

Abstract
Synthetic lethality is inviability of a double-mutant combination of two fully viable single mutants, commonly interpreted as redundancy at an essential metabolic step. The dut-1 defect in Escherichia coli inactivates dUTPase, causing increased uracil incorporation in DNA and known synthetic lethalities [SL(dut) mutations]. According to the redundancy logic, most of these SL(dut) mutations should affect nucleotide metabolism. After a systematic search for SL(dut) mutants, we did identify a single defect in the DNA precursor metabolism, inactivating thymidine kinase (tdk), that confirmed the redundancy explanation of synthetic lethality. However, we found that the bulk of mutations interacting genetically with dut are in DNA repair, revealing layers of damage of increasing complexity that uracil-DNA incorporation sends through the chromosomal metabolism. Thus, we isolated mutants in functions involved in (i) uracil-DNA excision (ung, polA, and xthA); (ii) double-strand DNA break repair (recA, recBC, and ruvABC); and (iii) chromosomal-dimer resolution (xerC, xerD, and ftsK). These mutants in various DNA repair transactions cannot be redundant with dUTPase and instead reveal “defect-damage-repair” cycles linking unrelated metabolic pathways. In addition, two SL(dut) inserts (phoU and degP) identify functions that could act to support the weakened activity of the Dut-1 mutant enzyme, suggesting the “compensation” explanation for this synthetic lethality. We conclude that genetic interactions with dut can be explained by redundancy, by defect-damage-repair cycles, or as compensation.
Isolation of synthetic lethals starting with a particular “bait” mutation [SL(baiT)] is a powerful tool for unraveling genetic interactions (30, 54, 71, 72). The phenomenon of synthetic lethality, when a double mutant of two fully viable single mutants is dead, illustrates genetic “buffering” (26) and is commonly interpreted in terms of either enzymatic, sequential, or functional redundancy (25, 26, 53) (Fig. 1A). In general, the redundancy explanation assumes that the two functions in question work in the same small segment of metabolism—that they are metabolically proximal. For example, the synthetic lethality of the uvrD rep double mutant (69) can be explained by the fact that the uvrD and rep genes in Escherichia coli code for two related DNA helicases with auxiliary roles in DNA replication/repair (27); in fact, Bacillus subtilis has only one helicase of this kind, PcrA, which is essential (55). Another example of synthetic lethality in E. coli is the combinations degP surA and skp surA, where all three genes code for the periplasmic chaperones, suggesting that the Skp, SurA, and DegP chaperones are functionally redundant (58). Finally, the synthetic lethality of suf isc combinations can be rationalized by the fact that both inactivated operons are involved in the synthesis of iron-sulfur clusters, found in catalytic centers of key enzymes of the central metabolism (68). The intuitive appeal of the redundancy explanation leads to the presumption of relatedness or metabolic proximity among the various synthetic lethals isolated with a particular bait (26, 53, 72).
FIG. 1.

Formal mechanisms behind synthetic lethality. Synthetic lethality describes the situation when both “a” and “b” single mutants are viable, while the “ab” double mutant is dead. (A) The redundancy explanation always signifies metabolic proximity of the “a” and “b” steps. S, substrate; P, product; encircling means either “poisonous” (substrate) or “essential” (product). In the first two examples, to observe synthetic lethality, either substrate has to be poisonous or the product has to be essential (both could be, but they do not have to be). At least three types of redundancy are possible. The most intuitive type (type 1) is “enzymatic redundancy,” when the same essential reaction is catalyzed by two (frequently related, paralogous) enzymes. Applied to Dut, which catalyzes the dUTP→dUMP conversion, this type of redundancy would mean a related dUTPase, a paralog of Dut. Another possibility (type 2) is “sequential redundancy,” usually cited when the null “a” and “b” mutants in two separate steps of the same essential pathway are lethal but partial mutants are viable; the combination of two partial mutants in this case may shut down the essential pathway completely. Applied to dUTPase, the second type of redundancy would mean a defect in the same pathway of dTTP biosynthesis (for example, inactivation of dcd or a partial defect in thyA [see below]). Finally, “functional redundancy” (possibility 3) is the most frequent: two unrelated reactions contribute to the formation of the same essential product (top) or to the removal of the same poisonous substrate (bottom). Applied to dUTPase, this third type of redundancy would mean either an alternative way of producing dUMP (see below) or an alternative dUTP-hydrolyzing activity (unrelated to Dut, for example, a nonspecific phosphatase) (34). (B) The D-D-R cycle signifies metabolic distance; the “a” and “b” steps are completely unrelated in this case. Starting from the “normal state,” a defect in “a” results either in accumulation of a poisonous substrate or in lack of an essential product, leading through “damage” in a major cellular component (for example, DNA) to a dysfunctional state. However, a repair system that addresses the symptom rather than the cause restores the functional state, completing the cycle.
However, when the synthetic-lethal analysis starts with a known nonredundant function in a known nonredundant pathway, like the homologous strand exchange protein RecA of recombinational repair, the isolated “synthetic lethal with recA” mutants, SL(recA) (37), are all unrelated and metabolically distant, so by necessity they fall outside of the redundancy paradigm. Indeed, all the SL(recA) mutations, instead of identifying alternative recombinational-repair pathways (as would be expected within the redundancy explanation), inactivate diverse chromosomal-fragmentation avoidance mechanisms, which leads to increased fragmentation of the E. coli chromosome (37). For example, there is increased oxidative DNA damage in fur mutants due to a higher free-iron concentration in their cytoplasm (32), there is increased excision repair in rdgB mutants due to a proposed contamination of the DNA precursor pools with noncanonical nucleotides (10), and there is unscheduled initiation of chromosomal replication in seqA mutants (44), with the consequent RecA dependence of all three mutants (37). Therefore, we propose that the SL(recA) mutants illustrate the power of synthetic-lethal analysis to identify “defect-damage-repair” (D-D-R) cycles (Fig. 1B), in which a particular metabolic defect leads to unrelated damage in a major cellular component (in this case, chromosomal fragmentation), which is counteracted by another dedicated activity (in this case, RecA-catalyzed recombinational repair, which reassembles fragmented chromosomes). Not only there is no redundancy between the RecA function on the one hand and the Fur, RdgB, and SeqA functions on the other, but these SL(recA) mutants identify functions that are clearly nonredundant and metabolically distant among themselves, as well.
One of the recently found SL(recA) mutations (34) is the missense mutation in the dut gene (29) that codes for dUTPase, a pyrophosphatase specific for the noncanonical DNA precursor dUTP (6, 24). Dut is an essential function (19) that limits the cellular levels of dUTP and provides a major source of dUMP, the key intermediate in dTTP biosynthesis (50), but the missense mutation in question, dut-1, apparently has enough residual activity to keep cells fully alive, even at 42°C, when essentially no enzymatic dUTPase activity is detected in vitro (29). At the same time, the dut-1 defect in E. coli leads to increased uracil incorporation in DNA and causes synthetic lethalities with base excision repair defects, xthA, polA, and ligA (70, 73), as well as with recombinational repair defects in recA, recBC, and ruvABC (34). There is no enzymatic or even functional redundancy between dUTPase on the one hand and various DNA repair functions on the other. In fact, all these known SL(dut) combinations are examples of the D-D-R cycles: the dUTPase defect leads to uracil incorporation into DNA with subsequent excision, and the resulting abasic sites and nicks, if left unrepaired, cause various DNA problems.
In order to define the applicability of the redundancy explanation versus the D-D-R cycle explanation for the results of synthetic-lethal analysis with the dut-1 mutation as bait, we systematically isolated SL(dut) mutants and evaluated the metabolic proximity/distance of the identified functions from Dut. Metabolic proximity or relatedness signifies redundancy, while metabolic distance or unrelatedness raises a suspicion of the D-D-R cycles. We report here that the isolated mutants comprise multiple examples of the D-D-R triads and two examples of functional redundancy and, unexpectedly, also reveal a novel type of genetic interaction, which we call “compensation.”
MATERIALS AND METHODS
Bacterial strains.
The E. coli strains, all K-12, are listed in Table 1. The dut-1 allele, a C→T transition at nucleotide 74 of the gene (34), was moved around using its linkage to the zic-4901::Tn10 marker and was scored on solid LB medium by sensitivity to 10 mM uracil (29, 34). The recA, recBC, ruvABC mutants were confirmed by their characteristic UV sensitivities. The deletion mutants ΔxerD::cat and ΔdegP::cat were created by replacing the open reading frames with the chloramphenicol resistance marker and were confirmed by PCR (16). The sequences of the primers used in the construction and verification are available upon request. The ΔxerD mutants were additionally confirmed by observing greatly elongated cells (∼10% of the total) in growing cultures. The ΔdegP mutants were additionally confirmed by their inability to grow in LB at 45°C. The similarly constructed deletion mutants Δtdk-6::kan, Δdgt::cat, and ΔxthA::cat were obtained from Ka Wai Kuong, Brian Budke, and Luciana Amado (our laboratory), respectively. The Δtdk mutants were confirmed by their sensitivity to trimethoprim. The Δdgt and ΔxthA mutants were confirmed by PCR.
TABLE 1.
E. coli strains used in the study
| Strain | Backgrounda | Relevant genotype (plasmid) | Source/reference/derivation |
|---|---|---|---|
| Previous studies | |||
| AB1157 | AB1157 | Rec+ | 4 |
| AK105 | AB1157 | dut-1 | 34 |
| AK106 | AB1157 | dut-1 recA200(Ts) | 34 |
| AK141 | AB1157 | Δtdk-6::kan | 37 |
| L-44 | DH5α recA+ | dut-1(pEAK12-4) | 34 |
| L-49 | L-44 | dut-1 recA938::cat(pEAK12-4) | 34 |
| L-50 | DH5α recA+ | dut-1recBCD3::kan(pEAK12-4) | 34 |
| L-80 | AB1157 | Δung-689::cat | 34 |
| DH5α::pir+ | DH5α | pir+ | 47 |
| This study | |||
| AAM1 | AB1157 | ΔthyA71::cat | Deletion-replacement |
| HT5 | DH5α recA+ | dut-1(pEAK12-4) (pHT2) | L-44(pHT2) |
| HT50 | AB1157 | ΔxthA::cat | Deletion-replacement |
| HT51 | AB1157 | ΔxthA::cat dut-1 | AK105 × P1 HT50 |
| HT81 | AB1157 | ΔdegP::cat | Deletion-replacement |
| HT82 | AB1157 | ΔdegP::cat dut-1 | AK105 × P1 HT81 |
| HT83 | AB1157 | ΔdegP::cat dut-1 Δung-689::cat | HT82 × P1 L-80 |
| HT84 | AB1157 | ΔdegP::cat Δung-689::cat | HT81 × P1 L-80 |
| HT122 | AB1157 | ΔxerD::cat | Deletion-replacement |
| HT123 | AB1157 | ΔxerD::cat dut-1 | AK105 × P1 HT122 |
| HT348 | AB1157 | Δtdk-6::kan dut-1 | AK105 × P1 AK141 |
The complete genotypes of the backgrounds listed are as follows: AB1157, F− λ− rac− thi-1 hisG4 Δ(gpt-proA)62 argE3 thr-1 leuB6 kdgK51 rfbD1 araC14 lacY1 galK2 xylA5 mtl-1 tsx-33 supE44(glnV44) rpsL31(StrR); DH5α, F′ F′ endA1 hsdR17(r− m+) glnV44 thi-1 recA1 relA1 Δ(lacIZYA-argF)U169 deoR(Φ80-ΔlacZM15) gyrA(NalR); DH5α::pir, F′ λ::pir endA1 hsdR17(r− m+) glnV44 thi-1 recA1 relA1 Δ(lacIZYA-argF)U169 deoR(Φ80-ΔlacZM15) gyrA(NalR).
Plasmids.
pHT2 is a derivative of pA-BCD (the p15A replicon) (75) in which the 4-kbp XmaI-XmaI fragment was replaced by the 3-kbp XmaI-NgoMIV fragment from pEAK14 (E. A. Kouzminova, unpublished data) containing the functional tdk gene. As a result, the plasmid carries the recB+, recC+, and tdk+ genes. The pHT3 plasmid is a derivative of pA-BCD, in which the 4-kbp XmaI-XmaI fragment was removed, and served as a vector-only control during the screening for pHT2. pHT4 resulted from ligation of MluI-cut pK80 (40) with the 4.3-kbp MluI-MluI fragment containing the degP gene from the E. coli chromosome. The pEAK12-4 plasmid, carrying dut+ and lacZ+ genes on an ori(Ts) replicon, was described previously (34). pEAK27 is a pCY566 cosmid (14) made on the basis of pMTL23p (11), into whose EcoRI and BamHI sites the [EcoRI-BglII 1.1-kbp] PCR-amplified chromosomal fragment carrying the ung+ gene was cloned. pEAK29 is pBR322 with the [EcoRV-BglII 1.3-kbp] fragment containing the ung+ gene from pEAK27 cloned into its EcoRV and BamHI sites.
Media and growth conditions.
Cells were grown in LB broth (10 g tryptone, 5 g yeast extract, 5 g NaCl per liter, pH ≤7.2, with NaOH) or on LB plates (15 g agar per liter of LB broth). When the cells were carrying plasmids, the media were supplemented with the required antibiotic: 100 μg/ml ampicillin, 50 μg/ml kanamycin, 10 μg/ml tetracycline, 10 μg/ml chloramphenicol, or 100 μg/ml spectinomycin. MacConkey (plus lactose) medium was used in the screen for Dut-dependent mutants (20 g Difco MacConkey agar per 400 ml H2O).
Color screen for Dut-dependent mutants.
The strain L-44 pEAK12-4 (later on with pHT2, as well) was mutagenized, plated on MacConkey-lactose plates supplemented with 10 μg/ml kanamycin to obtain about 200 to 300 transformants per plate, and incubated at 35.5°C for 36 to 48 h (37, 45). MacConkey medium allows Lac+ cells to form purple colonies and Lac− cells to form pale colonies. L-44 is dut+ lacZ+ and forms purple colonies when grown at 28°C. At 35.5°C, pEAK12-4 replicates less efficiently and is lost from cells at a rate of ∼5% per generation. Since it takes ∼25 generations to form a regular-size (3- to 4-mm) colony, this apparently very low rate of plasmid loss translates into a colony in which only ∼30% of the cells still keep the plasmid (0.9525 ∼ 0.3), which results in the sectoring phenotype of the colonies (a purple “star” in the center surrounded by pale borders) (37, 45). At 42°C, L-44 loses pEAK12-4 completely and forms pale colonies on MacConkey agar. However Dut-dependent mutants are unable to grow any further once the plasmid is lost, which results in small solid-purple colonies at 35.5°C and no growth at 42°C due to the loss of Dut.
From the initial screen, solid-purple colonies (primary candidates) were restreaked on a master plate (LB plus kanamycin at 28°C), as well as on a MacConkey agar plate without kanamycin, and incubated at 42°C for 20 h to test for the inability to grow. At 42°C, there were also big solid-purple colonies or colonies with a sectoring phenotype; they were ignored, because either they carried plasmids that lost temperature sensitivity or they were Dut-independent mutants. Overnight culture of the mutants that were inviable at 42°C, grown in LB plus ampicillin at 28°C to an optical density at 600 nm of 0.2, were diluted and spot tested for temperature sensitivity and also in a qualitative UV test (a general test for DNA repair). The former consisted of diluting the cells 104-fold in 1% NaCl and spotting them (10 μl) onto one LB-plus-ampicillin plate and two LB plates. One of the LB plates was developed at 28°C (the master plate), the other at 42°C (Dut dependence); the ampicillin plate at 42°C checked for the ability to lose the plasmid. For the qualitative UV test, the same cell cultures were spread evenly across a square LB plate using capillary tubes, dried, and irradiated with various doses of UV light.
UV-sensitive mutants were sequenced without further testing. The temperature-sensitive but UV-resistant mutants were selected for transfer into the AB1157 background. P1 lysates were prepared on these mutants, and the following three strains were transduced to kanamycin resistance: the original L-44 pEAK12-4 strain, AB1157 (the dut+ control), and AK105 (AB1157 dut-1). Three transductants of each strain were subjected to the same spotting test described above. Mutations conferring temperature-sensitive growth in the backgrounds of L-44 pEAK12-4 and AK105, but growing normally at 42°C in an AB1157 background, were considered Dut dependent and were identified by sequencing. Eventually, due to the increased frequency of Dut-dependent candidates in the DH5α background that failed to be confirmed in the AB1157 background, we switched to confirming our mutants in L-44 pEAK12-4 only.
Insertional mutagenesis and mutant sequencing.
To facilitate mutant identification, we used insertional mutagenesis. Our insertion module comes from pRL27, a plasmid carrying a hyperactive Tn5 transposase under the control of the tetA promoter, together with a separate insertion cassette that comprises a kanamycin resistance marker and the R6K pir-dependent origin of replication (41). Using pRL27 effectively limits the transposon to a single hop, because the transposase gene is outside of the insertional cassette. Also, our significant collection of the pRL27-induced mutations from this and other projects (10, 34, 37, 45, 61) demonstrates that the mutant phenotypes are not limited to complete gene inactivation, but also include polar effects on the downstream genes and antisense effects on the upstream genes. Infrequently, we even encounter inactivation of the regulatory C-terminal domains of proteins, as well as gene overexpression (a particular orientation of the insert in the promoter region).
Cells were mutagenized by electroporating them with 10 ng of pRL27 and plating them on MacConkey medium for kanamycin resistance. Reduced kanamycin (10 μg/ml) was used for the first plating after pRL27 electroporation. Once a desired candidate was identified for sequencing, total DNA from 2-ml overnight cultures was prepared as described previously (35); 1/20 of it was digested with MluI (the insertion cassette itself did not contain any MluI site), self-ligated, and electroporated into DH5α pir+ cells (47), in which the circularized piece of the chromosome was able to replicate in the presence of the Pir protein, with selection for kanamycin resistance. Four individual transformants of each candidate were picked, and their plasmids were isolated. The plasmid DNA was digested with MluI, and the smallest plasmid was selected for sequencing of the insertion-chromosome border. The primers for sequencing from pRL27 inserts were described previously (10).
RESULTS AND DISCUSSION
The expectations.
We expected to isolate Dut-dependent mutants in at least four major categories. The first two were of the redundancy type (Fig. 1A). (i) Mutants defective in DNA precursor metabolism; defects in this area (for example, an increase in the noncanonical DNA analog dUTP or a decrease in the canonical DNA nucleotides) may lead to a higher frequency of uracil misincorporation or to a greater defect in dTTP production. (ii) Mutants defective in other pathways of avoidance of chromosomal fragmentation (seqA, rdgB, and fur) (37); the Dut activity may be viewed as a way to avoid chromosomal fragmentation, but there are other systems working toward the same goal. A combined defect of two inactivated chromosomal-fragmentation avoidance pathways could be expected to overload the repair systems of the cell, killing it.
The two other expected categories of SL(dut) mutants were of the “D-D-R” type (Fig. 1B): (iii) mutants defective in the known steps of uracil excision repair downstream of uracil-DNA-glycosylase (UDG) (xthA, polA, and ligA) (70, 73), as well as in any unknown genes involved in this reaction, especially those coding for the postulated deoxyribophosphodiesterase (dRPase) (see below) (22), and (iv) mutants defective in recombinational repair of double-strand breaks (recA, recBC, and ruvABC) (34). We also hoped to isolate any unknown genes of recombinational repair.
The screen.
SL(dut) mutants were isolated using the color screen developed in our laboratory (37, 45) (Fig. 2). This screen identifies mutants that depend on a functional gene residing on a plasmid at the temperatures semipermissive for plasmid replication. To this end, we mutagenized a dut-1 lacZ mutant carrying an ori(Ts) plasmid that complemented both the dut and the lacZ defects and identified mutants unable to lose the plasmid (Fig. 2).
FIG. 2.
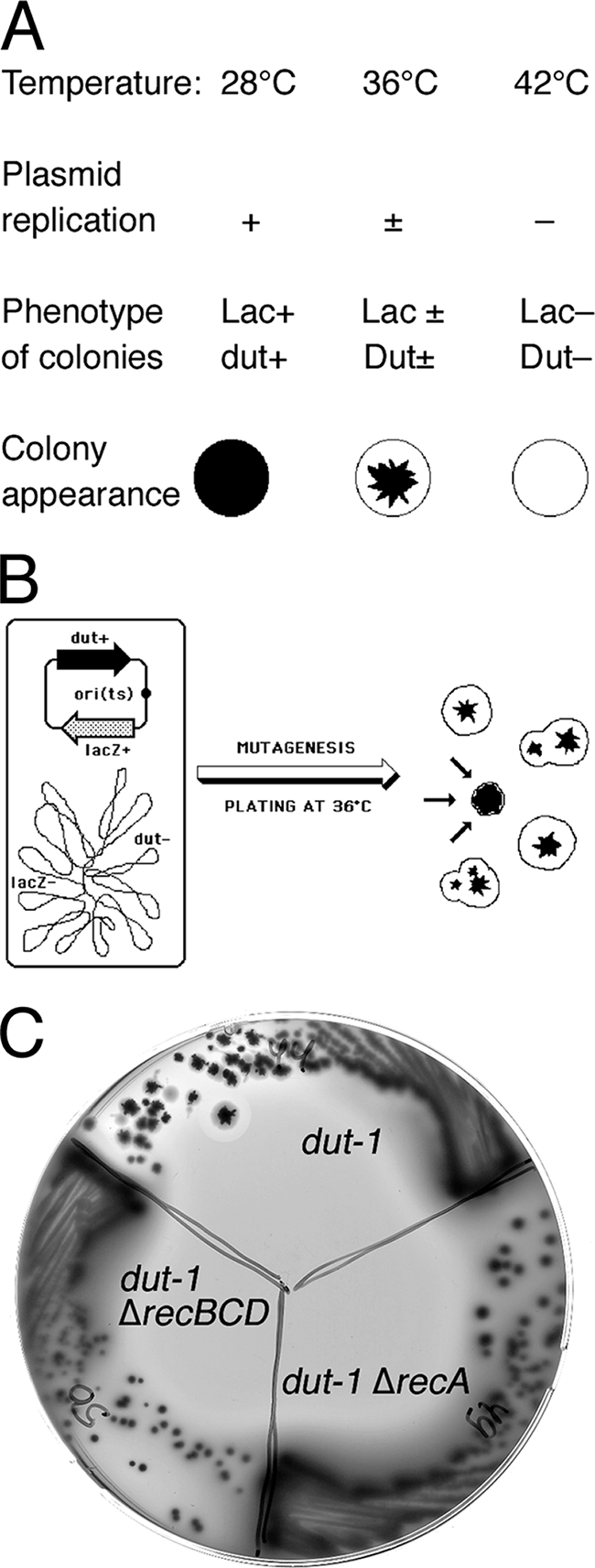
Color screen for Dut-dependent mutants. The screen looks for the inability to lose a temperature-sensitive plasmid carrying the dut+ gene. (A) Properties of the strain for the screen [lacZ dut p(ori-Ts)-lacZ+-dut+] at three temperatures, plated on MacConkey-lactose agar. (B) Scheme of the experimental strain and the expected colony phenotype of Dut-independent (sectoring) mutants, as well as a single Dut-dependent mutant (converging arrows). (C) Sectoring phenotype of the parental color screen strain [dut pdut+(Ts)] and the absence of sectoring in its two derivatives carrying either ΔrecA or ΔrecBCD mutations on MacConkey-lactose agar at 36°C (controls). The parental strain forms sectoring colonies, characteristic of Dut-independent strains, whereas the double mutants form smaller nonsectoring colonies, revealing their Dut dependence.
We completed a total of eight rounds of mutagenesis: the first four rounds were in the original system, while the remaining rounds were in the system modified to avoid isolation of the most frequent mutants of the first four rounds. According to the statistics of the first four rounds of mutagenesis (Table 2), we screened more than 107,000 mutants, checked 2,832 primary candidates for inability to grow at 42°C, performed UV and spot tests on 260 secondary candidates to verify their temperature sensitivity and to determine whether they were involved in DNA repair, directly sequenced 43 UV-sensitive mutants, and transduced 36 UV-resistant mutants into the AB1157 background to confirm their synthetic lethality with the dut defect. Upon confirmation of their Dut dependence in an AB1157 background, we sequenced 22 UV-resistant candidates.
TABLE 2.
Statistics of the screen
| Stage | No. for screen run:
|
Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| Colonies screened | 32,000 | 8,900 | 34,400 | 32,000 | 34,642 | 6,983 | 42,972 | 7,264 | ∼200,000 |
| Streaked at 42°C | 676 | 307 | 960 | 889 | 180 | 58 | 344 | 63 | 3,477 |
| UV/spot test | 156 | 57 | 345 | 321 | 32 | 16 | 39 | 20 | 986 |
| UVsusceptiblea | 10 | 3 | 9 | 17 | 7 | 4 | 22 | 6 | 78 |
| Transduced into AB1157 background | 19 | 15 | 1 | 1 | 6 | 4 | 11 | 6 | 63 |
| UV-resistant sequenced | 10 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 13 |
| UV-susceptible sequenced | 10 | 2 | 10 | 17 | 7 | 2 | 10 | 2 | 60 |
Even slightly.
From the first four rounds of mutagenesis, we had isolated repetitive hits (39 hits out of a total of 44) in either tdk, recB, or recC. To concentrate on the isolation of low-frequency and novel Dut-dependent mutants, we introduced a plasmid, pHT2, compatible with pEAK12 and carrying wild-type copies of the recBC and tdk genes, into L-44, the strain in which the mutagenesis was performed, to prevent future isolation of the corresponding mutants. Since the frequency of the Dut-dependent insertions was lower in the new setup and the contributions of spontaneous mutants had increased, we had to introduce another modification. Starting from screen 7, instead of directly sequencing UV-sensitive mutants, we first verified the cotransducibility of insertions with the UV-sensitive phenotype after back-crossing them into L-44 pEAK12. In runs 5 to 8 (Table 2), we screened almost 92,000 mutants, checked 645 primary candidates for inability to grow at 42°C, performed UV and spot tests on 107 secondary candidates to verify their temperature sensitivity and to determine whether they were involved in DNA repair, directly sequenced 22 UV-sensitive mutants, and transduced 10 UV-sensitive and 27 UV-resistant mutants into the original L-44 pEAK12 strain to confirm their synthetic lethality with the dut defect (in the end, we confirmed only one UV-resistant Dut-dependent mutant). In the discussion below, we present only the Dut-dependent mutants that either made sense without additional testing or were confirmed by additional tests.
Nucleotide metabolism: tdk.
There was one particular Dut-dependent mutation that we isolated continuously and in disproportionately high numbers, considering the small size of the affected gene (618 bp). In fact, after isolating close to 30 independent inserts in this gene in the first four rounds of mutagenesis, we had to introduce a plasmid with the wild-type copy of the gene to prevent it from distracting us from other Dut-dependent mutants. The gene was tdk (28), coding for a thymidine kinase that catalyzes the salvage pathway of dUMP/dTMP biosynthesis by phosphorylating either dU or dT (52) (Fig. 3A and B). Interestingly, dUMP was always thought to be produced mainly by the de novo pathway, via dUTP hydrolysis by Dut (Fig. 3A), with the salvage pathway being mentioned as an option whose importance was unclear (50, 51). The dut tdk synthetic lethality indicates that (i) the salvage pathway can take over dUMP production if the Dut route is unavailable and (ii) there are no other pathways of dUMP biosynthesis besides the two pathways controlled by Dut and Tdk. When both pathways are inactivated, the cells are likely to succumb to a form of thymineless death due to the inability to make dTTP.
FIG. 3.

SL(dut) no. 1: an example of a true redundancy. (A) De novo and salvage pathways of dTTP production. The steps are labeled by gene names. Question marks identify proposed steps for which no genes are currently known. (B) Positions and orientations of Dut-dependent inserts into the tdk gene. The inserts are shown as black flags pointing in the directions of their kanamycin genes. These are the first 17 inserts of about 30 that we isolated in screen runs 1 to 4. Multiple inserts at the 44-bp and 52-bp positions of the gene are all independent (nonsisters), suggesting hot spots for pRL27 insertion. The small arrow on a stem upstream of the gene indicates a promoter. The gene diagrams were generated with the help of the EcoCyc site (http://ecocyc.org/). (C) The poor growth of Δtdk dut-1 double mutants at 42°C is not rescued by supplementation with thymidine. Serial dilutions (10-μl) of rapidly growing cultures were spotted onto M9-plus-CAA minimal medium plates and incubated for 48 h at 28°C or for 24 h at 42°C. Additions were as follows: thymidine (thy) at 50 μg/ml and/or uracil (ura) at 10 mg/ml. Uracil is an indicator for the dut defect (see Materials and Methods). The strains were wild type, AB1157; dut, AK105; Δtdk, AK141; Δtdk dut, HT348; and ΔthyA, AAM1.
To test this idea, we spotted dut, tdk, and the double mutants, as well as the thyA mutant, on minimal plates with or without thymidine (and/or uracil) and incubated them at 28°C or at 42°C (since the dut-1 allele we used was a temperature-sensitive one) (Fig. 3C). We confirmed our previous observations and those of others (34, 70) that the dut mutant grew more slowly at 42°C without thymidine, suggesting that the salvage (Tdk) pathway cannot produce the same level of dUMP from internal sources as the Dut pathway does. The Tdk pathway took over completely in the presence of exogenous thymidine, as expected. Predictably, in the absence of both Dut and Tdk functions, thymidine supplementation did not make a difference, although the double mutant still showed some residual growth even in the absence of thymidine at 42°C, in contrast to the nongrowing thyA mutant (Fig. 3C). At 28°C, all the mutants behaved as expected, including the characteristic uracil sensitivity of the dut mutants (29, 34), except for the puzzling sensitivity of tdk mutants to the mixture of thymidine plus uracil.
The tdk dut lethality can be explained by functional redundancy: although Dut and Tdk catalyze enzymatically opposite reactions (dephosphorylation in the case of Dut versus phosphorylation in the case of Tdk), both reactions contribute to the formation of the same intermediates of dTTP biosynthesis (dUMP and dTMP) and can therefore be considered redundant. Assuming that synthetic-lethal interactions identify redundant functions, we expected isolation of more defects in the nucleotide metabolism; however, the tdk inactivation turned out to be the only SL(dut) defect in our collection that affected another gene of the DNA precursor metabolism.
Expected Dut-dependent mutants with defects in base excision repair: xthA, polA, and an unexpected ung mutant.
The toxicity of the dut defect comes from contamination of the DNA precursor pools with the noncanonical DNA precursor dUTP (77). Due to the similar chemical structures of dUTP and dTTP, DNA polymerases of E. coli readily misincorporate dUTP in place of dTTP (7, 62). In addition, uracil can also be formed in DNA by deamination of cytosine (43) (although calculations show that this process must be 105 times less productive [36]). However, no uracil was detected in the bulk DNA of wild-type or dut-1 mutant cells (35), due to the presence of a highly active enzyme, UDG (42) (the product of the ung gene [17]). UDG excises every uracil-DNA almost as soon as it is incorporated; this excision generates the short replication intermediates in the dut mutants (1, 74). In fact, it is the uracil excision that eventually kills, directly or indirectly, SL(dut) mutants, as all of them are known to be suppressed by ung inactivation (34, 70, 73).
The complete DNA repair reaction after uracil removal by UDG (22) includes nicking 5′ of the abasic site by exonuclease III (70) or endonuclease IV (15), removal of the dangling sugar-phosphate residue by the still-mysterious deoxyribophosphodiesterase (22, 56), filling in of the gap by DNA polymerase I, and, last, sealing of the nick by DNA ligase (73) (Fig. 4B). Not surprisingly, the dut mutations are synthetically lethal with xthA, polA, and ligA mutants, which all block the base excision repair pathway downstream of UDG (70, 73), demonstrating the lethality of unrepaired abasic sites or nicks.
FIG. 4.

SL(dut) no. 2: defects in uracil excision repair. (A) The positions and orientations of inserts are shown as black flags pointing in the directions of their kanamycin genes. Note that the scales are different for the three chromosomal regions. The small arrows on stems upstream of genes indicate promoters. (B) Hydrolysis of dUTP by Dut versus the alternative pathway of uracil-DNA incorporation with subsequent base excision repair. DNA-U, DNA with incorporated uracil; DNA-abs, DNA with abasic sites; DNA-nick-abs, DNA with nicks at abasic sites. The enzymes are identified by their corresponding genes: dut, dUTPase; ung, UDG; xthA, exonuclease III; polA, DNA polymerase I; ligA, DNA ligase. The stage catalyzed by a hypothetical enzyme, deoxyribophosphodiesterase, is indicated by the abbreviation dRPase. (C) Overproduction of UDG kills dut mutants. UDG was overproduced either due to the ung+ gene on a multicopy plasmid or due to the pRL27 insert upstream of the ung promoter. Serial dilutions of rapidly growing cultures (10 μl) were spotted onto LB plates and incubated for 36 h at 27°C and for 20 h at 37°C or at 42°C. The cells were either wild type (AB1157) or the dut-1 mutant (AK105). The p-ung+++ plasmid is pEAK27 (copy number, ∼600); the p-ung+ plasmid is pEAK29 (copy number, ∼50). The pRL27-ung+++ insert is from panel A. Since the uncomplemented AK105 pRL27-ung+++ construct was inviable at any temperature, the strain was constructed in the presence of the pEAK12-4 plasmid [ori(Ts) dut+], which is lost at 37°C and above.
We hoped that our systematic isolation of Dut-dependent mutants would identify an enzyme(s) catalyzing the “dRPase” step of the base excision reaction. However, we isolated only three mutants in the base excision repair of DNA-uracil. One of our Dut-dependent mutants had an antisense insertion in a gene with unknown function, ydjZ, downstream of xthA, which made us wonder if the actual cause of the synthetic lethality in this case was the xthA defect (Fig. 4A). To verify our interpretation, the ydjZ::kan and ΔxthA::cat mutations were individually transduced into the AB1157 dut mutant. To our surprise, ydjZ dut-1 proved to be viable in this background (unlike in the original DH5α background [data not shown]); however, we found the constructed ΔxthA dut-1 combination to be synthetically lethal in the AB1157 background, validating the published results (70).
A polA mutant with an antisense insert located in the middle of the gene was also isolated from the screen (Fig. 4A). The polA gene codes for DNA polymerase I, the abundant DNA repair polymerase of E. coli that finalizes all excision repair reactions (22). The enzyme has three major activities: the DNA polymerization activity and two exonuclease activities, 3′→5′ and 5′→3′ (33). The 5′→3′ exonuclease occupies the N-terminal one-third of the protein, and the 3′→5′ proofreading exonuclease is located in the middle, while the DNA polymerization activity is found in the C-terminal half of the protein (21). Therefore, our centrally located insert most likely inactivated the DNA polymerization activity of the enzyme and could also reduce the two exonuclease activities. Although E. coli strains carrying a complete deletion of the polA gene cannot grow on rich media, strains with a single defect in either DNA polymerase or 5′→3′ exonuclease can (31), explaining our isolation of the polA insert.
Both the dut xth and dut polA synthetic lethals cannot be explained by redundancy and instead exemplify defect-damage repair cycles. However, one more Dut-dependent mutant in excision repair that we have isolated can be explained by redundancy: an insert immediately upstream of the ung promoter (Fig. 4A) likely increases transcription of the gene, perhaps causing lethality through acceleration of uracil excision. Indeed, we have observed with the plasmid-cloned ung+ gene that overproduction of UDG is detrimental to wild-type cells but is even more detrimental to dut mutants (Fig. 4C). If dUTP hydrolysis on one hand and attenuation of the DNA-uracil excision on the other are both viewed as contributing to the reduction of chromosomal damage due to DNA-uracil excision, this can be considered an example of functional redundancy.
The expected Dut-dependent mutants with defective double-strand break repair: recA, recBC, and ruvABC.
The synthetic lethality of dut mutations with all known mutations inactivating double-strand DNA break repair (recA, recBC, and ruvABC) (34) indicates that excision of uracil from DNA somehow triggers chromosomal fragmentation (Fig. 5A). In fact, chromosomal fragmentation in dut mutants is perhaps the highest among known RecA-dependent mutants (34, 35, 37). We have isolated multiple inserts in all six genes whose products are specifically involved in double-strand break repair: recA, recBC, and ruvABC (Fig. 5B). This robust isolation of known Dut-dependent mutants proves that the screen works and also confirms the earlier findings that combinations of a recombinational defect with the dut defect are synthetically lethal (34). This also lends strong genetic support to the results of physical studies indicating that uracil incorporation and excision cause double-strand breaks in the E. coli chromosome (34, 35, 37). The number of isolated inserts, together with a similar number from the earlier search for RdgB-dependent mutants (45), also argues against the existence of other, nonessential genes involved in double-strand break repair in E. coli besides recA, recBC, and ruvABC. In fact, the combined density of inserts in these six genes from the two screens is 44 inserts/10,119 bp, or 1 insert per 230 bp, making failure of detection of any but a very small gene unlikely.
FIG. 5.

SL(dut) no. 3: defects in double-strand break repair. (A) Uracil-DNA incorporation, in a process that is still unclear but includes DNA replication (35, 36), leads to double-strand DNA breaks, which are mended by RecA-, RecBC-, and RuvABC-dependent recombinational repair. DNA-U, DNA with incorporated uracil; DNA-DSB, DNA with double-strand breaks. (B) Positions and orientations of inserts isolated as Dut-dependent mutants in this work and RdgB-dependent mutants in a previous work (45). The inserts are shown as black flags pointing in the directions of their kanamycin genes. The Dut-dependent inserts are shown above the genes, while RdgB-dependent inserts are below the genes. Note that the scales are different for the three chromosomal regions. The small arrows on stems upstream of genes indicate promoters, whereas small stem-loops downstream of genes indicate terminators. (C) Scheme of double-strand break repair in E. coli.
The recBC genes encode the two subunits of the heterotrimeric RecBCD enzyme, a powerful DNA helicase/nuclease (39). RecBCD binds double-stranded DNA ends and processes them to generate 3′ single-stranded DNA overhangs for the RecA protein to form filaments on (Fig. 5C). The RecA protein binds to the 3′ single-stranded DNA overhangs and promotes homologous pairing and strand exchange, forming a recombination intermediate (59). The points in the recombinational intermediates where homologous strands are exchanged between the two interacting DNA duplexes are called “Holliday junctions.” The RuvABC resolvasome binds, protects, translocates, and resolves Holliday junctions, completing recombinational repair (78) (Fig. 5C). Each enzyme (RecA, RecBC, and RuvABC) plays an essential role in the repair of chromosomal fragmentation; therefore, the absence of any of them in a mutant with an increased level of chromosomal fragmentation, like the dut mutant, should cause cell death. Finally, recombinational repair, like any other type of DNA repair except direct damage reversal, also depends on DNA Pol I and ligase functions, and the dut polA synthetic lethals (Fig. 4) could be also explained by their defect in recombinational repair, if they were not affected even more strongly at the earlier stage of excision repair.
The three recombinational-repair functions have no enzymatic redundancies even among themselves, so the redundancy idea does not explain the dut rec and dut ruv synthetic lethals. The dependence of dut mutants on recombinational-repair functions again points to the D-D-R cycles.
Partially Dut-dependent mutants defective in chromosomal-dimer resolution: xerCD and ftsK.
Whereas all the above-mentioned mutations cause synthetic lethality in combination with the dut defect in the AB1157 background, inserts in and around xerC, xerD, and ftsK (Fig. 6B) inhibit growth of the dut mutant at the nonpermissive temperature rather than completely blocking it. We sequenced these partially Dut-dependent mutants because of their modest UV sensitivity and because the xerC insert and the insert in front of ftsK were still synthetic lethal in combination with the dut defect in the DH5α background (not shown). We constructed a precise xerD deletion and confirmed the severely inhibited growth in combination with the dut defect in the AB1157 background (Fig. 6C). We noticed that, at least in the AB1157 background, both the ΔxerD mutant and the insert upstream of ftsK showed cold sensitivity at 28°C (Fig. 6C).
FIG. 6.

Synthetic inhibition in combination with dut: defects in chromosomal-dimer resolution. (A) Outline of chromosomal-dimer formation and resolution. The chromosome in late replication stages is shown with the terminus still unreplicated; duplex DNA is shown as a single line. The small open arrows designate the dif site. A double-strand break in the replicated portion of the chromosome is repaired with the help of the intact sister duplex, and DNA junctions are resolved to yield a crossover. A single crossover in a circular chromosome translates into a dimeric chromosome once replication is complete. Dimeric chromosomes cannot segregate into daughter cells. However, completion of replication also duplicates the dif site, allowing XerCD/FtsK-catalyzed site-specific recombination between the two copies of dif to split the chromosomal dimer into two monomers, which segregate into daughter cells. (B) The positions and orientations of inserts are shown as black flags pointing in the directions of their kanamycin genes. Note that the scales are different for the three chromosomal regions. The small arrows on stems upstream of genes indicate promoters. (C) Spot test showing temperature-independent synthetic inhibition of ΔxerD dut mutants. Serial dilutions of rapidly growing cultures (10 μl) were spotted onto LB plates and incubated for 36 h at 28°C, for 24 h at 34°C, or for 18 h at 42°C. The dut recA(Ts) mutant demonstrated synthetic lethality. The strains were wild type, AB1157; dut, AK105; dut recA(Ts), AK106; ΔxerD, HT122; and ΔxerD dut, HT123.
Circular chromosomes become dimeric when a single exchange (or any odd number of sister chromatid exchanges) occurs during replication (Fig. 6A). The cell cannot segregate a single dimeric chromosome into two daughter cells. To produce viable progeny after chromosomal dimerization, bacterial cells employ a specialized site-specific recombinase, XerCD, which separates the dimeric chromosome into monomers by acting at the dif sites in the replication terminus region with the help of the C-terminal domain of an essential cell division protein, FtsK (8, 9, 13, 38, 65). FtsK is positioned at the division septum and functions to coordinate chromosomal segregation with cell division via dimer resolution and also to facilitate sister chromatid decatenation by topoisomerase IV (3, 20).
Chromosomal dimerization happens on average once every six to seven generations in E. coli growing under the most favorable conditions (66). Dimer formation and resolution frequencies decrease in rec-deficient mutants. In contrast, hyper-rec mutations, like dut, polA, or uvrD, lead to an increase in the recombination frequency at the dif site as a result of a higher frequency of dimer formation and subsequent resolution (66). Hence, it is not surprising that the dut defect, with its attendant increase in double-strand breaks, has a synthetic phenotype with the defect in chromosomal-dimer resolution. However, the combination cannot be a synthetic-lethal one, as a maximum of half of the exchanges could be resolved to generate crossovers, and moreover, only odd numbers of crossovers result in dimeric chromosomes. Thus, the fraction of dividing cells with dimeric chromosomes cannot exceed 50% of all dividing cells, and therefore, the lethality of the dut mutants with disabled dimer resolution system cannot exceed 50%.
The synthetic phenotype of all inserts in this group can be explained by either direct inactivation or insufficient expression of the xerC, xerD, and ftsK genes. On the other hand, while inserts in xerC and xerD clearly inactivate the corresponding proteins, inserts in dapF and yigA might, in fact, act to overproduce XerC, and the insert upstream of the ftsK promoter is likely to overproduce FtsK. While it is unclear why XerC overproduction could be detrimental to the cell, overproduction of FtsK might titrate the recombinase away from the septum. Strong interaction between FtsK and XerD (but not XerC) is observed in vitro (79).
Isolation of inserts in and around xerCD and ftsK lends further support for the D-D-R explanation of synthetic lethality. It also shows that our color screen is sensitive enough to pick up synthetic combinations that are still viable, although grossly inhibited.
The partially Dut-dependent mutant with deregulated phosphate utilization: phoU.
We isolated a phoU insert as partially Dut dependent in the DH5α background (Fig. 7A); however, our attempts to confirm the phenotype in the AB1157 background using a ΔphoU mutant were unsuccessful due to the very low rate of growth of this mutant and rapid suppression, both reported previously (64). The synthetic defect of the dut phoU double mutant reveals yet another possible explanation for synthetic lethality and also explains the puzzling pstC and pstS suppressors of the dut rec synthetic lethality that we isolated previously (34). The three genes are in the same pstSCAB-phoU operon (Fig. 7A) (2), with PstC and PstS proteins being parts of the high-affinity phosphate-ABC transporter, while PhoU is one of the negative regulators of the pho regulon (49) to which the genes for the PstSABC transporter also belong (reviewed in reference 76). Additionally, there is genetic evidence that the PhoU protein directly utilizes the imported phosphate, because the severe growth inhibition of ΔphoU mutants is suppressed by inactivation of the pstSCAB genes (64). The suspected high concentration of the intracellular phosphate may explain the accumulation of polyphosphates in phoU mutants (48). dUTPase catalyzes hydrolysis of dUTP to dUMP and pyrophosphate, the latter being, in essence, the smallest “polyphosphate.” Therefore, a possible explanation of the synthetic inhibition of the phoU dut-1 double mutant at the nonpermissive temperature is that the intracellular increase in polyphosphates in general and pyrophosphate in particular makes the dUTP hydrolysis (a pyrophosphate-producing reaction) less favorable (Le Chatelier's principle) (Fig. 7B). Since the enzymatic activity of dUTPase is <5% in the dut-1 mutant, any additional inhibition of this reaction may turn cells into Δdut phenocopies, which is lethal. On the other hand, inactivation of the PstSCAB transporter may decrease the intracellular pyrophosphate concentration, helping the Dut-1 mutant enzyme and relieving the dut rec synthetic lethality (Fig. 7B).
FIG. 7.

SL(dut) no. 4: deregulation of phosphate metabolism. (A) The position and orientation of the insert that caused synthetic lethality with dut is shown as a black flag above the genes pointing in the direction of its kanamycin gene. The positions and orientations of the suppressors of synthetic lethality with dut are shown below the genes as open flags (dut degP suppressors [this work]) or hatched two-tailed flags (dut rec suppressors [34]). The small arrows on stems upstream of genes indicate promoters, whereas small stem-loops downstream of genes indicate terminators. (B) A possible explanation for the synthetic lethality of the dut-1 phoU combination and for the suppression of SL(dut) by inactivation of the pstSCAB genes. The dashed arrow indicates inefficient reaction; the thick arrow indicates robust reaction.
Remarkably, this time, neither the redundancy explanation, nor the D-D-R cycle can explain the dut phoU synthetic defect. Instead, this interaction can be best understood in terms of aggravation of the dut-1 defect from partial to almost complete by the unrelated phoU defect. In fact, since the dut gene is essential, partial dut mutants are expected to have this type of synthetic-lethal interaction, where the second mutation inactivates the function acting to compensate for the partial defect in an essential gene.
Synthetic lethality with a chaperone defect and its suppressor analysis: degP.
One of the strongest Dut-dependent mutants that we isolated has an insertion in degP (Fig. 8A). DegP (equivalent to HtrA) is a widely conserved heat shock protein essential for cell survival at temperatures of 44°C and higher (reviewed in reference 12). At low temperatures, the DegP protein functions predominantly as a periplasmic chaperone, but above 40°C, it acquires the additional function of an ATP-independent serine protease (63). Since Dut is a cytoplasmic protein, the known functions of DegP do not suggest an immediate explanation for the dut degP synthetic lethality, and we originally thought that our degP insertion killed dut mutants via an antisense effect on the upstream gene, dgt, which codes for deoxyguanosine 5′-triphosphate triphosphohydrolase (57). If Dgt could hydrolyze dUTP, it would partially complement the dut defect, becoming essential in the dut-1 background. However, we found that the Δdgt::cat mutant is Dut independent at any temperature in both AB1157 and DH5α backgrounds (not shown). We then confirmed, by constructing a ΔdegP allele, that the lethality was indeed due to degP inactivation (Fig. 8B). Both the ΔdegP mutant and our original degP insertion mutant are sensitive to temperatures in excess of 44°C (not shown), consistent with the known degP mutant properties (67). The cause of the synthetic lethality of the degP dut double mutant is unclear; if DegP is also active in the cytoplasm, as one report suggests (80), it could help stabilize the mutant Dut-1 protein at higher temperatures, alleviating the dut-1 defect (Fig. 8C). If this is true, then the dut degP synthetic lethality would clearly fall under the rubric of “aggravation,” as the degP defect would allow the partial dut-1 defect to become a complete one.
FIG. 8.

SL(dut) no. 5: inactivation of a chaperone/protease. (A) The position and orientation of the insert that caused synthetic lethality with dut is shown as a black flag above the genes pointing in the direction of its kanamycin gene. The small arrows on stems upstream of genes indicate promoters. (B) Spot test for synthetic lethality of ΔdegP dut mutants at 42°C and its suppression by inactivation of ung. Serial dilutions of rapidly growing cultures (10 μl) were spotted onto LB plates and incubated for 36 h at 28°C or for 16 h at 42°C. The dut recA(Ts) and dut recA(Ts) ung mutants were plated in parallel as controls. The strains were wild type, AB1157; dut, AK105; dut recA(Ts), AK106; dut recA(Ts) Δung, L-85; ΔdegP, HT81; ΔdegP dut, HT82; and ΔdegP dut Δung, HT83. (C) The explanation for the dut-1 degP synthetic lethality: aggravation through lack of support for the unstable mutant protein.
To gain insights into the mechanisms behind the dut degP synthetic lethality, we mutagenized the double mutants at the permissive temperature with pRL27 and plated for suppressors at 42°C. To verify the linkage of suppressors of the kan inserts, we reintroduced the potential suppressors into the original strain by P1 transduction. Initially, the transduction did not work in the degP mutants. Suspecting interference of the pleiotropic ΔdegP mutation with P1 development, we complemented all our suppressed strains with a plasmid carrying a functional degP+ gene, after which we had no problem with transduction.
Two suppressors of the dut degP lethality inactivated the pstC gene of the pstSCAB-phoU operon (see above) (Fig. 7A), confirming our earlier isolation of pstS and pstC suppressors of the dut rec synthetic lethality (34). Two more inserts landed in the 150-bp intergenic region between the pstA and pstB genes, which has been recently implicated in the regulation of stationary sigma expression during phosphate starvation (60). Interestingly, the dut degP pstC triple mutant was still unable to grow at 45°C (the phenotype of the ΔdegP mutation) while showing resistance to 10 mM uracil in the medium, suggesting that (i) it is the dut defect, rather than the degP defect, that is being suppressed by pstC and (ii) the dut defect is primarily responsible for the synthetic lethality of the dut degP combination at 42°C.
Two more suppressors of the dut degP lethality inactivated the glnD gene, coding for the uridylyl-transferase regulator of nitrogen metabolism (23). This enzyme modifies another protein with UMP groups using UTP in a reversible reaction. It is possible, if the enzyme was modified with some number of dUMP groups instead, that reversal of the reaction would generate dUTP, as well as UTP, exacerbating the synthetic phenotype of the dut degP mutants. Inactivation of the GlnD activity would then eliminate this spurious source of dUTP, which could be just enough to allow dut degP mutants to grow.
All Dut-dependent mutants can be made viable by inactivation of UDG.
Complete inactivation of the dut gene is lethal, even in the ung mutant backgrounds (19; E. A. Kouzminova and A. Kuzminov, unpublished data), which is sometimes explained in terms of dUTP hydrolysis by Dut, at least in cells grown in minimal medium, being the only way to produce dTMP, the key intermediate in the dTTP biosynthesis pathway (51). However, the thyA defect, which blocks the same pathway at the next step, although completely dead in the absence of thymidine supplementation, is completely rescued by exogenous thymidine (5), arguing against this explanation for the Δdut lethality. In fact, our isolation of tdk inactivation as SL(dut) mutation rules out this explanation, indicating the existence of an alternative pathway to produce dTMP. El-Hajj and associates had proposed that the reason for the lethality of Δdut ung double mutants, even if supplemented with thymidine, is accumulation of uracil in DNA to levels that disrupt gene regulation (18, 19). This interesting explanation awaits experimental testing.
Hochhauser and Weiss isolated a dut-1(Ts) allele that had 5% and <1% of the wild-type dUTPase activity at the permissive (25°C) and the nonpermissive (42°C) temperatures, respectively, and that was fully viable at both temperatures (29); however, Kouzminova and Kuzminov (34) failed to confirm the temperature sensitivity of the dut-1 allele in a different background and instead reported 2.2 to 3.3% of the wild-type in vitro activity at both temperatures. The dut-1 mutant cells are still apparently able to generate enough dUMP/dTMP even at the nonpermissive temperature, because their growth in rich media is not enhanced by addition of thymidine (34), while the dTTP pool sizes are even elevated threefold (77). One possible explanation proposed for this unexpected observation is that the residual dUTPase activity of the dut-1 mutant is compensated for by the 30-fold expansion of the dUTP pool (51). The only pernicious consequence of this arrangement is the increased incorporation of uracil in the DNA of dut mutants, with the resulting increased level of excision repair.
All known synthetic-lethal combinations with the dut defect are partially rescued by inactivation of UDG (34, 70, 73). Nevertheless, the ung mutants are never isolated in selections for suppressors of Dut-dependent mutants—they suppress only when introduced without selection, by P1 transduction. To determine whether the synthetic lethality of the novel Dut-dependent mutants is a result of DNA-uracil excision or is due to something else, we introduced Δung::cat into the synthetic-lethal or inhibited combinations and determined by a spot test whether the deficiency in DNA-uracil excision was able to suppress their poor colony-forming abilities at 42°C. We found that the polA, xthA, phoU, xerCD, and degP Dut-dependent mutants were indeed suppressed by ung inactivation (Fig. 8B and data not shown), suggesting that the primary reason for their inviability is uracil-DNA incorporation with subsequent excision. The only synthetic lethality that did not react to UDG inactivation was dut tdk (data not shown); the apparent reason was that, in the absence of dUMP and dTMP, the mutant could not synthesize its DNA in the first place.
Summary of our findings.
To test the two explanations of synthetic lethality (Fig. 1), we systematically isolated mutants of E. coli that were synthetically lethal with a dut-1 mutation in the dUTPase gene. We found that inactivation of the following 10 genes made E. coli Dut dependent: degP, polA, recA, recB, recC, ruvA, ruvB, ruvC, tdk, and xthA (Fig. 9). We also found one insertion upstream of the ung promoter that killed the dut mutants, likely due to overexpression of UDG (Fig. 4C). Finally, two groups of synthetically inhibited mutants were also isolated: (i) ftsK, xerC, and xerD and (ii) phoU. All the identified SL(dut) mutants, as well as suppressors of SL(dut), can be presented in a single metabolic scheme, in which the DNA precursor metabolism occupies a relatively small (left) part and the scheme is dominated by various DNA repair pathways (Fig. 9). Thus, we have examples of all four expected categories of Dut-dependent mutants, with multiple entries in “D-D-R” categories 3 and 4. There is a single entry for the tdk mutants in category 1 (functional redundancy for dUMP production) and a single entry for UDG overproduction in category 2 (other ways to avoid chromosomal fragmentation, in this case by downregulating uracil-DNA excision). Thus, systematically isolated SL(dut) mutants argue that genetic interactions mostly identify the “D-D-R” triads and only rarely functional redundancies (Fig. 1).
FIG. 9.

Metabolism of uracil in DNA precursor pools and in DNA. Shown from left to right are DNA precursor metabolism, base excision repair of DNA-uracils, recombinational repair of double-strand breaks, and, finally, resolution of chromosomal dimers. Dark purple, the dut step; orange, known SL(dut) mutations; yellow, mutations synthetically inhibited in combination with the dut defect; light blue, known suppressors of SL(dut) combinations. The ung gene is shown in both blue and orange, because its inactivation suppresses SL(dut) whereas its overproduction is suggested to lead to SL(dut). DNA is shown as a double line unless its chromosomal configuration is also indicated (top, bottom, and right), in which case DNA is shown as a single line.
Surprisingly, we also found at least two combinations, dut-1 degP and dut-1 phoU, that cannot be explained by any of the four above-mentioned categories, falling neither under the “D-D-R” idea nor under the “redundancy” umbrella. In both cases, the second mutation aggravates the dut-1 defect itself, either via (presumably) less cytoplasmic chaperone activity in the degP mutant or via an increased intracellular polyphosphate level in the phoU mutant (pushing the dUTP→dUMP-plus-PPi reaction backwards). Therefore, we propose a third explanation of synthetic lethality: inactivation of a compensating activity. This explanation is possibly limited to synthetic lethals in which the original bait mutation is a partial inactivation of an otherwise essential gene, like our dut-1 allele. The overall conclusion from our systematic analysis is that the current tendency to interpret genetic interactions only in terms of redundancy is an unnecessary oversimplification.
The implications.
Presentation of all the genes that we found in the SL(dut) screen and suppressor selections in a single metabolic network is instructive (Fig. 9), as it reveals how the increased intracellular dUTP concentration triggers DNA modifications (DNA with incorporated uracil), which are then transformed into lesions of increasing complexity: abasic sites, nicks, double-strand DNA breaks, and chromosomal dimers. These consequences of uracil-DNA incorporation are countered, consecutively, by base excision repair, recombinational-repair, and chromosomal-dimer resolution systems. Our interpretation of the corresponding synthetic lethals in terms of the “D-D-R” cycles is relevant to other cases of genetic interactions in both bacteria (37, 45, 46) and yeast (30, 54, 71), where defects in various activities broadly associated with DNA metabolism cause cell dependence on recombinational repair. On the other hand, our results also confirm true metabolic buffering (26), either in terms of the avoidance of chromosomal fragmentation (downregulation of UDG activity), as alternative means of dUMP production (tdk), or in terms of facilitating dUTP hydrolysis by a weakened Dut-1 protein, the last both at the protein level (degP) and at the level of the reaction by-product, pyrophosphate (phoU). Thus, systematic isolation of SL(dut) mutants and their suppressors not only reveals the maze of seemingly unrelated metabolic circuits connected to a common metabolic network (Fig. 9), but also argues for the need for more mechanistic interpretations to explain genetic interactions.
Acknowledgments
We thank Lisa Lukas for guidance at the initial stages of the project and all the members of our laboratory for providing strains and general support. B. Weiss (Emory) offered many helpful suggestions to improve the manuscript.
This work was supported by grant RSG-05-135-01-GMC from the American Cancer Society and by grant GM 073115 from the National Institutes of Health.
Footnotes
Published ahead of print on 27 June 2008.
REFERENCES
- 1.Amado, L., and A. Kuzminov. 2006. The replication intermediates in Escherichia coli are not the product of DNA processing or uracil excision. J. Biol. Chem. 28122635-22646. [DOI] [PubMed] [Google Scholar]
- 2.Amemura, M., K. Makino, H. Shinagawa, A. Kobayashi, and A. Nakata. 1985. Nucleotide sequence of the genes involved in phosphate transport and regulation of the phosphate regulon in Escherichia coli. J. Mol. Biol. 184241-250. [DOI] [PubMed] [Google Scholar]
- 3.Aussel, L., F. X. Barre, M. Aroyo, A. Stasiak, A. Z. Stasiak, and D. Sherratt. 2002. FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108195-205. [DOI] [PubMed] [Google Scholar]
- 4.Bachmann, B. J. 1987. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12, p. 1190-1219. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, DC.
- 5.Barner, H. D., and S. S. Cohen. 1954. The induction of thymine synthesis by T2 infection of a thymine requiring mutant of Escherichia coli. J. Bacteriol. 6880-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertani, L. E., A. Haggmark, and P. Reichard. 1963. Enzymatic synthesis of deoxyribonucleotides. II. Formation and interconversion of deoxyuridine phosphates. J. Biol. Chem. 2383407-3413. [PubMed] [Google Scholar]
- 7.Bessman, M. J., I. R. Lehman, J. Adler, S. B. Zimmerman, E. S. Simms, and A. Kornberg. 1958. Enzymatic synthesis of deoxyribonucleic acid. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acid. Proc. Natl. Acad. Sci. USA 44633-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakely, G., S. Colloms, G. May, M. Burke, and D. Sherratt. 1991. Escherichia coli XerC recombinase is required for chromosomal segregation at cell division. New Biol. 3789-798. [PubMed] [Google Scholar]
- 9.Blakely, G., G. May, R. McCulloch, L. K. Arciszewska, M. Burke, S. T. Lovett, and D. J. Sherratt. 1993. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75351-361. [DOI] [PubMed] [Google Scholar]
- 10.Bradshaw, J. S., and A. Kuzminov. 2003. RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol. Microbiol. 481711-1725. [DOI] [PubMed] [Google Scholar]
- 11.Chambers, S. P., S. E. Prior, D. A. Barstow, and N. P. Minton. 1988. The pMTL nic− cloning vectors. I. Improved pUC polylinker regions to facilitate the use of sonicated DNA for nucleotide sequencing. Gene 68139-149. [DOI] [PubMed] [Google Scholar]
- 12.Clausen, T., C. Southan, and M. Ehrmann. 2002. The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10443-455. [DOI] [PubMed] [Google Scholar]
- 13.Clerget, M. 1991. Site-specific recombination promoted by a short DNA segment of plasmid R1 and by a homologous segment in the terminus region of the Escherichia coli chromosome. New Biol. 3780-788. [PubMed] [Google Scholar]
- 14.Cronan, J. E. 2003. Cosmid-based system for transient expression and absolute off-to-on transcriptional control of Escherichia coli genes. J. Bacteriol. 1856522-6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham, R. P., S. M. Saporito, S. G. Spitzer, and B. Weiss. 1986. Endonuclease IV (nfo) mutant of Escherichia coli. J. Bacteriol. 1681120-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan, B. K., P. A. Rockstroh, and H. R. Warner. 1978. Escherichia coli K-12 mutants deficient in uracil-DNA glycosylase. J. Bacteriol. 1341039-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Hajj, H. H., L. Wang, and B. Weiss. 1992. Multiple mutant of Escherichia coli synthesizing virtually thymineless DNA during limited growth. J. Bacteriol. 1744450-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Hajj, H. H., H. Zhang, and B. Weiss. 1988. Lethality of a dut (deoxyuridine triphosphatase) mutation in Escherichia coli. J. Bacteriol. 1701069-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espeli, O., C. Lee, and K. J. Marians. 2003. A physical and functional interaction between Escherichia coli FtsK and topoisomerase IV. J. Biol. Chem. 27844639-44644. [DOI] [PubMed] [Google Scholar]
- 21.Freemont, P. S., D. L. Ollis, T. A. Steitz, and C. M. Joyce. 1986. A domain of the Klenow fragment of Escherichia coli DNA polymerase I has polymerase but no exonuclease activity. Proteins 166-73. [DOI] [PubMed] [Google Scholar]
- 22.Friedberg, E. C., G. C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. ASM Press, Washington, DC.
- 23.Garcia, E., and S. G. Rhee. 1983. Cascade control of Escherichia coli glutamine synthetase. Purification and properties of PII uridylyltransferase and uridylyl-removing enzyme. J. Biol. Chem. 2582246-2253. [PubMed] [Google Scholar]
- 24.Greenberg, G. R., and R. L. Somerville. 1962. Deoxyuridylate kinase activity and deoxyuridinetriphosphatase in Escherichia coli. Proc. Natl. Acad. Sci. USA 48247-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guarente, L. 1993. Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet. 9362-366. [DOI] [PubMed] [Google Scholar]
- 26.Hartman, J. L., B. Garvik, and L. Hartwell. 2001. Principles for the buffering of genetic variation. Science 2911001-1004. [DOI] [PubMed] [Google Scholar]
- 27.Heller, R. C., and K. J. Marians. 2007. Non-replicative helicases at the replication fork. DNA Repair 6945-952. [DOI] [PubMed] [Google Scholar]
- 28.Hiraga, S., K. Igarashi, and T. Yura. 1967. A deoxythymidine kinase-deficient mutant of Escherichia coli. I. Isolation and some properties. Biochim. Biophys. Acta 14541-51. [DOI] [PubMed] [Google Scholar]
- 29.Hochhauser, S. J., and B. Weiss. 1978. Escherichia coli mutants deficient in deoxyuridine triphosphatase. J. Bacteriol. 134157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, M. E., and R. D. Kolodner. 2005. A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol. Cell 17709-720. [DOI] [PubMed] [Google Scholar]
- 31.Joyce, C. M., and N. D. F. Grindley. 1984. Method for determining whether a gene of Escherichia coli is essential: application to the polA gene. J. Bacteriol. 158636-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keyer, K., and J. Imlay. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 9313635-13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornberg, A., and T. A. Baker. 1992. DNA replication. W. H. Freeman and Company, New York, NY.
- 34.Kouzminova, E. A., and A. Kuzminov. 2004. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol. Microbiol. 511279-1295. [DOI] [PubMed] [Google Scholar]
- 35.Kouzminova, E. A., and A. Kuzminov. 2006. Fragmentation of replicating chromosomes triggered by uracil in DNA. J. Mol. Biol. 35520-33. [DOI] [PubMed] [Google Scholar]
- 36.Kouzminova, E. A., and A. Kuzminov. 2008. Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-strand breaks. Mol. Microbiol. 68202-215. [DOI] [PubMed] [Google Scholar]
- 37.Kouzminova, E. A., E. Rotman, L. Macomber, J. Zhang, and A. Kuzminov. 2004. RecA-dependent mutants in E. coli reveal strategies to avoid replication fork failure. Proc. Natl. Acad. Sci. USA 10116262-16267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuempel, P. L., J. M. Henson, L. Dircks, M. Tecklenburg, and D. F. Lim. 1991. dif, a recA-independent recombination site in the terminus region of the chromosome of Escherichia coli. New Biol. 3799-811. [PubMed] [Google Scholar]
- 39.Kuzminov, A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63751-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuzminov, A., and F. W. Stahl. 1997. Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J. Bacteriol. 179880-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larsen, R. A., M. M. Wilson, A. M. Guss, and W. W. Metcalf. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178193-201. [DOI] [PubMed] [Google Scholar]
- 42.Lindahl, T. 1974. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. USA 713649-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindahl, T., and B. Nyberg. 1974. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 133405-3410. [DOI] [PubMed] [Google Scholar]
- 44.Lu, M., J. L. Campbell, E. Boye, and N. Kleckner. 1994. SeqA: a negative modulator of replication initiation. Cell 77413-426. [DOI] [PubMed] [Google Scholar]
- 45.Lukas, L., and A. Kuzminov. 2006. Chromosomal fragmentation is the major consequence of the rdgB defect in Escherichia coli. Genetics 1721359-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michel, B., G. Grompone, M. J. Florès, and V. Bidnenko. 2004. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. USA 10112783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 1702575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morohoshi, T., T. Maruo, Y. Shirai, J. Kato, T. Ikeda, N. Takiguchi, H. Ohtake, and A. Kuroda. 2002. Accumulation of inorganic polyphosphate in phoU mutants of Escherichia coli and Synechocystis sp. strain PCC6803. Appl. Environ. Microbiol. 684107-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakata, A., M. Amemura, and H. Shinagawa. 1984. Regulation of the phosphate regulon in Escherichia coli K-12: regulation of the negative regulatory gene phoU and identification of the gene product. J. Bacteriol. 159979-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neuhard, J., and R. A. Kelln. 1996. Biosynthesis and conversions of pyrimidines, p. 580-599. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 51.Neuhard, J., and P. Nygaard. 1987. Purines and pyrimidines, p. 445-473. In F. C. Neidhardt (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, DC.
- 52.Okazaki, R., and A. Kornberg. 1964. Deoxythymidine kinase of Escherichia coli. I. Purification and some properties of the enzyme. J. Biol. Chem. 239269-274. [PubMed] [Google Scholar]
- 53.Ooi, S. L., X. Pan, B. D. Peyser, P. Ye, P. B. Meluh, D. S. Yuan, R. A. Irizarry, J. S. Bader, F. A. Spencer, and J. D. Boeke. 2006. Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2256-63. [DOI] [PubMed] [Google Scholar]
- 54.Ooi, S. L., D. D. Shoemaker, and J. D. Boeke. 2003. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 35277-286. [DOI] [PubMed] [Google Scholar]
- 55.Petit, M. A., and D. Ehrlich. 2002. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 213137-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piersen, C. E., A. K. McCullough, and R. S. Lloyd. 2000. AP lyases and dRPases: commonality of mechanism. Mutat. Res. 45943-53. [DOI] [PubMed] [Google Scholar]
- 57.Quirk, S., S. K. Bhatnagar, and M. J. Bessman. 1990. Primary structure of the deoxyguanosine triphosphate triphosphohydrolase-encoding gene (dgt) of Escherichia coli. Gene 8913-18. [DOI] [PubMed] [Google Scholar]
- 58.Rizzitello, A. E., J. R. Harper, and T. J. Silhavy. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 1836794-6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roca, A. I., and M. M. Cox. 1997. RecA protein: structure, function, and role in recombinational DNA repair. Prog. Nucleic Acid Res. Mol. Biol. 56129-223. [DOI] [PubMed] [Google Scholar]
- 60.Schurdell, M. S., G. M. Woodbury, and W. R. McCleary. 2007. Genetic evidence suggests that the intergenic region between pstA and pstB plays a role in the regulation of rpoS translation during phosphate limitation. J. Bacteriol. 1891150-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi, I. Y., J. Stansbury, and A. Kuzminov. 2005. Defect in acetyl-CoA↔acetate pathway poisons recombinational repair-deficient mutants of Escherichia coli. J. Bacteriol. 1871266-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shlomai, J., and A. Kornberg. 1978. Deoxyuridine triphosphatase of Escherichia coli. Purification, properties, and use as a reagent to reduce uracil incorporation into DNA. J. Biol. Chem. 2533305-3312. [PubMed] [Google Scholar]
- 63.Spiess, C., A. Beil, and M. Ehrmann. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97339-347. [DOI] [PubMed] [Google Scholar]
- 64.Steed, P. M., and B. L. Wanner. 1993. Use of the rep technique for allele replacement to construct mutants with deletions of the pstSCAB-phoU operon: evidence of a new role for the PhoU protein in the phosphate regulon. J. Bacteriol. 1756797-6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steiner, W., G. Liu, W. D. Donachie, and P. Kuempel. 1999. The cytoplasmic domain of FtsK protein is required for resolution of chromosome dimers. Mol. Microbiol. 31579-583. [DOI] [PubMed] [Google Scholar]
- 66.Steiner, W. W., and P. L. Kuempel. 1998. Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. J. Bacteriol. 1806269-6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strauch, K. L., K. Johnson, and J. Beckwith. 1989. Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J. Bacteriol. 1712689-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takahashi, Y., and U. Tokumoto. 2002. A third bacterial system for the assembly of iron-sulfur clusters with homologs in archaea and plastids. J. Biol. Chem. 27728380-28383. [DOI] [PubMed] [Google Scholar]
- 69.Taucher-Scholtz, G., M. Abdel-Monem, and H. Hoffman-Berling. 1983. Functions of DNA helicases in Escherichia coli, p. 65-76. In N. R. Cozzarelli (ed.), Mechanisms of DNA replication and recombination. A. R. Liss, New York. NY.
- 70.Taylor, A. F., and B. Weiss. 1982. Role of exonuclease III in the base excision repair of uracil-containing DNA. J. Bacteriol. 151351-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tong, A. H., M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader, N. Pagé, M. Robinson, S. Raghibizadeh, C. W. Hogue, H. Bussey, B. Andrews, M. Tyers, and C. Boone. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2942364-2368. [DOI] [PubMed] [Google Scholar]
- 72.Tong, A. H., G. Lesage, G. D. Bader, H. Ding, H. Xu, X. Xin, J. Young, G. F. Berriz, R. L. Brost, M. Chang, Y. Chen, X. Cheng, G. Chua, H. Friesen, D. S. Goldberg, J. Haynes, C. Humphries, G. He, S. Hussein, L. Ke, N. Krogan, Z. Li, J. N. Levinson, H. Lu, P. Ménard, C. Munyana, A. B. Parsons, O. Ryan, R. Tonikian, T. Roberts, A. M. Sdicu, J. Shapiro, B. Sheikh, B. Suter, S. L. Wong, L. V. Zhang, H. Zhu, C. G. Burd, S. Munro, C. Sander, J. Rine, J. Greenblatt, M. Peter, A. Bretscher, G. Bell, F. P. Roth, G. W. Brown, B. Andrews, H. Bussey, and C. Boone. 2004. Global mapping of the yeast genetic interaction network. Science 303808-813. [DOI] [PubMed] [Google Scholar]
- 73.Tye, B.-K., and I. R. Lehman. 1977. Excision repair of uracil incorporated in DNA as a result of a defect in dUTPase. J. Mol. Biol. 117293-306. [DOI] [PubMed] [Google Scholar]
- 74.Tye, B. K., P. O. Nyman, I. R. Lehman, S. Hochhauser, and B. Weiss. 1977. Transient accumulation of Okazaki fragments as a result of uracil incorporation into nascent DNA. Proc. Natl. Acad. Sci. USA 74154-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang, J. C., R. Chen, and D. A. Julin. 2000. A single nuclease active site of the Escherichia coli RecBCD enzyme catalyzes single-stranded DNA degradation in both directions. J. Biol. Chem. 275507-513. [DOI] [PubMed] [Google Scholar]
- 76.Wanner, B. L. 1996. Phosphorus assimilation and control of the phosphate regulon, p. 1357-1381. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC.
- 77.Warner, H. R., B. K. Duncan, C. Garrett, and J. Neuhard. 1981. Synthesis and metabolism of uracil-containing deoxyribonucleic acid in Escherichia coli. J. Bacteriol. 145687-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West, S. C. 1997. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 31213-244. [DOI] [PubMed] [Google Scholar]
- 79.Yates, J., I. Zhekov, R. Baker, B. Eklund, D. J. Sherratt, and L. K. Arciszewska. 2006. Dissection of a functional interaction between the DNA translocase, FtsK, and the XerD recombinase. Mol. Microbiol. 591754-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoo, S. J., J. H. Seol, S. K. Woo, S. W. Suh, D. S. Hwang, D. B. Ha, and C. H. Chung. 1993. Hydrolysis of the IciA protein, an inhibitor of DNA replication initiation, by protease Do in Escherichia coli. FEBS Lett. 32717-20. [DOI] [PubMed] [Google Scholar]