5% CO2-95% humidified air, and bacteria were stored as EBs in SPG (0.0038 M KH2PO4, 0.0072 M K2HPO4, 0.0049 M l-glutamic acid, 0.218 M sucrose, pH 7.4) at −80°C as previously described (4). EBs were added to HeLa cells at the indicated multiplicities of infection (MOIs), and infections were synchronized by centrifugation at 1,600 × g for 25 min at 4°C. Infected cells were placed at 37°C for 30 min, the medium was replaced, and infections were allowed to proceed for the indicated times.
Generation of anti-Pls1 and -Pls2 antibodies.
Glutathione S-transferase (GST) fusions to Pls1 and Pls2 were generated by subcloning of the CT049 and CT050 coding sequences into a modified pGEX-4T-1 vector (GE Healthcare) with compatible restriction enzyme recognition sites. Recombinant proteins were produced in Escherichia coli BL21 (Stratagene) after a 4-h induction with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 30°C and were purified by affinity chromatography on glutathione-coated Sepharose beads (GE Healthcare). Rabbits were immunized with recombinant proteins, and immunoglobulin G (IgG) antibodies were purified by binding to protein A-coated Sepharose beads (GE Healthcare). Bound antibodies were eluted with 0.2 M glycine, neutralized with 1 M K2HPO4, and depleted of anti-GST antibodies by repeated passage over a GST-Sepharose column. Whole serum was used for immunoblots and microinjections, while protein A-purified and GST-depleted antisera were used for immunofluorescence microscopy. The specificities of the anti-Pls1 and anti-Pls2 antibodies were confirmed by blotting against green fluorescent protein (GFP)-tagged Pls1 and Pls2 expressed in yeast (39) or by preincubation with recombinant GST-Pls1 and GST-Pls2 (see Fig. 2C). The following commercial antibodies were used: mouse anti-GFP monoclonal antibody (StressGen), mouse monoclonal anti-Omp2 (RDI), rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Abcam), and mouse monoclonal anti-tubulin (Sigma). Mouse monoclonal anti-IncA and rabbit polyclonal anti-RpoD were generous gifts from D. Rockey (Oregon State University) and M. Tan (University of California, Irvine), respectively. Rabbit polyclonal antibodies to MOMP, Hsp60, and CdsJ were obtained from K. Fields (University of Miami). Mouse monoclonal antibodies to chlamydial lipopolysaccharide (LPS) and MOMP were a gift from H. Caldwell (RML/NIH).
FIG. 2.

Pls protein expression in C. trachomatis-infected cells. (A) Rabbits were immunized with purified GST-CT049 (Pls1) or GST-CT050 (Pls2), and specificities of the resulting antisera were determined by screening GFP-tagged CT049 and CT050 expressed in yeast. Expression of recombinant proteins was monitored with anti-GFP antibodies. (B) Pls1 and Pls2 expression during infection was assessed by immunoblot analysis with anti-Pls1 and -Pls2 antisera, using lysates from HeLa cells infected with C. trachomatis L2 for 12 to 48 h. The level of RpoD was monitored to assess the expression of chlamydial proteins, and the host marker GAPDH was used as a loading control. (C) Pls1 and Pls2 antisera (red in merged images) specifically labeled bright punctate structures within the inclusion. HeLa cells were infected with L2 and immunostained with Pls antisera in the presence of excess GST-Pls1 or -Pls2. Note that intra-inclusion bright punctate structures were no longer detected by the antisera in the presence of the corresponding blocking antigen. Bacterial and host DNAs were detected with TOPRO-3 (blue). Closer inspection of stained inclusions reveals background staining of Pls1 (D) and Pls2 (E) with TOPRO-3-positive bacteria, in addition to bright Pls-positive aggregates.
Analysis of Pls1 and Pls2 expression.
HeLa cell monolayers were infected with L2 at an MOI of ∼1. Infections were synchronized at 4°C, and total proteins were collected at 0, 12, 24, 36, and 48 h postinfection by lysis in 1% Triton X-100 in the presence of protease inhibitor cocktail (Complete Mini, EDTA-free; Roche), 2 mM phenylmethylsulfonyl fluoride (Sigma), and 10 μM clasto-lactacystin β-lactone (Calbiochem), followed by sonication and treatment with 4 mM dithiothreitol. Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes (Bio-Rad), blocked in 5% nonfat milk diluted in Tris-buffered saline (0.5 M Tris, 1.5 M NaCl, pH 7.4) with 0.1% Tween 20 (TBST), and incubated with primary antibodies diluted in 5% milk-TBST for 3 h. The blots were washed extensively in TBST and incubated with horseradish peroxidase-linked secondary antibodies (GE Healthcare). Bound antibodies were detected with a Supersignal Pico HRP detection kit (Pierce).
Immunofluorescence microscopy.
HeLa cells were grown on glass coverslips and infected with L2 at an MOI of 1 as described above. At the indicated times postinfection, the cells were fixed with 100% ice-cold methanol and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4). After being blocked in 2% bovine serum albumin in PBS, cells were incubated with primary antibodies diluted in 2% bovine serum albumin-PBS, followed by secondary fluorophore-conjugated anti-rabbit or anti-mouse IgG (Molecular Probes). Cells were washed extensively with PBS, stained with 1 μM TOPRO-3 (Molecular Probes) to detect nucleic acids, and mounted on microscope slides. Infected cells were imaged with a Leica TCS SL confocal microscope under a 63× oil immersion objective (2.5-μm-thick sections). Images were processed with Leica software. To inhibit cell wall function, infected cells were treated with 10 μg/ml ampicillin (Amp; Sigma). To inhibit the TTS system of C. trachomatis, infected cells were treated with 70 μM C1 [N-(3,5-dibromo-2-hydroxybenzylidene)-4-nitrobenzohydrazide] (ChemBridge).
Membrane fractionation.
Five T-175 flasks (Sarstedt) of HeLa cells at 80% confluence were infected with L2 at an MOI of ∼1 for 40 h, and cells were lysed with 60 strokes in a Dounce homogenizer in the presence of protease inhibitor cocktail (Complete Mini, EDTA-free; Roche) and 10 μM lactacystin and spun at 500 × g to remove nuclei. Cytoplasmic components in the postnuclear supernatants (PNS) were separated from membranes and bacteria by ultracentrifugation at 100,000 × g for 1 h in a TLA-55 rotor in a Beckman Coulter Optima MAX-E ultracentrifuge. The membrane fraction was resuspended in 30% (vol/vol) iodixanol (Optiprep Axis-Shield, Norway) in PBS and overlaid with 2 ml 25% Optiprep-PBS, 1.5 ml 20% Optiprep-PBS, and 1 ml 5% Optiprep-PBS. Membranes were separated by flotation after ultracentrifugation at 100,000 × g for 2 h. Membrane fractions were collected at the 5 to 20% and 20 to 25% interphases. The bacterial pellet was collected from the bottom of the tube, resuspended in PBS, and treated with 10 μg/ml DNase (Roche) at 37°C for 1 h. Proteins were denatured in sample buffer and subjected to immunoblot analysis as described above.
Microinjection of anti-Pls1 and -Pls2 antibodies.
HeLa cells were grown in 35-mm poly-d-lysine-coated glass-bottomed culture dishes (MatTek) and infected with lymphogranuloma venereum serovar L2 at an MOI of ∼0.5. Infections were synchronized at 4°C, and nonadherent bacteria were removed by replacing the medium. At 4 h postinfection, ∼300 cells were microinjected with a combination of anti-Pls1 and -Pls2 antisera or preimmune serum, using an Eppendorf capillary femtotip needle (Fischer Scientific). Crude antiserum was diluted 1:10 in microinjection buffer (48 mM K2HPO4, 14 mM NaHsHPO4, KH2PO4, pH 7.2) prior to injection. The injections were performed with an Eppendorf FemtoJet/TransferMan NK2 microinjection system equipped with a Nikon TE2000 fluorescence microscope. Infected cells were incubated for another 26 h (30 h total), fixed with 3% formaldehyde, permeabilized with 0.1% Triton X-100, and stained with Alexa 555-conjugated anti-rabbit antibodies (Molecular Probes). Bacterial and nuclear DNAs were identified by staining with Hoechst 33258 (Molecular Probes), and the inclusion size was determined by analysis of images acquired in a Zeiss Axioskop 2 Mot Plus epifluorescence microscope equipped with an Orca ER Hamamatsu charge-coupled device camera. Images of ∼170 infected cells microinjected with either a combination of anti-Pls1 and anti-Pls2 sera or preimmune serum were acquired, and the largest axis of each inclusion was measured at the focal plane where the boundary of each inclusion was in focus. The inclusion size of each individual microinjected cell was then measured in pixels, using Axiovision software (Zeiss). The distribution of inclusion size, median, standard deviation, two-tailed t test statistical analysis, and P values were calculated with SAS System Local XP Pro software (SAS Institute).
RESULTS
Identification of Pls proteins CT049 and CT050.
In a screen based on the reactivity of recombinant chlamydial proteins to antisera generated against density gradient-purified inclusion membranes, we identified the proteins encoded by the C. trachomatis serovar D ORFs CT049 and CT050 as putative inclusion membrane-associated proteins (39). CT049 and CT050 encode nonglobular, acidic proteins with predicted molecular masses of 50.7 kDa and 56.7 kDa, respectively, that lack the bilobed hydrophobic motif of classical Inc proteins or any putative transmembrane domains. These proteins are weakly homologous to each other, with 22.5% overall sequence identity. Similarity searches against chlamydial proteins indicated that ORF CT051 is also paralogously related to CT049 and CT050 (Fig. 1A). Based on genome-wide transcriptional profiles, CT049, CT050, and CT051 are first transcribed at 8 h postinfection (2).
FIG. 1.

Genomic arrangement of Pls (Pmp-like secreted) genes. (A) Organization of the genes encoding Pls1 (CT049), Pls2 (CT050), and Pls3 (CT051). Pls2 is paralogously related to Pls1 and Pls3. The percentages of identity (Id) and similarity (Sm) based on BLAST searches are shown. (B) Pls proteins are paralogously related to the passenger domain of the predicted autotransporter polymorphic outer membrane protein PmpC. Regions of similarity between Pls proteins and PmpC were identified by PSI-BLAST. Pls proteins share limited homology to two distinct regions within the passenger domain and to the putative Chlamydia polymorphic middle protein signature domain (ChlamPMP).
PSI-BLAST searches of chlamydial sequences for related proteins revealed that the CT049, CT050, and CT051 proteins are related to the passenger domain of the autotransporter protein PmpC, a member of the highly polymorphic Pmp family of outer membrane proteins (Fig. 1B). Like the genes encoding Pmps, CT049 and CT050 display an unusually high degree of single-nucleotide polymorphisms among C. trachomatis reference strains (14), suggesting that their encoded proteins may be under selective pressure to undergo antigenic variation. Furthermore, because homologues of CT049, CT050, and CT051 are found in Chlamydia muridarum and Chlamydophila sp., we postulate that the functions of these genes are conserved among the chlamydiae. Given their relatedness to Pmp proteins and their identification as putative secreted proteins, we refer to CT049 to -051 as Pls1 to -3.
To characterize Pls proteins, we raised antisera against these proteins by immunizing rabbits with recombinant GST-tagged Pls1 and Pls2. We could not express recombinant Pls3 in E. coli, and therefore this protein was not characterized further. The specificities of the polyclonal anti-Pls1 and -Pls2 antisera were confirmed by immunoblot analysis of GFP-tagged CT049 and CT050 expressed in yeast. By SDS-PAGE, both GFP-tagged CT049 and CT050 migrated as significantly higher-molecular-mass species than their predicted sizes (78.7 kDa and 84.7 kDa, respectively), possibly as a result of the highly acidic nature of these proteins (pIs of 4.62 and 4.7, respectively) (Fig. 2A).
We assessed the expression profile of Pls1 and Pls2 by infecting HeLa cells with C. trachomatis L2 and detecting Pls proteins at different stages during infection by immunoblot analysis. Pls1 and Pls2 antisera detected multiple immunoreactive bands by 36 h and 24 h postinfection, respectively, and these bands increased in intensity throughout the infectious cycle (Fig. 2B). Anti-Pls1 antibodies detected immunoreactive bands of 80 and 60 kDa and a high-molecular-mass species of >150 kDa, which accumulated throughout infection. The anti-Pls2 antibodies specifically recognized a >250-kDa high-molecular-mass species and three specific, 75-kDa, 50-kDa, and 35-kDa bands. The abundance of the 75-kDa band decreased after 24 h postinfection, concomitant with an increase in the 50-kDa and 35-kDa forms. Based on the abnormal migration of GFP-tagged Pls1 and Pls2 in SDS-PAGE, we speculate that the lower-molecular-mass bands constitute processed forms of full-length Pls1 and Pls2 that migrate as 80-kDa and 75-kDa bands, respectively. Preimmune antisera did not recognize any immunoreactive material in infected cells by either Western blotting or immunofluorescence microscopy (not shown).
The abundance of the high-molecular-mass Pls1 and Pls2 protein complexes (>200 kDa) was sensitive to the levels of reducing agents in the SDS-PAGE sample buffer and to heat denaturation, suggesting that these proteins may form stable, disulfide-cross-linked oligomeric aggregates. Both Pls1 and Pls2 were also very sensitive to postlysis proteolysis, and the relative abundance of each protein fragment was variable among samples unless excess lactacystin was included in the lysis buffer. These proteins were also unstable when expressed in E. coli, with multiple proteolytic fragments accumulating upon overexpression of the GST-tagged proteins (not shown).
Pls1 and Pls2 localize to non-bacterium-associated structures in the lumen of the inclusion.
To determine the subcellular localization of Pls1 and Pls2 during infection, we infected HeLa cells with C. trachomatis L2 for 24 h and immunostained cells with anti-Pls1 and anti-Pls2 antibodies. Bacterial and host DNAs were detected with the dye TOPRO-3. Both Pls1 and Pls2 recognized scattered punctate structures within the inclusion lumen that did not appear to significantly overlap with bacterial DNA (Fig. 2C). These antibodies were specific for their target antigens, since anti-Pls antibodies failed to detect any such structures in uninfected cells and the recognition of immunoreactive material in infected cells was blocked by preincubation of antibodies with excess GST-Pls1 and -Pls2 (Fig. 2C). Closer inspection of these infected cells revealed that in addition to bright clusters, Pls1 and Pls2 also weakly stained punctate structures associated with the surfaces of TOPRO-3-positive bacteria (Fig. 2D and E).
We assessed the expression pattern and subcellular localization of Pls1 and Pls2 in greater detail by performing a time course of infection. HeLa cells were infected with L2 for 6 to 30 h and processed for indirect immunofluorescence microscopy with anti-Pls1 and -Pls2 antibodies. In addition, anti-MOMP and -IncA antibodies were used to detect bacterial and inclusion membranes, respectively. Consistent with our immunoblot results, immunoreactive material was not detectable prior to 12 h postinfection. At 18 h, the Pls1 and Pls2 staining was weak and mostly associated with RBs (Fig. 3). By 24 h, however, anti-Pls1 and -Pls2 antibodies prominently labeled intra-inclusion globular structures in the inclusion lumen. These Pls-positive globular structures showed limited association with MOMP (Fig. 3) or Omp2 (Fig. 4). Similarly, the Pls-positive intra-inclusion globular structures did not colocalize with IncA-positive vesicles within inclusions (8), suggesting that these structures are distinct. In addition, we observed an intimate association of a subset of these globular structures with the inclusion membrane, including instances where these structures may be extruded across the inclusion membrane (Fig. 3, arrowheads). Because methanol and paraformaldehyde fixation did not affect the morphology of these structures, it is unlikely that these immunostaining patterns are artifacts of fixation (not shown). Overall, since Pls1 and Pls2 in the inclusion lumen did not colocalize with bacterial markers (Omp2 and MOMP), we postulate that the punctate and globular Pls-positive structures constitute secreted forms of these proteins.
FIG. 3.

Subcellular localization of Pls1 and Pls2 in C. trachomatis-infected cells. HeLa cells were left untreated or infected with L2 at an MOI of ∼1 for 18 or 24 h. Cells were processed for indirect immunofluorescence microscopy with either anti-Pls1 (A) or -Pls2 (B) antibodies (red) and imaged by laser scanning confocal microscopy. Bacterial outer membranes and inclusion membranes were detected with monoclonal antibodies against MOMP and IncA, respectively (green). Note the presence of Pls1 and Pls2 as extrabacterial globular aggregates in the inclusion lumen and in close association with the inclusion membrane. Potential extrusion sites of Pls-positive material are shown by arrowheads.
FIG. 4.

The accumulation of Pls proteins in the inclusion lumen is sensitive to Amp. (A) HeLa cells were infected with L2 for 16 h and either left untreated or treated with 10 μg/ml Amp or 70 μM of the TTS inhibitor C1 for an additional 8 h. At 24 h postinfection, cells were processed for immunofluorescence microscopy, and Pls1 or Pls2 was detected with specific antisera. In addition, bacteria were detected with anti-Omp2 and anti-LPS antibodies. Inclusion membranes were detected with anti-IncA antibodies. Note the accumulation of Pls1 and Pls2 in the cytoplasm of enlarged RBs and the lack of Pls-positive intra-inclusion globular structures in Amp-treated cells. (B) Pls1 and Pls2 transiently localize to the surfaces of enlarged RB outer membranes after recovery from Amp treatment. HeLa cells were infected in duplicate with L2 for 16 h and treated with 10 μg/ml Amp for 8 h. One set of cells was fixed, and the other was allowed to recover from Amp for an additional 24 h (Amp + Rec.). Pls1 and -2 and MOMP were detected by indirect immunofluorescence, as described above. Note the reduced levels of Pls1 and Pls2 expression in Amp-treated cells (middle panels) and the association of Pls1 and Pls2 at the surfaces of enlarged RBs (arrowheads) in cells recovering from Amp treatment. Host and bacterial DNAs were stained with TOPRO-3 (blue).
Pls1 and Pls2 localization to the inclusion lumen requires a functional cell wall.
Bioinformatic searches for secretion signal peptides in Pls1 and Pls2 with SignalP 3.0 (3) revealed that these proteins lack classical sec-dependent secretion signals and therefore may be targets of nonclassical secretion. We considered the possibility that Pls1 and Pls2 could be targeted to the inclusion lumen, and possibly the inclusion membrane, by the chlamydial TTS system. To test the role of TTS in the secretion of these proteins, we used C1, a small-molecule inhibitor of TTS (30). C1 disrupts the developmental cycle of C. trachomatis, arrests inclusion expansion, and inhibits the translocation of Inc proteins to the inclusion membrane (29). We infected HeLa cells with L2 for 16 h, followed by treatment with dimethyl sulfoxide (solvent-only control) or 70 μM C1 for 8 h. Infected cells were processed for indirect immunofluorescence with anti-Pls antibodies and anti-Omp2 or anti-IncA antibody. As previously reported, C1 treatment arrested inclusion growth and partially inhibited IncA transport to the inclusion membrane (45). However, C1 did not significantly inhibit the accumulation of Pls1 globular structures in the inclusion (Fig. 4A). Pls2 secretion appeared to be more sensitive to C1 treatment, since there was greater overlap between Pls2 and IncA/Omp2. Nonetheless, a significant amount of Pls2 remained at globular structures within the inclusion lumen. Based on these findings, we speculate that either Pls proteins are a TTS cargo that is relatively insensitive to C1 treatment or the chlamydial TTS system does not play a major role in Pls secretion.
Given the homology of Pls proteins to the passenger domain of PmpC, we hypothesized that Pmps and Pls may have similar secretion requirements. Recently, it was reported that the translocation of PmpD to the bacterial cell surface is inhibited by β-lactam antibiotics (20), presumably as a result of inhibition of signal peptidase I activity at the cytoplasmic membrane (22). In Chlamydia-infected cells, low-level β-lactam antibiotic treatment prevents bacterial division, leading to the formation of enlarged RBs that morphologically resemble persistent forms (26). However, these enlarged RBs are unlikely to represent “true” persistent forms because they display transcriptional profiles identical to those of unperturbed RBs (31). We infected HeLa cells with L2 for 16 h, added Amp (10 μg/ml) for 8 h, and assessed the subcellular localization of Pls proteins. Interestingly, Amp treatment shifted the localization of Pls1 and Pls2 from the inclusion lumen to the cytoplasm of enlarged RBs (Fig. 4A). In contrast, IncA remained at the inclusion membrane and intra-inclusion vesicles. Next, we tested the effect of removing Amp on the recovery of Pls1 and Pls2 secretion. Within 6 h after Amp removal, Pls1 and Pls2 no longer localized to the cytoplasm of RBs but, instead, were seen in association with the outer surfaces of enlarged RBs, where they extensively colocalized with MOMP (Fig. 4B, insets). We postulate that the Pls proteins at the bacterial surfaces following removal of Amp represent secretion intermediates.
We observed that the intensity of Pls1 and Pls2 staining was significantly lower in Amp-treated cells than in untreated cells. To assess the levels of Pls proteins in cells treated with various inhibitors of protein secretion, we infected HeLa cells with L2 for 30 h and either left them untreated or pretreated them with Amp (10 μg/ml) or C1 (70 μM) for 2, 6, and 10 h prior to harvest. Total protein was collected, and the steady-state levels of bacterial proteins were assessed by immunoblot analysis. While levels of the chlamydial protein RpoD remained fairly constant throughout inhibitor treatment, we observed distinct changes in the levels of Pls proteins in Amp-treated cells (Fig. 5). For example, there was a decrease in the level of the 60-kDa species of Pls1. The effect of Amp treatment on the Pls2 level was more pronounced, with a stepwise loss of the 75-kDa, 50-kDa, and 35-kDa forms throughout antibiotic treatment. By 10 h of Amp treatment, only trace amounts of the 35-kDa form of Pls2 remained. In contrast, the steady-state levels of other chlamydial proteins, including Inc proteins and Omp2, steadily increased. Interestingly, C1 treatment did not appreciably change the steady-state levels of Inc proteins, Omp2, or Pls proteins. Overall, we interpret these results as evidence that despite the nonclassical secretion of Pls proteins, the general secretory pathway and/or cell wall function is required for their secretion into the inclusion lumen and that inhibition of transport from the bacterial cell leads to the degradation of Pls proteins.
FIG. 5.
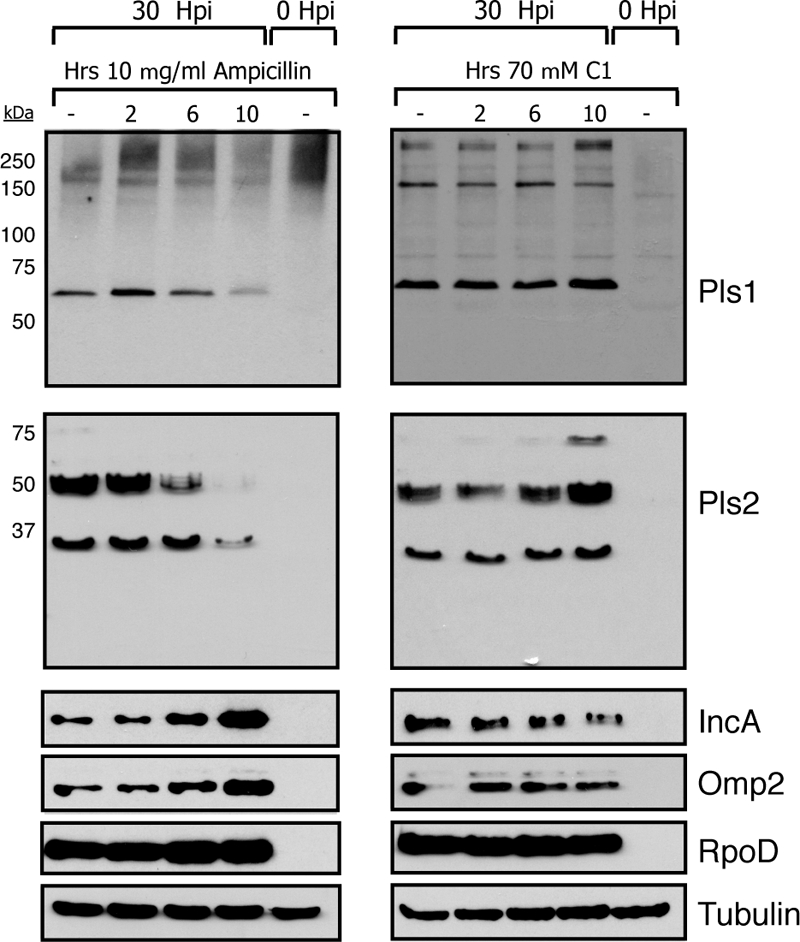
Pls1 and Pls2 are differentially processed in Amp-treated cells. HeLa cells were infected with L2 at an MOI of ∼1 and harvested at 30 h postinfection. Prior to cell harvesting, parallel samples were treated with either 10 μg/ml Amp or 70 μM C1 for 0, 2, 6, or 10 h. Total protein lysates were generated and subjected to immunoblot analysis with anti-Pls1, -Pls2, -Omp2, -IncA, and -RpoD antibodies. Tubulin levels are shown as a loading control. Note the loss of Pls1 and Pls2 during prolonged Amp treatment, despite the accumulation of other bacterial markers.
Pls1 and Pls2 associate with membranes.
Because Pls1 and Pls2 were identified as putative inclusion membrane-associated proteins, we wanted to determine if the Pls1- and Pls2-positive globular structures that are present in the inclusion lumen associate with membranes. HeLa cells were infected with L2 for either 24 (not shown) or 48 h and lysed by Dounce homogenization, and intact bacteria and total membranes were separated from host cytoplasmic components (high-speed supernatants [HSS]) by differential centrifugation. Under the conditions used, the bacterial membrane proteins MOMP and Omp2, the periplasmic lipoprotein CdsJ, and the cytoplasmic proteins Hsp60 and RpoD sedimented after ultracentrifugation (high-speed pellet [HSP]), indicating that no significant bacterial lysis had occurred. In contrast, the low-molecular-weight (low-MW) fragments of Pls2 partitioned with the host cytoplasmic protein GAPDH in the soluble HSS fraction, suggesting that this processed form of Pls2 is soluble. However, the majority of Pls1, Pls2, and their processed forms fractionated with the bacterial/membrane pellets (HSP) (Fig. 6). Next, we resuspended the bacterial/membrane pellets in 30% Optiprep, and total membranes (HSP) were separated by flotation on isopycnic density gradients. A significant proportion of Pls1 and Pls2 and their processed forms floated up with membranes of intermediate buoyancy that partially overlapped with IncA-positive inclusion membranes. In contrast, the bacterial membrane-associated markers MOMP, Omp2, and CdsJ were largely restricted to the bottom of the gradient, suggesting that Pls-positive membranes have distinct buoyancy properties from those of bulk bacterial membranes (not shown) (Fig. 6).
FIG. 6.
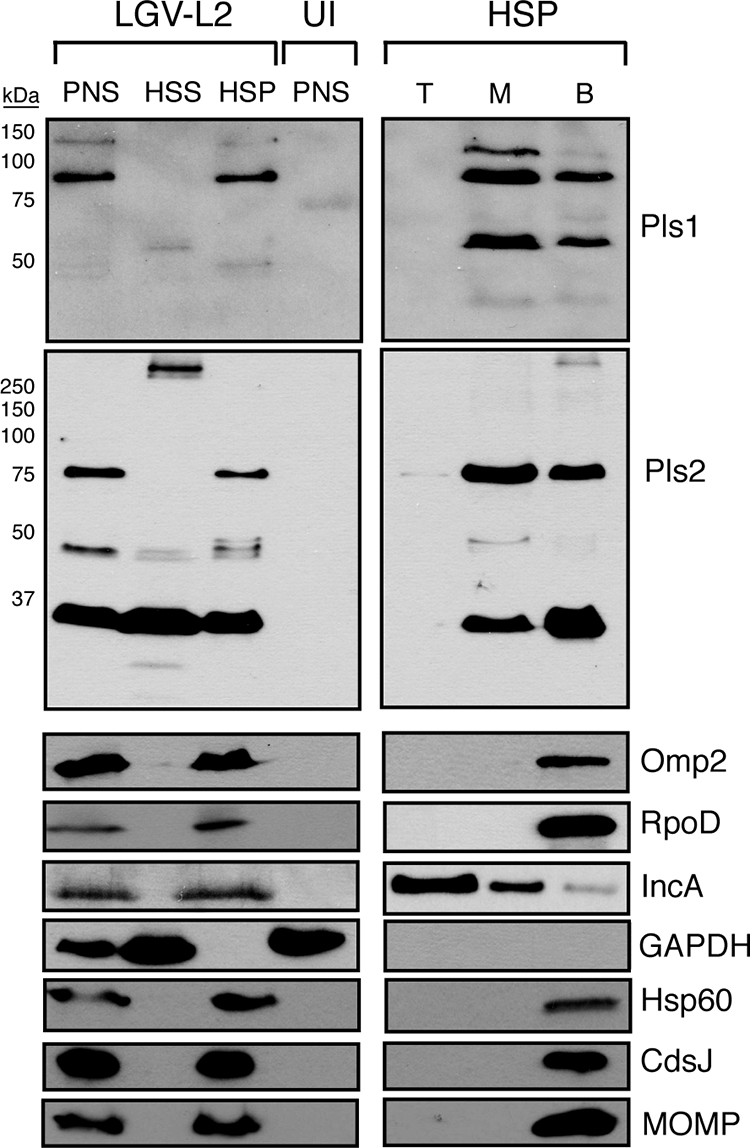
Pls1 and Pls2 cofractionate with membranes. HeLa cells infected with L2 at an MOI of ∼1 were harvested at 48 h postinfection. The PNS was ultracentrifuged to generate cytosol-enriched HSS and HSP. Total membranes and intact bacteria partitioned with the HSP. Membranes in the HSP were further separated by density gradient centrifugation, and samples were collected from the top (T), middle (M), and bottom (B) fractions. The bottom fraction consisted mostly of intact bacteria. PNS and membrane fractions were solubilized in SDS sample buffer, boiled under reducing conditions, and subjected to immunoblot analysis. Note the partitioning of low-MW processed forms of Pls1 and Pls2 with cytosolic fractions and the association of larger processed Pls forms with middle membrane fractions. Other bacterial membrane proteins (MOMP and Omp2) remained at the bottom of the gradient.
Overall, these findings led us to conclude that a significant proportion of Pls1 and Pls2 is secreted into the inclusion lumen and that Pls1 and Pls2 are processed into species of various MWs that display differential association with membranes.
Pls1 and Pls2 functions are required for efficient inclusion expansion.
We observed Pls-positive globular structures in close apposition to the inclusion membranes, including areas were these globular structures appear to be extruded from the inclusion lumen (Fig. 3, arrowheads). We hypothesized that Pls proteins may perform functions at the inclusion membrane or in the host cytoplasm that are important to the pathogenesis of C. trachomatis. To address this possibility, we neutralized the function of Pls1 and Pls2 by microinjecting infected HeLa cells (at 4 h postinfection) with Pls1 and Pls2 antisera. In addition, to address the possibility that these proteins perform overlapping and redundant functions, we also microinjected infected cells with a mix of both antisera. L2-infected HeLa cells were fixed and stained with anti-rabbit IgG to detect microinjected cells, and the size and morphology of inclusions were determined at 30 h postinfection. Neither the preimmune serum nor anti-Pls1 or -Pls2 alone had any effect on inclusion size or morphology. In contrast, microinjections of anti-Pls1 and -Pls2 combined led to significant and reproducible reductions in inclusion size (Fig. 7). These results suggest that Pls1 and Pls2 contribute to the expansion of the inclusion.
FIG. 7.

Microinjection of Pls1 and Pls2 antisera inhibits inclusion expansion. HeLa cells were infected with L2 at an MOI of ∼1. Infected cells were then microinjected with Pls1 and Pls2 preimmune sera or Pls1 and Pls2 sera at 4 h postinfection. At 30 h, cells were fixed, and microinjected cells were detected with fluorophore-conjugated anti-rabbit IgG and Hoechst 33258. The sizes of ∼170 inclusions in microinjected cells from each experiment were measured in pixels, and the distributions of inclusion size were analyzed. The rectangular box corresponds to data points between the 25th and 75th percentiles of the total distribution. The horizontal line denotes the median. The individual dots correspond to data points above the 95th and below the 5th percentile of the total distribution. A two-tailed t test was performed for each data set. The average size of inclusions from cells microinjected with a combination of Pls1 and Pls2 antisera was 35.2% smaller than that for cells injected with preimmune sera. Parts A and B represent results from two independent experiments.
DISCUSSION
In this study, we characterized Pls1 and Pls2, two chlamydial proteins that localize to structures within the inclusion lumen and adjacent to the inclusion membrane. Pls1 and Pls2 show limited homology to two distinct regions in the passenger domain of PmpC (CT414), a member of the polymorphic outer membrane protein (Pmp) family (40, 42). Pmps are autotransporter membrane proteins containing a signal peptide, a cleavable passenger domain, and a β-barrel pore that facilitates presentation of the passenger domain on the bacterial surface (17). Because the Pmp family of proteins are significantly expanded in C. muridarum and Chlamydophila species and display an unusually high degree of polymorphism, these proteins are thought to be under selective pressure from the immune system (15, 24, 32, 35). This is supported by the finding that PmpD is a target for neutralizing antibodies (9) and by recent evidence that Pmps are efficient substrates for major histocompatibility complex class I presentation (16). Interestingly, like the Pmps, Pls1 and Pls2 are among the most polymorphic proteins in the sequenced C. trachomatis reference strains (5, 14). Because our findings indicate that significant proportions of Pls1 and Pls2 are secreted into the inclusion lumen, where they can associate with inclusion membranes and potentially the cytoplasm, we speculate that these proteins are under selective pressure to remain polymorphic, possibly to evade cytoplasmic innate immune surveillance mechanisms or antigen presentation pathways. While the molecular function of these proteins remains unknown, it is likely that they are important for chlamydial pathogenesis, since microinjection of anti-Pls antibodies hindered the ability of the inclusion to expand.
Based on membrane fractionation, Pls1 and Pls2 can be found as both soluble and membrane-bound forms. The low-MW processed forms of Pls2 preferentially partitioned with the soluble fraction of infected cells. These soluble forms of Pls2 are unlikely to be derived from lysed Chlamydia, because the release of bacterial cytoplasmic or membrane components was minimal under the isolation conditions used. In addition, a significant proportion of Pls1, Pls2, and their processed forms associated with membranes that had buoyancy properties distinct from those of Omp2- and MOMP-positive outer membranes (Fig. 6). Similarly, IncA-positive inclusion membranes showed only a partial overlap with Pls-positive membranes. Overall, these findings suggest that Pls-containing membranes, bacterial membranes, and inclusion membranes are distinct.
Given the lack of a secretion signal, it is not clear how Pls proteins are exported from the bacterial cytoplasm. We used the prediction program Secretome 2.0, which compares the amino acid compositions, secondary structures, and disordered regions of secreted proteins, to determine if Pls proteins share structural similarities with nonclassical secreted proteins (3). These prediction programs gave SecP scores for Pls1 (NN = 0.89), Pls2 (NN = 0.73), and Pls3 (NN = 0.89) that were significantly higher than the normal threshold (NN = 0.5). For comparison, the known autotransporter PmpC had a SecP score of 0.91. These results suggest that the structures of Pls proteins are consistent with those of proteins that reside outside the bacterial cell.
By immunofluorescence microscopy, we first detected Pls1 and Pls2 in association with RBs (<18 h), followed by prominent localization to punctate and globular structures in the inclusion lumen. Consistent with our membrane fractionation experiments, these globular structures displayed limited colocalization with MOMP or Omp2 (Fig. 3 and 4), suggesting that these represent extrabacterial material. It should be noted that ∼50% of Pls1 and Pls2 was found at the bottom of the density gradient (Fig. 6). This material likely represents Pls proteins that remained associated with bacteria (Fig. 2D and E).
Although the lack of classical signal peptides in Pls1 and Pls2 indicated that these proteins may be potential targets of TTS, the localization of Pls1, and to a lesser extent Pls2, to globular structures was insensitive to the TTS inhibitor C1. In contrast, treatment with low levels of β-lactam antibiotics led to the retention of Pls proteins in the bacterial cytoplasm (Fig. 4). Concomitant with this redistribution of Pls proteins, we observed a marked decrease in the intensity of antibody staining, presumably as a result of increased protein degradation with Amp treatment (Fig. 5). In addition to their role in blocking transpeptidases responsible for cross-linking peptidoglycan, β-lactam antibiotics also inhibit signal peptidase I (22). As a result, β-lactams inhibit the proteolytic processing of autotransporters such as PmpD, presumably by preventing their release from the cytoplasmic membrane and transport to the outer membrane (20). Upon removal of Amp, Pls proteins transiently colocalized with MOMP at the surfaces of enlarged RBs (Fig. 4), suggesting that the pathway for Pls secretion involves association with the bacterial surface. In addition, Pls1 and Pls2 display characteristics of proteins that are translocated to the chlamydial outer membrane, including extensive proteolytic processing into discrete fragments and incorporation into SDS-resistant and dithiothreitol-sensitive high-MW complexes (>200 kDa) (Fig. 2B).
Potential pathways for secretion include atypical signal peptides or a novel mechanism of sec-dependent secretion. Although we cannot exclude a role for TTS in the export of these proteins, the specific effect of Amp treatment on the localization and stability of Pls proteins led us to favor a secretion pathway involving a periplasmic intermediate. How periplasmic Pls1 and Pls2 are then transported across the outer membrane is also unclear. Given the homology of Pls proteins to the passenger domain of PmpC, we speculate that Pls1 and Pls2 may gain access to the cell surface in a manner analogous to that of autotransporters. We envision a scenario wherein Pls proteins are recognized by the translocation domain of Pmps and are coexported to the outer membrane, perhaps aided by cofactors like Omp85 (10). As a result, Pls1 and Pls2 secretion would be analogous to that observed in two-partner secretion systems (27). A similar transport pathway may be used by the C. pneumoniae protein Cpn0797, which also shares homology to autotransporters but which is considerably shorter than typical autotransporters (11). Whether the cognate outer membrane translocator is provided by recycling a cleaved Pmp transporter or by a gene product encoded elsewhere in the chromosome remains to be determined.
Alternatively, both soluble and membrane-bound Pls proteins may follow a more exotic secretion route. Recently, outer membrane vesicles have been postulated to represent a significant route for the secretion of proteins to the extracellular milieu (21, 25). These membrane vesicles bud off from outer membranes and can deliver LPS, outer membrane proteins, and periplasmic proteins to target cells (21). Indeed, ultrastructural studies by the Wyrick and Matsumoto groups have revealed that such vesicles exist in Chlamydia-infected cells, especially under conditions of cellular stress (13, 26). In addition, immunofluorescence and electron microscopy studies indicate that PmpD associates with extrabacterial vesicles in the inclusion lumen, further supporting a potential role for these vesicles in protein secretion (44). Given the parallels between PmpD and Pls secretion and the localization of Pls proteins to extrabacterial membranes, we propose that Pls1 and Pls2 may utilize outer membrane vesicles to access the inclusion lumen, inclusion membrane, and possibly the host cytoplasm. However, unlike the case for PmpD, where the bulk of protein remains associated with bacterial surfaces (20), Pls proteins are associated mostly with intraluminal vesicles.
The extent to which Pls proteins secreted into the inclusion lumen access the cytoplasm of the host cell is unclear. We have observed Pls-positive globular structures in close association with the cytoplasmic and luminal sides of the inclusion membrane (Fig. 3) and occasionally in the cytoplasm of infected cells (not shown). The inhibitory effects on inclusion expansion by microinjection of anti-Pls antibodies further suggest that these proteins are accessible to antibodies at the inclusion membrane or in the cytoplasm of the infected cell. The mechanism underlying the inhibition of inclusion growth is unclear, but the fact that both Pls1 and Pls2 needed to be neutralized is consistent with potential redundancy in the functions of these proteins.
Chlamydiae are highly adapted to living within host cells. As such, they have evolved a variety of efficient mechanisms to deliver proteins into the host cellular environment to promote bacterial replication and dissemination. In this study, we characterized Pls1 and Pls2, two proteins that are secreted into the inclusion lumen and associate with inclusion membranes. In general, how these obligate pathogens deliver proteins to the inclusion lumen and the range of biological activities occurring within the luminal space of the inclusion are unknown. Future work aimed at defining the biochemical processes occurring within the inclusion lumen will significantly further our understanding of chlamydial biology and pathogenesis.
Acknowledgments
We thank H. Caldwell, K. Fields, M. Tan, and D. Rockey for antibodies and strains and Jordan Cocchiaro and Joseph St. Geme III for helpful comments on the manuscript.
This work was supported by funds from the Burroughs Wellcome Program in Infectious Diseases and the National Institute for Allergy and Infectious Diseases (AI061538).
Editor: R. P. Morrison
Footnotes
Published ahead of print on 30 June 2008.
REFERENCES
- 1.Bannantine, J. P., R. S. Griffiths, W. Viratyosin, W. J. Brown, and D. D. Rockey. 2000. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 235-47. [DOI] [PubMed] [Google Scholar]
- 2.Belland, R. J., G. Zhong, D. D. Crane, D. Hogan, D. Sturdevant, J. Sharma, W. L. Beatty, and H. D. Caldwell. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 1008478-8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340783-795. [DOI] [PubMed] [Google Scholar]
- 4.Caldwell, H. D., J. Kromhout, and J. Schachter. 1981. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 311161-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson, J. H., S. F. Porcella, G. McClarty, and H. D. Caldwell. 2005. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect. Immun. 736407-6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chellas-Gery, B., C. N. Linton, and K. A. Fields. 2007. Human GCIP interacts with CT847, a novel Chlamydia trachomatis type III secretion substrate, and is degraded in a tissue-culture infection model. Cell. Microbiol. 92417-2430. [DOI] [PubMed] [Google Scholar]
- 7.Clifton, D. R., K. A. Fields, S. S. Grieshaber, C. A. Dooley, E. R. Fischer, D. J. Mead, R. A. Carabeo, and T. Hackstadt. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. USA 10110166-10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cocchiaro, J., Y. Kuma, E. Fischer, and R. H. Valdivia. 2008. Cytoplasmic lipid droplets are translocated into the inclusion lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. USA 1059379-9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crane, D. D., J. H. Carlson, E. R. Fischer, P. Bavoil, R. C. Hsia, C. Tan, C. C. Kuo, and H. D. Caldwell. 2006. Chlamydia trachomatis polymorphic membrane protein D is a species-common pan-neutralizing antigen. Proc. Natl. Acad. Sci. USA 1031894-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dautin, N., and H. D. Bernstein. 2007. Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 6189-112. [DOI] [PubMed] [Google Scholar]
- 11.Dong, F., R. Flores, D. Chen, J. Luo, Y. Zhong, Z. Wu, and G. Zhong. 2006. Localization of the hypothetical protein Cpn0797 in the cytoplasm of Chlamydia pneumoniae-infected host cells. Infect. Immun. 746479-6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fields, K. A., E. R. Fischer, D. J. Mead, and T. Hackstadt. 2005. Analysis of putative Chlamydia trachomatis chaperones Scc2 and Scc3 and their use in the identification of type III secretion substrates. J. Bacteriol. 1876466-6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giles, D. K., J. D. Whittimore, R. W. LaRue, J. E. Raulston, and P. B. Wyrick. 2006. Ultrastructural analysis of chlamydial antigen-containing vesicles everting from the Chlamydia trachomatis inclusion. Microbes Infect. 81579-1591. [DOI] [PubMed] [Google Scholar]
- 14.Gomes, J. P., W. J. Bruno, A. Nunes, N. Santos, C. Florindo, M. J. Borrego, and D. Dean. 2007. Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 1750-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomes, J. P., A. Nunes, W. J. Bruno, M. J. Borrego, C. Florindo, and D. Dean. 2006. Polymorphisms in the nine polymorphic membrane proteins of Chlamydia trachomatis across all serovars: evidence for serovar Da recombination and correlation with tissue tropism. J. Bacteriol. 188275-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grotenbreg, G. M., N. R. Roan, E. Guillen, R. Meijers, J. H. Wang, G. W. Bell, M. N. Starnbach, and H. L. Ploegh. 2008. Discovery of CD8+ T cell epitopes in Chlamydia trachomatis infection through use of caged class I MHC tetramers. Proc. Natl. Acad. Sci. USA 1053831-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson, I. R., and A. C. Lam. 2001. Polymorphic proteins of Chlamydia spp.—autotransporters beyond the proteobacteria. Trends Microbiol. 9573-578. [DOI] [PubMed] [Google Scholar]
- 18.Ho, T. D., and M. N. Starnbach. 2005. The Salmonella enterica serovar Typhimurium-encoded type III secretion systems can translocate Chlamydia trachomatis proteins into the cytosol of host cells. Infect. Immun. 73905-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hybiske, K., and R. S. Stephens. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 10411430-11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiselev, A. O., W. E. Stamm, J. R. Yates, and M. F. Lampe. 2007. Expression, processing, and localization of PmpD of Chlamydia trachomatis serovar L2 during the chlamydial developmental cycle. PLoS ONE 2e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuehn, M. J., and N. C. Kesty. 2005. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 192645-2655. [DOI] [PubMed] [Google Scholar]
- 22.Kuo, D., J. Weidner, P. Griffin, S. K. Shah, and W. B. Knight. 1994. Determination of the kinetic parameters of Escherichia coli leader peptidase activity using a continuous assay: the pH dependence and time-dependent inhibition by beta-lactams are consistent with a novel serine protease mechanism. Biochemistry 338347-8354. [DOI] [PubMed] [Google Scholar]
- 23.Lad, S. P., J. Li, J. da Silva Correia, Q. Pan, S. Gadwal, R. J. Ulevitch, and E. Li. 2007. Cleavage of p65/RelA of the NF-kappaB pathway by Chlamydia. Proc. Natl. Acad. Sci. USA 1042933-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longbottom, D., M. Russell, S. M. Dunbar, G. E. Jones, and A. J. Herring. 1998. Molecular cloning and characterization of the genes coding for the highly immunogenic cluster of 90-kilodalton envelope proteins from the Chlamydia psittaci subtype that causes abortion in sheep. Infect. Immun. 661317-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mashburn-Warren, L. M., and M. Whiteley. 2006. Special delivery: vesicle trafficking in prokaryotes. Mol. Microbiol. 61839-846. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto, A., and G. P. Manire. 1970. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J. Bacteriol. 101278-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazar, J., and P. A. Cotter. 2007. New insight into the molecular mechanisms of two-partner secretion. Trends Microbiol. 15508-515. [DOI] [PubMed] [Google Scholar]
- 28.Moulder, J. W. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 55143-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muschiol, S., L. Bailey, A. Gylfe, C. Sundin, K. Hultenby, S. Bergstrom, M. Elofsson, H. Wolf-Watz, S. Normark, and B. Henriques-Normark. 2006. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 10314566-14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nordfelth, R., A. M. Kauppi, H. A. Norberg, H. Wolf-Watz, and M. Elofsson. 2005. Small-molecule inhibitors specifically targeting type III secretion. Infect. Immun. 733104-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouellette, S. P., T. P. Hatch, Y. M. AbdelRahman, L. A. Rose, R. J. Belland, and G. I. Byrne. 2006. Global transcriptional upregulation in the absence of increased translation in Chlamydia during IFNgamma-mediated host cell tryptophan starvation. Mol. Microbiol. 621387-1401. [DOI] [PubMed] [Google Scholar]
- 32.Pedersen, A. S., G. Christiansen, and S. Birkelund. 2001. Differential expression of Pmp10 in cell culture infected with Chlamydia pneumoniae CWL029. FEMS Microbiol. Lett. 203153-159. [DOI] [PubMed] [Google Scholar]
- 33.Peters, J., D. P. Wilson, G. Myers, P. Timms, and P. M. Bavoil. 2007. Type III secretion a la Chlamydia. Trends Microbiol. 15241-251. [DOI] [PubMed] [Google Scholar]
- 34.Pirbhai, M., F. Dong, Y. Zhong, K. Z. Pan, and G. Zhong. 2006. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 28131495-31501. [DOI] [PubMed] [Google Scholar]
- 35.Read, T. D., R. C. Brunham, C. Shen, S. R. Gill, J. F. Heidelberg, O. White, E. K. Hickey, J. Peterson, T. Utterback, K. Berry, S. Bass, K. Linher, J. Weidman, H. Khouri, B. Craven, C. Bowman, R. Dodson, M. Gwinn, W. Nelson, R. DeBoy, J. Kolonay, G. McClarty, S. L. Salzberg, J. Eisen, and C. M. Fraser. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 281397-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schachter, J. 1999. Infection and disease epidemiology, p. 139-169. In R. S. Stephens (ed.), Chlamydia: intracellular biology, pathogenesis and immunity. American Society for Microbiology, Washington, DC.
- 37.Scidmore-Carlson, M. A., E. I. Shaw, C. A. Dooley, E. R. Fischer, and T. Hackstadt. 1999. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol. Microbiol. 33753-765. [DOI] [PubMed] [Google Scholar]
- 38.Shaw, A. C., B. B. Vandahl, M. R. Larsen, P. Roepstorff, K. Gevaert, J. Vandekerckhove, G. Christiansen, and S. Birkelund. 2002. Characterization of a secreted Chlamydia protease. Cell. Microbiol. 4411-424. [DOI] [PubMed] [Google Scholar]
- 39.Sisko, J. L., K. Spaeth, Y. Kumar, and R. H. Valdivia. 2006. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol. Microbiol. 6051-66. [DOI] [PubMed] [Google Scholar]
- 40.Stephens, R. S., S. Kalman, C. Lammel, J. Fan, R. Marathe, L. Aravind, W. Mitchell, L. Olinger, R. L. Tatusov, Q. Zhao, E. V. Koonin, and R. W. Davis. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282754-759. [DOI] [PubMed] [Google Scholar]
- 41.Subtil, A., C. Delevoye, M. E. Balana, L. Tastevin, S. Perrinet, and A. Dautry-Varsat. 2005. A directed screen for chlamydial proteins secreted by a type III mechanism identifies a translocated protein and numerous other new candidates. Mol. Microbiol. 561636-1647. [DOI] [PubMed] [Google Scholar]
- 42.Tanzer, R. J., D. Longbottom, and T. P. Hatch. 2001. Identification of polymorphic outer membrane proteins of Chlamydia psittaci 6BC. Infect. Immun. 692428-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandahl, B. B., A. Stensballe, P. Roepstorff, G. Christiansen, and S. Birkelund. 2005. Secretion of Cpn0796 from Chlamydia pneumoniae into the host cell cytoplasm by an autotransporter mechanism. Cell. Microbiol. 7825-836. [DOI] [PubMed] [Google Scholar]
- 44.Wehrl, W., V. Brinkmann, P. R. Jungblut, T. F. Meyer, and A. J. Szczepek. 2004. From the inside out—processing of the chlamydial autotransporter PmpD and its role in bacterial adhesion and activation of human host cells. Mol. Microbiol. 51319-334. [DOI] [PubMed] [Google Scholar]
- 45.Wolf, K., H. J. Betts, B. Chellas-Gery, S. Hower, C. N. Linton, and K. A. Fields. 2006. Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol. Microbiol. 611543-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong, G., P. Fan, H. Ji, F. Dong, and Y. Huang. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193935-942. [DOI] [PMC free article] [PubMed] [Google Scholar]