

Abstract
Yersinia pseudotuberculosis uses a plasmid (pYV)-encoded type III secretion system (T3SS) to translocate a set of effectors called Yops into infected host cells. YopJ functions to induce apoptosis, and YopT, YopE, and YopH act to antagonize phagocytosis in macrophages. Because Yops do not completely block phagocytosis and Y. pseudotuberculosis can replicate in macrophages, it is important to determine if the T3SS modulates host responses to intracellular bacteria. Isogenic pYV-cured, pYV+ wild-type, and yop mutant Y. pseudotuberculosis strains were allowed to infect bone marrow-derived murine macrophages at a low multiplicity of infection under conditions in which the survival of extracellular bacteria was prevented. Phagocytosis, the intracellular survival of the bacteria, and the apoptosis of the infected macrophages were analyzed. Forty percent of cell-associated wild-type bacteria were intracellular after a 20-min infection, allowing the study of the macrophage response to internalized pYV+ Y. pseudotuberculosis. Interestingly, macrophages restricted survival of pYV+ but not pYV-cured or ΔyopB Y. pseudotuberculosis within phagosomes: only a small fraction of the pYV+ bacteria internalized replicated by 24 h. In addition, ∼20% of macrophages infected with wild-type pYV+ Y. pseudotuberculosis died of apoptosis after 20 h. Analysis of yop mutants expressing catalytically inactive effectors revealed that YopJ was important for apoptosis, while a role for YopE, YopH, and YopT in modulating macrophage responses to intracellular bacteria could not be identified. Apoptosis was reduced in Toll-like receptor 4-deficient macrophages, indicating that cell death required signaling through this receptor. Treatment of macrophages harboring intracellular pYV+ Y. pseudotuberculosis with chloramphenicol reduced apoptosis, indicating that the de novo bacterial protein synthesis was necessary for cell death. Our finding that the presence of a functional T3SS impacts the survival of both bacterium and host following phagocytosis of Y. pseudotuberculosis suggests new roles for the T3SS in Yersinia pathogenesis.
Three species in the genus Yersinia are pathogenic for humans: the genetically closely related Y. pseudotuberculosis and Y. pestis and the more distantly related Y. enterocolitica (1, 52). These bacteria share a common tropism for lymphoid tissues and the ability to resist the host protective innate immune responses, even though their routes of transmission are quite different. Y. pseudotuberculosis and Y. enterocolitica are transmitted through contaminated food or water, while Y. pestis is transmitted through flea bites or aerosols (32, 52). A plasmid-encoded type III secretion system (T3SS) and a set of secreted toxins called Yops are required for the virulence of these bacterial pathogens (10). Contact with a host cell initiates the T3SS to inject a set of six Yop effector proteins, namely, YopE, YopH, YopJ, YopT, YopM, and YpkA, into the cytosol of the eukaryotic cell (10, 50). YopB and YopD have been implicated in the formation of a transmembrane channel, or translocon, that functions to deliver Yop effectors across the host plasma membrane (10, 13, 50). Upon delivery into a host cell via a T3SS translocon, Yop effectors modulate eukaryotic signaling pathways for the benefit of the bacterial pathogen (50). T3SSs are found in a number of other gram-negative pathogens, such as Salmonella enterica, where they play key roles in promoting bacterial pathogenesis (13, 19).
Expression and secretion of the Yops is regulated by temperature and other environmental signals. Upon shift of the growth temperature from 26°C to 37°C, the transcription of genes that encode the T3SS and the Yops is upregulated (7, 12). This gene expression increases further once the bacteria come into contact with eukaryotic cells, and only then are the Yop effector proteins secreted (33). At 37°C in vitro, decreasing the Ca2+ concentration to a micromolar level stimulates Yop secretion into growth media (10). This low-Ca2+ condition has been adopted by many researchers to maximally induce the production of Yops to assess their roles in pathogenesis. However, this condition may not reflect what happens in vivo. The total Ca2+concentration in serum is as high as 2.5 mM and it could be much higher in the small intestine. Millimolar levels of Ca2+ do not allow maximum Yop production and inhibit their secretion (9). Therefore, before Yersinia encounters host cells such as macrophages in vivo, it is unlikely that Yops are maximally expressed or secreted.
Several Yops, including YopH, YopE, YopT, and YpkA, interfere with the proper function of the actin cytoskeleton and therefore inhibit the phagocytosis of Yersinia (2, 15, 36, 37). YopH is a protein tyrosine phosphatase (5, 16, 57). YopE is a GTPase-activating protein; it inactivates the Rho family of small GTPases (4, 51). YopT is a cysteine protease that cleaves at the C termini of the Rho GTPases, thereby preventing membrane anchoring (42). YpkA contains a serine/threonine kinase domain and a GDI domain interacting with members of the Rho family GTPases (3, 23, 34). Because the Rho family of small GTPases are key regulators of the actin cytoskeleton, YopE, YopT, and YpkA/YopO all function to perturb actin cytoskeleton dynamics and interfere with bacterial uptake by macrophages. However, even when Yops are maximally expressed before infection, one-third to one-half of the bacteria that come in contact with cultured macrophages are internalized (15, 36, 37). Thus, Yop-expressing Yersinia reduces, but does not completely block, their uptake by macrophages.
In addition to countering phagocytic mechanisms, Yops are important for reducing cytokine production by macrophages. Tumor necrosis factor α (TNF-α) is a multifunctional proinflammatory cytokine secreted by macrophages following infection. YopJ is required by Yersinia to inhibit the production of TNF-α in macrophages infected in vitro (6, 30). Furthermore, YopJ, YopE, and YopT function in concert to inhibit the production, maturation, and secretion of interleukin-1β by macrophages (41). In this context, YopT and YopE catalytic activities have been shown to inhibit pore formation by the T3SS translocon (50). In the absence of YopT and YopE, pore formation in macrophages infected with Y. enterocolitica at a high multiplicity of infection (MOI) can result in the activation of caspase-1, the maturation and secretion of interleukin-1β, and cell death (41, 43).
YopJ has acetyltransferase activity and acetylates the Ser and Thr residues critical for the activation of the mitogen-activated protein (MAP) kinase kinases and the inhibitor κB kinase β. This activity allows YopJ to repress signaling through the MAP kinase and nuclear factor κB (NF-κB) pathways and to inhibit the production of proinflammatory cytokines (28). YopJ is also required by Yersinia to induce the apoptosis of infected macrophages (24, 26, 27, 39, 56). However, for YopJ-induced apoptosis to occur, a signal is required to activate the intrinsic apoptotic machinery in macrophages (29, 54). Infection with gram-negative bacteria activates both a proinflammatory pathway and an apoptotic pathway of the macrophage; for example, Toll-like receptor 4 (TLR4) can initiate both of these two pathways (18, 53, 55). Normally, the activation of MAP kinases and NF-κB, two key components of the proinflammatory pathway that leads to the secretion of TNF-α, counteracts the apoptotic pathway; since YopJ blocks these protective pathways, macrophages rapidly undergo apoptosis by default following infection with Yersinia (56). Thus, YopJ does not induce apoptosis per se, but through inhibiting the survival pathway, YopJ allows the intrinsic apoptotic pathway of macrophages to prevail.
Y. pseudotuberculosis is generally considered an extracellular pathogen in vivo (45), and therefore most studies have examined the function of the T3SS and Yops in promoting pathogenesis from outside the host cell. However, recent studies have shown that Y. pseudotuberculosis shares with Y. pestis the ability to survive and replicate in murine macrophages (14, 35). Studies analyzing the survival of Y. pseudotuberculosis and other Yersinia species in macrophages have generally utilized bacteria grown at 26°C, and therefore the T3SS is not primed to translocate Yops during phagocytosis (14, 35). Because Yersinia grown at 37°C can be phagocytosed by macrophages (15, 36, 37), it is important to determine if T3SS and Yop activities modulate host responses to the intracellular bacteria.
Roy et al. obtained evidence that Y. pseudotuberculosis primed to express the T3SS could survive in macrophages following phagocytosis (38). However, the intracellular survival of Y. pseudotuberculosis was significantly enhanced in macrophages defective for a calcium-regulated phagosome-lysosome fusion pathway (38). It was proposed that the permeabilization of plasma membrane by the T3SS translocon during bacterial phagocytosis promotes calcium influx, increased lysosome fusion with phagosomes, and decreased intracellular survival of Y. pseudotuberculosis in murine macrophages (38). It is also possible that the T3SS and the Yops could function to counteract host responses and thus promote pathogenesis when the bacterium is within an intracellular location.
Here we investigated how the deployment of the T3SS and Yops during phagocytosis alters the macrophage response to intracellular Y. pseudotuberculosis. Our results show that the presence of a functional T3SS during phagocytosis decreases the survival of the internalized bacteria but at the same time allows intracellular Yersinia to kill the host cell by a TLR4 and YopJ-dependent mechanism. We discuss how the outcome of this interaction could influence Yersinia pathogenesis.
MATERIALS AND METHODS
Bacterial strains and infection conditions.
The Y. pseudotuberculosis serogroup I strain 32777 (previously known as IP2777 [44]) and its derivatives were used in this study. Standard two-step allelic recombination techniques were used to introduce specific codon substitutions into the coding sequences for YopJ, YopT, YopE, and YopH in 32777. A suicide plasmid, pSB890-YopJC172A (56), was conjugated into 32777 by use of Escherichia coli strain S17λpir. Integration of this plasmid into the virulence plasmid was selected on Yersinia selection medium (Oxoid) supplemented with tetracycline. The loss of pSB890 and the replacement of the wild-type locus with yopJ(C172A) were achieved by selecting for resistance to sucrose. The maintenance of the virulence plasmid was verified on magnesium oxalate and Congo red agar. Clones containing yopJ(C172A) were identified by PCR amplification of the entire coding region, followed by restriction analysis and sequencing as described before (56). The resulting strain was designated mJ. Similarly, pSB890 plasmids containing the coding sequences of YopTC139A, YopER144A, and YopHR409A were used sequentially to generate multiple-yop-mutant strain 32777 mJTEH. To construct pSB890-YopTC139A, the coding sequence of YopTC139A was released with BglII and BamHI from plasmid pET28a-YopTC139A (42) and cloned into pSB890 at the BamHI site. To confirm the presence of the resulting yopT(C139A) mutation, the coding sequence of YopT was PCR amplified with primer T4 (5′-CGGATCCCTGAGTCACCTATTGG-3′) and T3 (5′-TATGGTACCGCGCATATGATTGTTT-3′) (49) and digested with BbvCI. Cleavage of the PCR product is diagnostic for the presence of the mutation. To construct pSB890-YopER144A, the coding sequence of YopER144A was released from pGEX2T-YopER144A (4) with BamHI and ligated into the BamHI site of pSB890. To identify the resulting yopE(R144A) mutation, the coding sequence of YopE was PCR amplified with primer YopE prom (5′-GTCGACTATCAACTTAACCAAAGCACT-3′) and YopE2 (5′-CGGGATCCCCATATCACATCAATGACA-3′) (49) and digested with EarI. A lack of cleavage is diagnostic for the presence of the mutation. The plasmid pSB890-YopHR409A and the method used to confirm the presence of the yopH(R409A) mutation have been described before (22). The ΔyopB strain in the 32777 background was created and identified as described before (30). All of the mutations were further confirmed with sequencing following PCR amplification. Plasmid p67GFP3.1 was introduced into these strains through conjugation as described before (35).
Infection conditions.
To prepare bacteria for infection, overnight cultures grown at 26°C in Luria-Bertani broth (LB) were diluted to an optical density at 600 nm (OD600) of 0.1 in LB broth supplemented with 2.5 mM CaCl2. The cultures were shaken at 37°C for 2 h. Subsequently, all solutions coming in contact with bacteria were prewarmed to 37°C. Bacteria were washed once and resuspended in Hanks balanced salt solution (HBSS) and diluted into cell culture medium (see next section) to infect macrophages at a MOI of 10. After centrifugation for 5 min at 200 × g to bring the bacteria into contact with the macrophages, an incubation was performed at 37°C for 15 min; therefore, a total of 20 min was allowed for the cells to take up the bacteria. Next, the cells were washed with phosphate-buffered saline (PBS) gently and incubated in cell culture medium containing gentamicin (8 μg/ml) for 1 h to kill extracellular bacteria. The cells were washed again with PBS and incubated in medium containing gentamicin (4.5 μg/ml) until the time indicated. Where indicated, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at 0.5 mM for 1 h to induce green fluorescent protein (GFP) expression.
Macrophage cultures.
C57BL/6 and C57BL/10ScNJ mice defective for the expression of TLR4 (Tlr4lps-del mice) were obtained from Jackson Laboratory. Bone marrow-derived macrophages (BMDMs) were obtained as previously described (8). Twenty-four hours before infection, the cells were seeded into multiwell plates or chambers in Dulbecco's modified Eagle's medium containing 15% L-cell conditioned medium, 10% fetal bovine serum (Gibco), 1 mM pyruvate, and 2 mM glutamate. To analyze the percentage of bacterial phagocytosis, BMDMs were seeded at a density of 0.9 × 105 cells/chamber into eight-chamber Lab-Tek Permanox slides (Nunc Inc.). For electron microscopy, BMDMs were seeded on Rinzyl in six-well plates at a density of 1 × 106 cells/well. For all other assays, the BMDMs were seeded at a density of 1.5 × 105 cells/well in 24-well tissue culture plates.
To quantify the percentages of BMDMs carrying live bacteria at different times postinfection, after GFP expression was induced for 1 h, sequential images of green fluorescence and phase contrast were taken from a random field by use of a Zeiss Axiovert S100 microscope with a 32× objective. The numbers of cells that carry no bacteria, that carry one to five bacteria, and that carry more than five bacteria were enumerated after the images were assembled in Adobe Photoshop.
Immunofluorescence microscopy.
To analyze the percentage of bacterial phagocytosis, GFP expression was induced in bacterial cultures before infection to allow the detection of total bacteria. At the end of a 20-min uptake period, the cells were washed and fixed in 4% paraformaldehyde in PBS at room temperature for 30 min. Extracellular bacteria were then immunolabeled with rabbit anti-Yersinia antiserum SB349 followed by goat anti-rabbit antibody conjugated to Alexa Fluor 594 (Molecular Probes) as described previously, except that the permeabilization step was omitted (14). The slides were examined by epifluorescence microscopy using a Zeiss Axioplan2 microscope equipped with a 100× oil immersion objective. In each experiment, about 100 BMDM-associated bacteria were counted from merged images of phase contrast and green and red fluorescence captured with a Spot camera (Diagnostic Instruments, Inc.) and assembled in Adobe Photoshop.
To analyze cell death, after infection and fixation, macrophage membranes were permeabilized in 0.1% Triton X-100 in PBS for 10 min, followed by staining with rabbit polyclonal anti-cleaved caspase-3 antibody (Trevigen) and secondary antibody goat anti-rabbit immunoglobulin G conjugated to Alexa Fluor 594 (Molecular Probes). Alternatively, the cells were labeled with an in situ cell death detection kit, TMR red (Roche), according to the manufacturer's instructions. Images of phase contrast and green (for GFP-positive bacteria) and red fluorescence were taken from a random field to count apoptotic cells. Most of the fields contained more than 250 cells.
Viable count assay.
After BMDMs were infected for the indicated times, the medium was aspirated carefully and the cells were lysed with 500 μl of 0.1% Triton X-100 in HBSS to release intracellular bacteria. After removal of the lysate, the wells were then washed a second time with 500 μl of LB. These lysate and wash solutions were combined and serially diluted to enumerate CFU on LB plates.
Western blot analysis.
Monoclonal antibodies to YopJ were produced by immunizing mice with Y. pseudotuberculosis YopJ produced in insect cells (a generous gift of Kim Orth, University of Texas Southwestern Medical Center). Hybridomas were selected by screening YopJ-transfected 293T cells by immunoblotting and flow cytometry.
To detect the expression of YopJ in bacteria, bacterial lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and analyzed by immunoblotting with antibodies against YopJ (clones C2 and D3) and DnaK (clone 8E2/2; Stressgen). Secondary antibody was IRDye800-conjugated anti-mouse immunoglobulin G (Rockland). The membrane was then scanned with an Odyssey VI scanner (Li-Cor Biosciences).
TNF-α secretion.
At the indicated times postinfection, tissue culture medium was collected and subjected to centrifugation for 10 min at 200 × g. TNF-α concentration in the supernatant was measured with Quantikine mouse TNF-α immunoassay from R&D Systems, Inc., following the manufacturer's instructions.
LDH release.
Lactate dehydrogenase (LDH) content in the supernatant collected from infected wells or from wells left uninfected was measured in triplicate with CytoTox 96 nonradioactive cytotoxicity assay (Promega) following the manufacturer's instructions. Total LDH was determined from separate uninfected wells that had been lysed by a freeze-thaw cycle in the medium. The percentage of LDH released was calculated with the following formula: LDHreleased/LDHtotal.
Electron microscopy.
After infection, the cells were washed with PBS and fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in PBS at room temperature for 30 min. This was followed by an additional wash step with PBS. Thin-section transmission electron microscopy was carried out by the Central Microscopy Imaging Center at Stony Brook University. Digital images were acquired with an FEI BioTwinG2 transmission electron microscope.
Statistical analysis.
Percentage data was first transformed with the formula log[−log(percentage)] to achieve Gaussian distribution. Statistical analysis was performed with InStat software. The results were analyzed with repeated one-way analysis of variance followed by the Tukey-Kramer multiple comparisons test. A P value of less than 0.05 was considered significant.
RESULTS
Phagocytosis of plasmid-cured, wild-type, and Yop mutant Y. pseudotuberculosis by BMDMs.
YopE, YopH, YopT, and YpkA all function to antagonize the phagocytosis of Yersinia by macrophages (15, 36, 37). However, the inhibition of phagocytosis by these Yops is not complete. For example, using the serogroup III strain YPIII of Y. pseudotuberculosis, Rosqvist et al. showed that under conditions in which the expression of the Yops was maximally induced before infection, a significant percentage (∼34%) of bacteria were phagocytosed (36). To better understand the impact of the T3SS and Yops on the host response to intracellular Y. pseudotuberculosis, we first needed to determine the percentage of bacteria that would be phagocytosed by BMDMs under our experimental conditions. As a wild-type strain we used the pYV+ 32777 serogroup I strain, which has been shown to survive and replicate in macrophages (14). Derivatives of 32777 encoding catalytically inactive YopJ (mJ) or catalytically inactive YopJ, YopT, YopE, and YopH (mJTEH) were constructed and studied in parallel to the wild-type strain to determine the importance of these Yops on intracellular pathogenesis (see Materials and Methods). A pYV-cured derivative of 32777 (32777c) was used as a T3SS null control. To mimic an in vivo infection condition, we preconditioned the bacteria by growth at 37°C in LB containing 2.5 mM CaCl2, which is sufficient to inhibit Yop secretion at 37°C (46). As shown by the results from immunoblotting whole-cell lysates (Fig. 1A), bacteria prepared under these conditions expressed low levels of YopJ (lane 1), which were similar to the levels after the bacteria were incubated in tissue culture medium (lane 2) but were dramatically lower than when bacteria were cultured under maximally inducing conditions (lane 3).
FIG. 1.

Phagocytosis of Y. pseudotuberculosis pregrown under moderate Yop-inducing conditions. (A) Expression of YopJ protein in Y. pseudotuberculosis strain 32777 under different growth conditions. Lanes 1 and 2, overnight 32777 cultures were diluted into fresh LB with 20 mM CaCl2 to an OD600 of 0.1 and cultured at 37°C for 2 h. Aliquots were washed once in HBSS and then resuspended and cultured for an additional 20 min in fresh LB-CaCl2 (lane 1) or tissue culture medium (as described in Materials and Methods) (lane 2). Lane 3, overnight culture was diluted into LB with 20 mM MgCl2 and 20 mM sodium oxalate to an OD600 of 0.1 and cultured at 26°C for 1 h and 37°C for additional 1.5 h. The equivalent of 5 × 107 CFU bacteria was lysed in 1× Laemmli buffer and analyzed by immunoblotting using monoclonal anti-YopJ antibody and anti-DnaK antibody. (B) BMDMs were infected at a MOI of 10 for 20 min with GFP-expressing 32777c, 32777, mJ, or mJTEH. Percentages of extracellular and partially phagocytosed bacteria were identified after immunolabeling without permeabilization. From each experiment, about 100 BMDM-associated bacteria were counted after sequential microscopic pictures of green and red fluorescent and phase-contrast pictures were taken. Data are presented as the percentages of intracellular bacteria from total cell-associated bacteria. Values represent means and standard deviations determined from three independent experiments. The percentage of intracellular 32777c was significantly different (P < 0.01) from those for 32777 and mJ.
To determine the percentages of bacteria that would be phagocytosed, the bacteria were used to infect BMDMs at a MOI of 10 for 20 min, after which the numbers of cell-associated and internalized bacteria were enumerated by immunofluorescence microscopy. To facilitate the detection of bacteria, cultures of the various strains used were induced to express GFP before infection. As shown in Fig. 1B, 68% of cell-associated 32777c was phagocytosed by BMDMs. In comparison, the wild-type 32777 and mJ strains were more resistant to phagocytosis, as 39% and 42%, respectively, of the bacteria coming in contact with BMDMs became intracellular (Fig. 1B). The multiple Yop mutant mJTEH displayed an intermediate phenotype, with 56% internalization. These results confirmed that YopJ is not involved in antagonizing phagocytosis, while YopT, YopE, and YopH do participate in this process (15). Most importantly for this study, the fact that ∼40 to 56% of the pYV+ bacteria were phagocytosed allowed us to study the impact of Yops on the response of macrophages to intracellular Yersinia.
The presence of functional T3SS decreased the survival of intracellular Y. pseudotuberculosis in BMDMs.
Next, the fate of the intracellular bacteria was determined. After initial contact of non-GFP-expressing bacteria with BMDMs for 20 min, gentamicin was included continuously in the medium at a low concentration to prevent the growth of extracellular Yersinia (35). At different times after the initial contact, IPTG was added for 1 h to induce the expression of GFP from live intracellular bacteria (GFP induction assay) (35) and the samples were examined by microscopy. One hour after the initial contact, BMDMs contained from 1 to >10 GFP-positive bacteria per cell, depending on the antiphagocytosis ability of the strain (Fig. 2A to D). At this time point, the BMDMs infected with either wild-type 32777 or the mJ strain displayed a round shape (Fig. 2B and C), which was less prominent in the cells infected with 32777c or mJTEH. This rounding phenotype had been linked to the effects of YopE and/or YopT on the actin cytoskeleton (21, 37). As the incubation time was extended, the number of BMDMs containing GFP-positive bacteria decreased, while in those BMDMs that remained infected, the number of bacteria increased (Fig. 2E to L). In some cells, after 24 h, the intracellular bacteria formed a cluster in which individual bacteria could no longer be distinguished (Fig. 2I to L). The decreases in the numbers of infected BMDMs were similar for the cells infected with 32777, mJ, and mJTEH, while a greater number of BMDMs infected with 32777c contained GFP-positive bacteria (Fig. 2A, E, and I).
FIG. 2.

Survival and replication of Y. pseudotuberculosis inside BMDMs, as determined by fluorescence microscopy. BMDMs were infected with the indicated strains that carried pGFP at a MOI of 10 for 1 h (A to D), 6 h (E to H), or 24 h (I to L). The survival of extracellular bacteria was prevented by the use of gentamicin as described in Materials and Methods. GFP expression was induced from the surviving intracellular bacteria with IPTG for 1 h before microscopic observation. Overlay images of phase contrast and green fluorescence from one experiment representative of three are shown. Arrowheads in panels B and C indicate cells displaying cytotoxicity, as shown by round shapes.
As a more quantitative approach to measure the intracellular survival of the different Y. pseudotuberculosis strains, the numbers of bacterial CFU in macrophages at different times postinfection were determined. Consistent with the results of the GFP induction assay, a small decrease in CFU for 32777c was seen at 4 h postinfection; however, after 24 h, the number of CFU was similar to that seen at the 1-h time point (Fig. 3). The number of CFU recovered from the cells infected with 32777 or mJ continued to decrease with time, and only 6% of the original inoculums were recovered at 24 h postinfection (Fig. 3). The mJTEH mutant displayed an intermediate phenotype at 4 h, consistent with the fact that more bacteria of this strain were phagocytosed (Fig. 1). However, after 24 h, only 5% of the inoculum was recovered (Fig. 3). Collectively, the results in Fig. 2 and 3 indicated that, compared to what was seen for the plasmid-cured 32777c strain, more of the pYV+ bacteria phagocytosed were unable to survive and replicate. As these restrictions on intramacrophage survival were similar for the wild type and the two Yop mutant strains, it appeared that the presence of a functional T3SS was the major determinant of reduced survival of intracellular bacteria.
FIG. 3.

Survival of Y. pseudotuberculosis inside BMDMs, as determined by viable count assay. BMDMs were infected with the indicated strains for 1, 4, or 24 h, as described for Fig. 2. At indicated times, the infected BMDMs were lysed and the CFU of intracellular bacteria determined by a viable count assay, as described in Materials and Methods. Results shown are the means and standard deviations of log(CFU) from three independent experiments with triplicate infection wells. P values are greater than 0.05 if not indicated.
To investigate whether a functional T3SS was indeed causing a survival decrease for the intracellular bacteria, a ΔyopB strain was generated in the background of 32777. As expected, this mutant strain still secretes other Yop proteins under low-calcium conditions (data not shown; similar to what is described for strain YP18 in reference 30). When tested under the intracellular infection conditions for 24 h, the ΔYopB strain replicated to the same extent as 32777c (Fig. 4A and C) and occupied the BMDMs to a percentage similar to that seen for 32777c (Fig. 4A and B). The progressions of intracellular replication are also similar between the ΔYopB strain and 32777c but are drastically different from that of 32777 (Fig. 4A and B). One hour postinfection, the following majorities of BMDMs infected with any strain contained one to five bacteria: 51%, 60%, and 53% in 32777c-, 32777-, and ΔYopB strain-infected samples, respectively. At 5 h postinfection, in 32777c- and ΔYopB-infected cells, the majority contained more than five bacteria, reflecting the fact that more bacteria replicate intracellularly. In contrast, in 32777-infected BMDMs, while the percentage of cells carrying one to five bacteria decreased from 60% to 28%, the percentage of cells carrying more than five bacteria increased only slightly, from 11% to 15%. In the meantime, the percentage of bacterium-free cells increased from 30% to 57% (Fig. 4B), suggesting that in wild-type 32777-infected samples, more BMDMs were able to restrict and clear the intracellular bacteria. In contrast, from 1 h to 5 h postinfection, in 32777c-infected BMDMs the percentage of cells that contained no bacteria increased only slightly, from 8% to 14%. The corresponding quantity for ΔYopB strain-infected cells is from 18% to 21%, a marginal increase as well. This result supports the idea that the presence of a functional T3SS decreases the survival of intracellular Y. pseudotuberculosis.
FIG. 4.

Survival and replication of Y. pseudotuberculosis inside BMDMs, as determined by GFP induction and viable count assays. BMDMs were infected with the indicated strains for 1, 4, or 24 h, as described for Fig. 2A. (A) Overlaid images of green fluorescence and phase contrast. (B) From the random images in panel A, the numbers of BMDMs containing no bacteria (none), one to five bacteria per cell (1-5), or more (>5) were enumerated and converted into percentages of total cells. Results shown are the means and standard deviations from two (for 1-h) or three (for 5- and 24-h) experiments. More than 50 cells were counted for each condition of each experiment. (C) Twenty-four hours postinfection, the CFU of intracellular bacteria was determined as described for Fig. 3. Results shown are the means and standard deviations of log(CFU) from three independent experiments with duplicate infection wells.
pYV-containing Y. pseudotuberculosis resided in a smaller phagosome in BMDMs than did the plasmid-cured strain.
Thin sections of macrophages infected for 5 h were examined by electron microscopy to determine if there were morphological differences in the phagosomes containing pYV-cured, wild-type, and Yop mutant Y. pseudotuberculosis. Macrophages infected with 32777c contained large phagosomes typically occupied by multiple bacteria (Fig. 5A and B). Macrophages infected with wild-type 32777 harbored bacteria that were present in moderately sized or tight-fitting phagosomes (Fig. 5C and D). The phagosome morphologies observed in BMDMs infected with the other pYV+ strains, mJ (Fig. 5E and F) and mJTEH (Fig. 5G and H), were similar to those seen for 32777-infected cells. The moderately sized phagosomes containing pYV+ bacteria contained from one to several organisms (Fig. 5C to H). Some of the pYV+ bacteria in moderately sized phagosomes appeared to be degraded (Fig. 5C and F). Even though it cannot be ruled out that the BMDMs ingested some dead bacteria killed by gentamicin in the medium, the presence of partially degraded bacteria in BMDMs infected with the pYV+ bacteria but not pYV-cured bacteria in the EM pictures, as well as the observed morphological differences between phagosomes containing pYV-cured and pYV+ bacteria, is consistent with the idea that the insertion of the T3SS translocon could result in more-efficient restriction of the survival of the internalized bacteria (38).
FIG. 5.
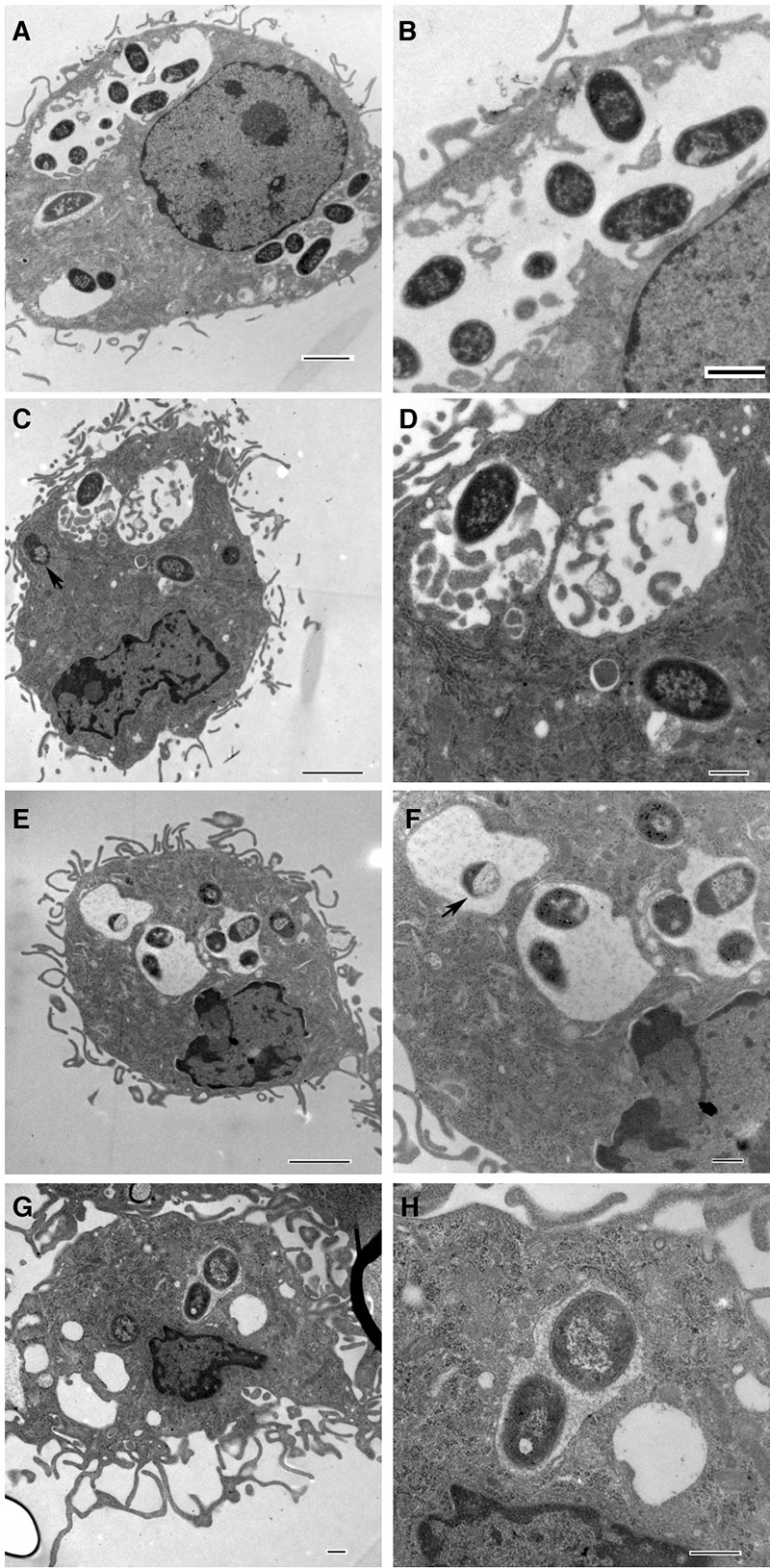
Morphology of the BMDMs and intracellular bacteria, as determined by thin-section electron microscopy. BMDMs were infected for 5 h as described for Fig. 2 with 32777c (A and B), 32777 (C and D), mJ (E and F), or mJTEH (G and H). Panels B, D, F, and H are enlargements of phagosomes from panels A, C, E, and G, respectively. Bars = 2 μm (A, C, and E), 1 μm (B), and 500 nm (D, F, G, and H). Arrows in panels C and F indicate partially degraded bacteria.
Apoptosis in BMDMs containing wild-type Y. pseudotuberculosis required YopJ and TLR4.
We next investigated the responses of the macrophages under these infection conditions. First, we determined whether BMDMs undergo cell death while harboring intracellular Y. pseudotuberculosis. Initially, cell death was measured by the amount of LDH released during the course of infection. LDH is a cytosolic enzyme that is released at the late stages of apoptosis, when the cell membrane is sufficiently damaged. After BMDMs were infected with wild-type 32777 for 21 h, ∼24% of total LDH was released (Fig. 6A). This amount is significantly different (P < 0.01) from the amounts of LDH released from the BMDMs infected with 32777c, mJ, and mJTEH. Moreover, macrophage cell death seems to have little impact on the initial survival of the intracellular bacteria, as the release of LDH was not detectable until ∼8 h postinfection (data not shown). In addition, the decrease of intracellular bacteria is independent of the cell death, because both mJ and 32777 survive to the same extents intracellularly (Fig. 3) and only 32777 causes significant macrophage death (Fig. 6A). The result obtained with mJ indicates that YopJ catalytic activity is important for the death of macrophages containing intracellular Y. pseudotuberculosis. Furthermore, the result obtained with mJTEH indicates that the absence of YopE and YopT catalytic activities did not result in measurable cell lysis due to pore formation (43), most likely due to the low MOI used.
FIG. 6.

Cell death of infected BMDMs requires YopJ and TLR4. BMDMs were infected with the indicated strains as described for Fig. 2 or were left uninfected. At 21 h postinfection, LDH released in the medium was determined with a CytoTox 96 assay. Total LDH was determined from two independent uninfected wells, in which all the cells were lysed by a freeze-thaw cycle. (A) Values represent means and standard deviations of percentages of LDH released from six experiments with duplicate infection wells. The level of LDH released from cells infected with 32777 (indicated with *) is significantly different from those seen for cells infected with other strains or left uninfected (UI) but otherwise treated similarly (P < 0.01). (B) BMDMs prepared from C57BL/6 or C57BL/10ScNJ (Tlr4lps-del) mice were infected with 32777 or mJ. Data shown are the means and standard deviations of results from six infection wells from two independent experiments. The P value was as indicated.
Signaling through TLR4 is important for YopJ-induced death of macrophages during extracellular Yersinia infection (18, 55). An LDH release assay was performed on infected BMDMs from mice defective for expression of TLR4 (Tlr4lps-del) to determine if TLR4 signaling is important for macrophage cell death in response to intracellular Y. pseudotuberculosis. The level of LDH release observed for Tlr4lps-del BMDMs was reduced compared to that for control BMDMs after infection with wild-type 32777 (Fig. 6B). Similar levels of apoptosis were seen for Tlr4lps-del and control BMDMs infected with mJ. This result indicated that TLR4 was necessary for cell death in macrophages infected with intracellular Y. pseudotuberculosis.
As a more specific marker of apoptosis, the activation of caspase-3 in macrophages infected with Y. pseudotuberculosis was determined by immunofluorescence microscopy using an antibody specific for cleaved caspase-3. In parallel, the presence of live bacteria in the BMDMs was determined by a GFP induction assay (Fig. 7A). Six hours after infection, 14% of BMDMs infected with wild-type 32777 were positive for the cleaved caspase-3 (Fig. 7C). This level is significantly different (P < 0.01) from those seen for the BMDMs infected with the other strains (Fig. 7C). GFP-positive bacteria could be detected in BMDMs that were positive for cleaved caspase-3 (Fig. 7B). As a second marker of apoptosis, the fragmentation of genomic DNA was assessed with terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL). Six hours postinfection, 8.5% of BMDMs infected with wild-type 32777 were TUNEL positive; again, this level was significantly different (P < 0.05) from the levels of TUNEL-positive BMDMs that were infected with other strains tested (Fig. 6D). Similarly, live bacteria could also be detected in cells that were TUNEL positive (data not shown). Overall, these results indicate that a fraction (∼10% to 20%) of BMDMs harboring wild-type Y. pseudotuberculosis underwent apoptosis in a YopJ-dependent manner.
FIG. 7.

Apoptosis of BMDMs, as measured by anti-cleaved caspase-3 immunostaining or TUNEL. BMDMs were infected with the indicated strains for 6 h and GFP expression was induced during the last hour as described in the legend for Fig. 2. Cleaved caspase-3 was identified with fluorescent microscopy after immunolabeling. Green color indicates the surviving bacteria and red the apoptotic cells. Images shown are representative of three experiments. (A) Top, phase-contrast image of the infected cells; bottom, overlay of red and green fluorescent signals. (B) Localization of intracellular 32777 with activated caspase-3 in the same cell. (C) Percentages of cleaved caspase-3 positive cells. *, The P value is significantly different (P < 0.01), comparing to all the others. (D) Percentages of TUNEL-positive cells, as determined by fluorescence microscopy. *, P value of <0.05, comparing to all the others. For panels C and D, the percentages of apoptotic cells were determined after counting the red apoptotic cell or nuclei among the total cells in a random field after sequential pictures were taken. Values shown are the means and standard deviations from three independent experiments.
Bacterial protein synthesis after phagocytosis is required for apoptosis of BMDMs containing intracellular Y. pseudotuberculosis.
The above-described results indicate that macrophages containing wild-type 32777 undergo YopJ- and TLR4-dependent apoptosis; however, they do not address whether the intracellular bacteria contribute to the cell death. It was possible that during phagocytosis, the translocation of YopJ and the stimulation of TLR4 from the extracellular bacteria were sufficient to trigger apoptosis under these infection conditions. To address this possibility, we asked if de novo protein synthesis by intracellular Y. pseudotuberculosis was required for apoptosis. To inhibit bacterial protein synthesis after phagocytosis, chloramphenicol was added to the infected macrophages 20 min after infection, at the same time as gentamicin. Chloramphenicol treatment prevented the replication of intracellular Y. pseudotuberculosis (data not shown) and did not induce LDH release from uninfected BMDMs (Fig. 8A). Importantly, chloramphenicol treatment reduced LDH release from wild-type 32777-infected BMDMs to the level seen for uninfected BMDMs (Fig. 8A). These results indicated that de novo protein synthesis by intracellular Y. pseudotuberculosis was required to induce macrophage apoptosis.
FIG. 8.

Role of bacterial protein synthesis in BMDM apoptosis and TNF-α secretion. BMDMs were left uninfected (UI) or were infected with the indicated strains for 21 h, as described for Fig. 2. When used, chloramphenicol (Cam) was added together with gentamicin (Gm). (A) LDH released in the medium was determined with a CytoTox 96 assay. P values of the pairs being compared are as indicated. (B) Secretion of TNF-α from infected BMDMs, as measured by enzyme-linked immunosorbent assay. Values represent means and standard deviations from seven (Gm-only) or three (Gm-and-Cam) independent experiments with duplicate infection wells. The TNF-α concentrations from the BMDMs left uninfected ranged from undetectable to 0.043 ng/ml.
YopJ and bacterial protein synthesis have opposite effects on TNF-α secretion from BMDMs containing intracellular Y. pseudotuberculosis.
TNF-α is an important cytokine in host defense against Yersinia infection (31). YopJ is required for Yersinia to inhibit TNF-α secretion from infected macrophages (6, 30). To clarify the role of intracellular Yersinia in TNF-α secretion, we used an enzyme-linked immunosorbent assay to measure levels of TNF-α secreted by infected BMDMs. As shown in Fig. 8B, high levels of TNF-α were secreted from the BMDMs infected for 21 h with 32777c (∼10.6 ng/ml) compared to what was seen for 32777 (∼3.8 ng/ml). High levels of TNF-α were also secreted from BMDMs infected with mJ (∼6.9 ng/ml) or mJTEH (∼7.1 ng/ml) compared to what was seen for the wild-type 32777-infected BMDMs (Fig. 8B), indicating that YopJ activity is mainly responsible for inhibiting TNF-α secretion under these conditions. Furthermore, among the three strains that lack YopJ, the virulence plasmid-cured strain 32777c induced the highest level of TNF-α secretion from the infected BMDMs, and the difference was statistically significant (P < 0.01) (Fig. 8B). The addition of chloramphenicol after phagocytosis reduced the levels of TNF-α secreted by BMDMs infected with 32777c, mJ, and mJTEH to the level seen for BMDMs infected with 32777. Collectively, these results suggest that de novo bacterial protein synthesis after phagocytosis was required to stimulate proinflammatory cytokine production and that YopJ functions to inhibit TNF-α production.
DISCUSSION
Y. pseudotuberculosis is considered primarily an extracellular pathogen, and the plasmid-encoded T3SS has been studied largely with respect to its role in promoting the extracellular phase of Y. pseudotuberculosis pathogenesis (50). However, an early intracellular stage may be important for Y. pseudotuberculosis pathogenesis (14, 35, 47). Our results confirmed that phagocytosis inhibition by Yops is not complete (15, 36), as roughly 40% of the wild-type bacteria that came in contact with macrophages were taken up (Fig. 1). We found that the presence of a fully functional T3SS strongly restricted the intracellular survival of the bacteria, as only a small percentage of the pYV+ wild-type Y. pseudotuberculosis internalized under these conditions survived and replicated. Although the presence of the T3SS appeared to restrict the intracellular survival of the bacteria, it also allowed the bacteria to translocate YopJ and induce macrophages to undergo apoptosis. These findings expand our understanding of how the T3SS modulates the immune response to Yersinia infection.
Previous studies have shown that pYV-containing strains of Y. pseudotuberculosis can survive and replicate in murine macrophages in vitro (14, 38, 47). In these studies, the bacteria were grown at 28°C to stationary phase prior to infection, so the T3SS and the Yops were not efficiently translocated during phagocytosis. Under this infection condition, the majority of the intracellular bacteria survived inside the macrophage for several hours without obvious survival restriction before they started to replicate (14, 47). Our study was carried out with bacteria grown at 37°C to log phase in the presence of Ca2+ to prime moderate T3SS and Yop expression (Fig. 1A), and we used a low MOI (10) in an attempt to mimic bacterial growth and infection conditions at early stages of in vivo pathogenesis. The fact that a high number of mJTEH bacteria were phagocytosed (∼70%) compared to what was seen for wild-type 32777 or mJ (∼40%) (Fig. 1B) confirmed that the growth and infection conditions used were sufficient to allow functional levels of antiphagocytic Yops to be translocated during uptake. Interestingly, bacteria internalized under these growth conditions experienced a significant survival restriction if they carried a functional T3SS but not when they lacked the virulence plasmid (Fig. 2 and 3) or YopB (Fig. 4). Nevertheless, a small fraction of the internalized pYV+ bacteria survived the restriction and replicated within macrophages (Fig. 3 and 4). We also found that 32777 grown to log phase at room temperature has an impaired ability to induce apoptosis, even though it still induces significantly more LDH release than seen for the BMDMs infected with 32777c or left uninfected (data not shown). Overall, these results suggest that the extent to which Y. pseudotuberculosis will replicate in macrophages in vivo depends on the growth condition and the level of T3SS and Yop expression at the time of contact with macrophages. The survival of Legionella pneumophila inside phagocytes also depends on the growth condition prior to infection (25). We hypothesize that the surviving pYV+ intracellular bacteria could contribute to pathogenesis by subsequently escaping the macrophage and entering into the extracellular phase of growth that is essential for virulence, for example.
Roy et al. studied whether the deployment of a T3SS translocon by a bacterium during phagocytosis could be responsible for triggering increased phagosome-lysosome fusion via a process controlled by synaptotagmin VII (38). In that study, wild-type BMDMs were infected with a ΔyopEHT mutant of Y. pseudotuberculosis, and results of a GFP induction assay showed a decrease in intracellular bacterial viability at 6 h postinfection. This decrease in viability was not observed when BMDMs from mice deficient in synaptotagmin VII were infected with the ΔyopEHT mutant or when Y. pseudotuberculosis lacking a functional T3SS (ysc mutant) was used to infect wild-type macrophages (38). Based upon this result it was suggested that synaptotagmin VII-mediated phagosome-lysosome fusion might require pore formation, an activity apparent in ΔyopEHT mutants but not in wild-type or ysc mutant strains at a high MOI (≥100) (38, 49). Using a low MOI and conditions that did not result in measurable pore formation or LDH release by macrophages infected with the mJTEH mutant (Fig. 6A), we found that the wild-type pYV+ strain and the mJTEH mutant underwent similar intracellular survival restrictions (Fig. 2, 3, and 5), but not the ΔYopB strain, which is defective in forming the translocon (Fig. 4). This is an important result, as it suggests that the normal process of translocon channel insertion and Yop delivery across nascent phagosomal membrane by wild-type Y. pseudotuberculosis is sufficient to generate the calcium signal sensed by the macrophage, resulting in increased phagosome-lysosome fusion (38).
In addition to restricting the intracellular survival of pYV+ bacteria, a portion of the macrophages infected with wild-type Y. pseudotuberculosis underwent apoptosis in a YopJ- and TLR4-dependent manner (Fig. 6B and 7). It is possible that amounts of YopJ sufficient to allow the subsequent initiation of apoptosis are translocated during the 20-min period of phagocytosis. Alternatively, the intracellular bacteria may actively contribute to apoptosis. To distinguish between these two possibilities, chloramphenicol, which blocks bacterial protein synthesis and inhibits intracellular bacterial replication, was added to infected macrophages. Interestingly, chloramphenicol treatment reduced the apoptosis of cells infected with wild-type Y. pseudotuberculosis (Fig. 8A). It is possible that chloramphenicol treatment decreases the expression and translocation of YopJ after the completion of phagocytosis. Our preliminary evidence suggests that YopJ can be translocated across the phagosomal membrane when it is ectopically expressed within intracellular Y. pseudotuberculosis (Y. Zhang, unpublished observations). Our current hypothesis is that under the normal operating conditions of the T3SS, YopJ is delivered during phagocytosis and shortly after the completion of phagocytosis to reach levels sufficient to trigger apoptosis.
Previously, it was found that TLR4 was important for Yersinia-induced macrophage apoptosis under infection conditions where Yops were maximally induced before infection and the extracellular bacteria were allowed to grow for several hours after infection (18, 55). However, TLR4-deficient macrophages were not completely resistant to apoptosis, suggesting that other mechanisms also activate the cell death pathways. In contrast, under the intracellular infection condition, TLR4 seemed to be the main receptor involved in activating the apoptotic pathway (Fig. 6B). It is interesting that the requirement for TLR4 in macrophage apoptosis by intracellular Y. pseudotuberculosis is similar to that observed in studies of delayed macrophage cell death induced by S. enterica serovar Typhimurium. Intracellular S. enterica serovar Typhimurium can kill macrophages by a delayed mechanism of cell death that requires a T3SS encoded in Salmonella pathogenicity island 2 (48) as well as TLR4 (17).
It is becoming increasingly clear that T3SS effectors delivered during the phagocytosis of pathogenic bacteria can play important functional roles at later stages of the host-pathogen interaction. T3SS-1 in S. enterica serovar Typhimurium is expressed by extracellular bacteria, promotes bacterial invasion into host cells, and is subsequently downregulated in the intracellular environment (40). SopB, a phosphatidylinositol phosphatase that is delivered into host cells by T3SS-1, contributes to bacterial uptake but also participates in an early step of phagosome maturation and continues to stimulate host signaling pathways long after the invasion step is complete (11, 20, 40). We have presented evidence that YopJ, delivered during the initial stages of bacterium-host cell interaction, impacts the intracellular pathogenesis of Y. pseudotuberculosis by decreasing the synthesis of TNF-α and by inducing macrophages to undergo apoptosis. Even if the majority of internalized Y. pseudotuberculosis is killed by the macrophages and cannot benefit directly from these activities, it is possible that they impair the host immune responses sufficiently to promote pathogenesis by the entire population. For example, the apoptosis of macrophages might decrease the class II presentation of antigens derived from degraded Y. pseudotuberculosis in phagolysosomes.
Acknowledgments
We thank Susan von Horn and Guo-Wei Tian at the Central Microscopy Imaging Center at Stony Brook University for assistance with electron microscopy and confocal microscopy, respectively; Kim Orth at the University of Texas Southwestern Medical Center for providing YopJ proteins used for antibody production; Wei-Xing Zong for stimulating discussion and suggestions; Hana Fukuto, Joseph McPhee, Céline Pujol, Adrianus van der Velden, and Gloria Viboud for comments and suggestions; and Galina Romanov for excellent technical assistance.
This work is supported by grants from the National Institutes of Health (AI043389) awarded to J.B.B., by NIAID National Institutes of Health intramural biodefense research grant Y2-AI-3739 to R.M.S., and by NIAMS intramural research funding to R.M.S.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 30 June 2008.
REFERENCES
- 1.Achtman, M., K. Zurth, G. Morelli, G. Torrea, A. Guiyoule, and E. Carniel. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 9614043-14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aepfelbacher, M., and J. Heesemann. 2001. Modulation of Rho GTPases and the actin cytoskeleton by Yersinia outer proteins (Yops). Int. J. Med. Microbiol. 291269-276. [DOI] [PubMed] [Google Scholar]
- 3.Barz, C., T. N. Abahji, K. Trulzsch, and J. Heesemann. 2000. The Yersinia Ser/Thr protein kinase YpkA/YopO directly interacts with the small GTPases RhoA and Rac-1. FEBS Lett. 482139-143. [DOI] [PubMed] [Google Scholar]
- 4.Black, D. S., and J. B. Bliska. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37515-527. [DOI] [PubMed] [Google Scholar]
- 5.Bliska, J. B., K. L. Guan, J. E. Dixon, and S. Falkow. 1991. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc. Natl. Acad. Sci. USA 881187-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boland, A., and G. R. Cornelis. 1998. Role of YopP in suppression of tumor necrosis factor alpha release by macrophages during Yersinia infection. Infect. Immun. 661878-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolin, I., and H. Wolf-Watz. 1988. The plasmid-encoded Yop2b protein of Yersinia pseudotuberculosis is a virulence determinant regulated by calcium and temperature at the level of transcription. Mol. Microbiol. 2237-245. [DOI] [PubMed] [Google Scholar]
- 8.Celada, A., P. W. Gray, E. Rinderknecht, and R. D. Schreiber. 1984. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 16055-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornelis, G. R., T. Biot, C. Lambert de Rouvroit, T. Michiels, B. Mulder, C. Sluiters, M.-P. Sory, M. Van Bouchaute, and J.-C. Vanooteghem. 1989. The Yersinia yop regulon. Mol. Microbiol. 31455-1459. [DOI] [PubMed] [Google Scholar]
- 10.Cornelis, G. R., A. Boland, A. P. Boyd, C. Geuijen, M. Iriarte, C. Neyt, M. P. Sory, and I. Stainier. 1998. The virulence plasmid of Yersinia, an antihost genome. Microbiol. Mol. Biol. Rev. 621315-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drecktrah, D., L. A. Knodler, K. Galbraith, and O. Steele-Mortimer. 2005. The Salmonella SPI1 effector SopB stimulates nitric oxide production long after invasion. Cell. Microbiol. 7105-113. [DOI] [PubMed] [Google Scholar]
- 12.Forsberg, A., and H. Wolf-Watz. 1988. The virulence protein Yop5 of Yersinia pseudotuberculosis is regulated at transcriptional level by plasmid-plB1-encoded trans-acting elements controlled by temperature and calcium. Mol. Microbiol. 2121-133. [DOI] [PubMed] [Google Scholar]
- 13.Galan, J. E., and H. Wolf-Watz. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444567-573. [DOI] [PubMed] [Google Scholar]
- 14.Grabenstein, J. P., M. Marceau, C. Pujol, M. Simonet, and J. B. Bliska. 2004. The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect. Immun. 724973-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grosdent, N., I. Maridonneau-Parini, M. P. Sory, and G. R. Cornelis. 2002. Role of Yops and adhesins in resistance of Yersinia enterocolitica to phagocytosis. Infect. Immun. 704165-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan, K. L., and J. E. Dixon. 1990. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science 249553-556. [DOI] [PubMed] [Google Scholar]
- 17.Guiney, D. G. 2005. The role of host cell death in Salmonella infections. Curr. Top. Microbiol. Immunol. 289131-150. [DOI] [PubMed] [Google Scholar]
- 18.Haase, R., C. J. Kirschning, A. Sing, P. Schrottner, K. Fukase, S. Kusumoto, H. Wagner, J. Heesemann, and K. Ruckdeschel. 2003. A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J. Immunol. 1714294-4303. [DOI] [PubMed] [Google Scholar]
- 19.He, S. Y., K. Nomura, and T. S. Whittam. 2004. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim. Biophys. Acta 1694181-206. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez, L. D., K. Hueffer, M. R. Wenk, and J. E. Galan. 2004. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 3041805-1807. [DOI] [PubMed] [Google Scholar]
- 21.Iriarte, M., and G. Cornelis. 1998. YopT, a new Yersinia Yop effector protein, affects the cytoskeleton of host cells. Mol. Microbiol. 29915-929. [DOI] [PubMed] [Google Scholar]
- 22.Ivanov, M. I., J. A. Stuckey, H. L. Schubert, M. A. Saper, and J. B. Bliska. 2005. Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol. Microbiol. 551346-1356. [DOI] [PubMed] [Google Scholar]
- 23.Juris, S. J., A. E. Rudolph, D. Huddler, K. Orth, and J. E. Dixon. 2000. A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc. Natl. Acad. Sci. USA 979431-9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills, S. D., A. Boland, M. P. Sory, P. van der Smissen, C. Kerbourch, B. B. Finlay, and G. R. Cornelis. 1997. Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc. Natl. Acad. Sci. USA 9412638-12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molofsky, A. B., and M. S. Swanson. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 5329-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monack, D. M., J. Mecsas, D. Bouley, and S. Falkow. 1998. Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J. Exp. Med. 1882127-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monack, D. M., J. Mecsas, N. Ghori, and S. Falkow. 1997. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc. Natl. Acad. Sci. USA 9410385-10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee, S., G. Keitany, Y. Li, Y. Wang, H. L. Ball, E. J. Goldsmith, and K. Orth. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 3121211-1214. [DOI] [PubMed] [Google Scholar]
- 29.Navarre, W. W., and A. Zychlinsky. 2000. Pathogen-induced apoptosis of macrophages: a common end for different pathogenic strategies. Cell. Microbiol. 2265-273. [DOI] [PubMed] [Google Scholar]
- 30.Palmer, L. E., S. Hobbie, J. E. Galan, and J. B. Bliska. 1998. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol. Microbiol. 27953-965. [DOI] [PubMed] [Google Scholar]
- 31.Parent, M. A., L. B. Wilhelm, L. W. Kummer, F. M. Szaba, I. K. Mullarky, and S. T. Smiley. 2006. Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infect. Immun. 743381-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 1035-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pettersson, J., R. Nordfelth, E. Dubinina, T. Bergman, M. Gustafsson, K. E. Magnusson, and H. Wolf-Watz. 1996. Modulation of virulence factor expression by pathogen target cell contact. Science 2731231-1233. [DOI] [PubMed] [Google Scholar]
- 34.Prehna, G., M. I. Ivanov, J. B. Bliska, and C. E. Stebbins. 2006. Yersinia virulence depends on mimicry of host rho-family nucleotide dissociation inhibitors. Cell 126869-880. [DOI] [PubMed] [Google Scholar]
- 35.Pujol, C., and J. B. Bliska. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 715892-5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosqvist, R., I. Bolin, and H. Wolf-Watz. 1988. Inhibition of phagocytosis in Yersinia pseudotuberculosis: a virulence plasmid-encoded ability involving the Yop2b protein. Infect. Immun. 562139-2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosqvist, R., A. Forsberg, M. Rimpilainen, T. Bergman, and H. Wolf-Watz. 1990. The cytotoxic protein YopE of Yersinia obstructs the primary host defense. Mol. Microbiol. 4657-667. [DOI] [PubMed] [Google Scholar]
- 38.Roy, D., D. R. Liston, V. J. Idone, A. Di, D. J. Nelson, C. Pujol, J. B. Bliska, S. Chakrabarti, and N. W. Andrews. 2004. A process for controlling intracellular bacterial infections induced by membrane injury. Science 3041515-1518. [DOI] [PubMed] [Google Scholar]
- 39.Ruckdeschel, K., O. Mannel, K. Richter, C. A. Jacobi, K. Trulzsch, B. Rouot, and J. Heesemann. 2001. Yersinia outer protein P of Yersinia enterocolitica simultaneously blocks the nuclear factor-kappa B pathway and exploits lipopolysaccharide signaling to trigger apoptosis in macrophages. J. Immunol. 1661823-1831. [DOI] [PubMed] [Google Scholar]
- 40.Schlumberger, M. C., and W. D. Hardt. 2006. Salmonella type III secretion effectors: pulling the host cell's strings. Curr. Opin. Microbiol. 946-54. [DOI] [PubMed] [Google Scholar]
- 41.Schotte, P., G. Denecker, A. Van Den Broeke, P. Vandenabeele, G. R. Cornelis, and R. Beyaert. 2004. Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J. Biol. Chem. 27925134-25142. [DOI] [PubMed] [Google Scholar]
- 42.Shao, F., P. O. Vacratsis, Z. Bao, K. E. Bowers, C. A. Fierke, and J. E. Dixon. 2003. Biochemical characterization of the Yersinia YopT protease: cleavage site and recognition elements in Rho GTPases. Proc. Natl. Acad. Sci. USA 100904-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin, H., and G. R. Cornelis. 2007. Type III secretion translocation pores of Yesinia enterocolitica trigger maturation and release of pro-inflammatory IL-1beta. Cell. Microbiol. 92893-2902. [DOI] [PubMed] [Google Scholar]
- 44.Simonet, M., and S. Falkow. 1992. Invasin expression in Yersinia pseudotuberculosis. Infect. Immun. 604414-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simonet, M., S. Richard, and P. Berche. 1990. Electron microscopic evidence for in vivo extracellular localization of Yersinia pseudotuberculosis harboring the pYV plasmid. Infect. Immun. 58841-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torruellas, J., M. W. Jackson, J. W. Pennock, and G. V. Plano. 2005. The Yersinia pestis type III secretion needle plays a role in the regulation of Yop secretion. Mol. Microbiol. 571719-1733. [DOI] [PubMed] [Google Scholar]
- 47.Tsukano, H., F. Kura, S. Inoue, S. Sato, H. Izumiya, T. Yasuda, and H. Watanabe. 1999. Yersinia pseudotuberculosis blocks the phagosomal acidification of B10. A mouse macrophages through the inhibition of vacuolar H(+)-ATPase activity. Microb. Pathog. 27253-263. [DOI] [PubMed] [Google Scholar]
- 48.van der Velden, A. W., S. W. Lindgren, M. J. Worley, and F. Heffron. 2000. Salmonella pathogenicity island 1-independent induction of apoptosis in infected macrophages by Salmonella enterica serotype Typhimurium. Infect. Immun. 685702-5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Viboud, G. I., and J. B. Bliska. 2001. A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J. 205373-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viboud, G. I., and J. B. Bliska. 2005. Yersinia outer proteins: role in modulation of host cell signalling responses and pathogenesis. Annu. Rev. Microbiol. 5969-89. [DOI] [PubMed] [Google Scholar]
- 51.Von Pawel-Rammingen, U., M. V. Telepnev, G. Schmidt, K. Aktories, H. Wolf-Watz, and R. Rosqvist. 2000. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol. Microbiol. 36737-748. [DOI] [PubMed] [Google Scholar]
- 52.Wren, B. W. 2003. The yersiniae-a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 155-64. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301640-643. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, Y., and J. B. Bliska. 2005. Role of macrophage apoptosis in the pathogenesis of Yersinia. Curr. Top. Microbiol. Immunol. 289151-173. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, Y., and J. B. Bliska. 2003. Role of Toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection. Infect. Immun. 711513-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang, Y., A. T. Ting, K. B. Marcu, and J. B. Bliska. 2005. Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J. Immunol. 1747939-7949. [DOI] [PubMed] [Google Scholar]
- 57.Zhang, Z. Y., J. C. Clemens, H. L. Schubert, J. A. Stuckey, M. W. Fischer, D. M. Hume, M. A. Saper, and J. E. Dixon. 1992. Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 26723759-23766. [PubMed] [Google Scholar]