

Abstract
The bacterial single-stranded DNA-binding protein (SSB) and the archaeal/eukaryotic functional homolog, replication protein A (RPA), are essential for most aspects of DNA metabolism. Structural analyses of the architecture of SSB and RPA suggest that they are composed of different combinations of a module called the oligonucleotide/oligosaccharide-binding (OB) fold. Members of the domains Bacteria and Eukarya, in general, contain one type of SSB or RPA. In contrast, organisms in the archaeal domain have different RPAs made up of different organizations of OB folds. Interestingly, the euryarchaeon Methanosarcina acetivorans harbors multiple functional RPAs named MacRPA1 (for M. acetivorans RPA 1), MacRPA2, and MacRPA3. Comparison of MacRPA1 with related proteins in the publicly available databases suggested that intramolecular homologous recombination might play an important role in generating some of the diversity of OB folds in archaeal cells. On the basis of this information, from a four-OB-fold-containing RPA, we engineered chimeric modules to create three-OB-fold-containing RPAs to mimic a novel form of RPA found in Methanococcoides burtonii and Methanosaeta thermophila. We further created two RPAs that mimicked the RPAs in Methanocaldococcus jannaschii and Methanothermobacter thermautotrophicus through fusions of modules from MacRPA1 and M. thermautotrophicus RPA. Functional studies of these engineered proteins suggested that fusion and shuffling of OB folds can lead to well-folded polypeptides with most of the known properties of SSB and RPAs. On the basis of these results, different models that attempt to explain how intramolecular and intermolecular homologous recombination can generate novel forms of SSB or RPAs are proposed.
The bacterial single-stranded DNA (ssDNA)-binding protein (SSB) and the archaeal/eukaryotic replication protein A (RPA) play multiple and essential roles in almost every aspect of nucleic acid metabolism, including DNA replication, repair, and recombination (14, 25). The SSBs and RPAs across the three domains of life share a common and conserved module called the oligonucleotide/oligosaccharide-binding fold (OB fold) (16). Structurally, the common OB folds consist of five-stranded β-sheets coiled to form a closed β-barrel structure usually capped by an alpha-helix (13, 16). Most of the bacterial SSBs have a single OB fold per polypeptide that oligomerizes in solution to form a homotetramer (14). However, in Deinococcus radiodurans and Thermus aquaticus, SSB polypeptides that contain two OB folds have been reported, and as expected, the functional form of the protein is a homodimer that mimics the homotetrameric bacterial SSB (2). In eukaryotes, RPA is a heterotrimeric complex composed of RPA70, RPA32, and RPA14 proteins of approximately 70, 32, and 14 kDa, respectively. Each subunit harbors at least one OB fold, and the largest subunit RPA70 contains a zinc finger motif at the C-terminal region (25). The OB folds in archaeal RPAs show high sequence and structural similarity to eukaryotic RPAs (7, 10). However, the number and organization of OB folds in the RPAs of archaea differ from those of their eukaryotic counterparts. In the Crenarchaeota, the Sulfolobus solfataricus RPA (SsoRPA) is the only well-characterized protein. The SsoRPA contains a single OB fold, and while it has been reported to oligomerize to form a homotetramer (6), a different report found that the protein exists as monomers (23). In the Euryarchaeota, RPAs display an unusual diversity and distribution. Single-subunit RPAs containing multiple OB folds and a putative zinc finger motif have been characterized for Methanocaldococcus jannaschii (7) and Methanothermobacter thermautotrophicus (9). The M. jannaschii RPA (MjaRPA) and M. thermautotrophicus RPA (MthRPA) contain four and five OB folds, respectively, with each protein harboring a putative Zn finger motif at the C-terminal region. The RPA in Pyrococcus furiosus exists as a stable heterotrimer consisting of three subunits, RPA41, RPA32, and RPA14. Each of the P. furiosus RPA subunits contains an OB fold, and in addition, RPA41 contains a zinc finger-like motif (12). The presence of three functional RPAs in the mesophilic archaeon Methanosarcina acetivorans has been demonstrated. Unlike the multiple RPAs found in P. furiosus, each M. acetivorans RPA (MacRPA) can act as a distinct SSB (19).
Modular rearrangements have been a well-known mechanism of protein evolution (24). Domains are basic evolutionary units of proteins. Therefore, via homologous or nonhomologous recombination events, proteins can evolve by domain shuffling, domain fusion, and domain fission, yielding new modular building blocks (17). In relation to this observation, an evolutionary model for ssDNA-binding proteins has been suggested. In this model, SSB and RPA are predicted to originate from a common ancestral ssDNA-binding protein that diverged through evolution by domain duplications, insertions, and deletions (3). Although the domains are usually the units that are deleted or duplicated, multi-OB-fold-containing RPAs provide a unique case, where recombination during replication can easily result in chimeric domains. Hence, the euryarchaeotes, which include the methanogens, might have harnessed these molecular mechanisms to create new gene structures (15) that encode RPAs of unprecedented diversity.
In the order Methanosarcinales, the three Methanosarcina species (M. acetivorans, M. mazei, and M. barkeri) have a RPA with four OB folds, which is similar to those of M. jannaschii and M. thermautotrophicus, except that the Methanosarcina protein lacks a zinc finger module (19). We hypothesized that the M. jannaschii and M. thermautotrophicus proteins evolved through gain of genetic information by a progenitor that was similar to the methanosarcinal protein. Also interestingly, Methanococcoides burtonii and Methanosaeta thermophila, a psychrophilic member and a thermophilic member, respectively, of the Methanosarcinales have genes encoding a three-OB-fold-containing RPA. Each protein exhibited high amino acid sequence identity to the four-OB-fold-containing RPA found in other members of the Methanosarcinales (Fig. 1A). On the basis of insights gained from the sequence alignment, we hypothesized that the three-OB-fold RPAs in M. burtonii and M. thermophila evolved from the four-OB-fold RPAs in the Methanosarcina spp., most likely through a recombination event that fused two adjacent OB folds.
FIG. 1.



Alignment, schematic representation of MacRPA1 and related RPA or RPA-like proteins, and homology modeling of naturally occurring and artificially synthesized OB folds. (A) Alignment of MacRPA1 and related proteins. The proteins are M. acetivorans RPA1 (MacRPA1) (GenBank accession no. AAM07979), M. mazei RPA1 (MmaRPA1) (GenBank accession no. NP_633323), M. barkeri RPA1 (MbaRPA1) (GenBank accession no. ZP_00542400), Methanococcoides burtonii RPA1-like protein (MbuRPA1) (GenBank accession no. ZP_00563288), Methanosaeta thermophila RPA1-like protein (MtheRPA) (GenBank accession no. YP_843314), Haloarcula marismortui RPA1-like protein (HmaRPA) (GenBank accession no. AAV47126), Halobacterium sp. strain NRC-1 RPA1-like protein (HspRPA) (GenBank accession no. NP_279274), and Natronomonas pharaonis RPA1-like protein (NphRPA) (GenBank accession no. YP_325819). The amino acid sequences for the different OB folds in MacRPA1 are delineated with the differently shaded lines and are labeled with the letters A, B, C, and D. Identical amino acids (white letters on a black background), conserved amino acids (black letters on gray background), and gaps introduced to maximize sequence alignment (dashes) are indicated. (B) Schematic representations showing the construction of MacRPA1 chimeras (MacRPA1 chimera-1, MacRPA1 chimera-2, and MacRPA1 chimera-3) from wild-type (WT) MacRPA1. The schematic representations of the MacRPA1 chimeras show OB folds A to D. (C) Schematic representations showing the construction of M. jannaschii RPA-like protein (MjaRPA-like protein). The first three OB folds (folds A to C) and the N-terminal third of the fourth OB fold of M. acetivorans RPA1 were fused to the indicated modules from M. thermautotrophicus RPA through fusion of adjacent OB folds. (D) Schematic representations showing the construction of MthRPA-like protein. The same fragment from wild-type MacRPA1 for construction of the MjaRPA-like protein as shown in panel B was used, but the fusion fragment from wild-type (WT) MthRPA was from the fourth OB fold (instead of the fifth) to the end of the polypeptide. The full-length and chimeric (fused) OB folds are shown by differently shaded boxes with letters A, B, C, D, and E. The boxes denoted by Zn represent zinc-binding domains with their sequences (x represents any amino acid residue). The motifs are not drawn to scale, and the exact points of fusion are described in Materials and Methods. (E) The highly conserved structures of archaeal OB folds allow for functional fusions. (i) The top row shows homology models for the four MacRPA1 OB folds. The four aromatic residues (depicted by stick representation) expected to form critical interactions with ssDNA were identified on the basis of the comparison of amino acid sequence alignment and structural alignment between MacRPA1 OB folds and that of the SsoSSB. Note that OB fold D contains a leucine in the position occupied by tyrosine or phenylalanine in most known OB folds. The number by each residue identifies it in the alignment shown in panel ii. The middle row shows fusion OB folds. The elements of each fusion fold corresponding to the original OB folds are shown in the faded parental colors. The site of the fusion is indicated by the arrow. The bottom row shows superimposition of the parental and fusion folds. (ii) Amino acid sequence alignment of individual MacRPA1 OB folds (MacRPA1_OB_A to MacRPA1_OB_D), chimeric OB folds (MacRPA1_OB_AB to MacRPA1_OB_CD), and SsoSSB protein (SsoSSB_1O7I.A). Numbers above the alignment indicate residues proposed to interact with ssDNA. Black arrows indicate the position of the fusion. Elements of the secondary structure are indicated under the alignment. Sequences were aligned using Muitalin 5.4.1 (4) by comparison with a table constructed using the Blosum62 substitution matrix with a gap opening weight of 7 and gap extension weight of 1.
Here we describe experiments that tested the two hypotheses described above, and we use biochemical and biophysical methods to demonstrate that each protein engineered to mimic the naturally occurring RPA homologs is highly soluble and can discriminate ssDNA from double-stranded DNA (dsDNA). Some of the engineered proteins were, however, not as efficient as the wild-type proteins were. We further tested the engineered RPAs for their effects on the activities of archaeal DNA replication proteins, including M. acetivorans flap endonuclease 1 (MacFEN1) and M. acetivorans DNA polymerase BI (MacPolBI) (13, 19).
Besides RPA/SSB proteins, OB folds play important roles in a diverse set of proteins (22). In fact, the OB fold has been found in at least 15 nonhomologous families of proteins (5). OB folds are thus found in proteins, such as Saccharomyces cerevisiae and Escherichia coli aspartyl-tRNA synthetases (22), the cold shock protein CspB (20), the E. coli lysyl-tRNA synthetases, staphylococcal nuclease, and the cold shock protein CspA (1). The work presented here provides important insights into how this diversity might have been achieved in nature. Furthermore, we propose an evolutionary model that explains how intramolecular and intermolecular homologous recombination processes might have led to domain deletion, gene fusion, and internal insertion of OB folds to generate the diverse RPA homologs found in extant euryarchaeotes.
MATERIALS AND METHODS
Gene fusion to create RPAs containing three OB folds (MbuRPA-like protein).
Methanococcoides burtonii belongs to the order Methanosarcinales. Similar to its relatives in the genus Methanosarcina, the genome of M. burtonii contains orthologs of the methanosarcinal RPA2 and RPA3 (19). However, instead of a four-OB-fold-containing RPA1, the genome contains an interesting derivative of this protein. Bioinformatic analysis in the present study suggests that a three-OB-fold RPA in M. burtonii evolved through the fusion of the two middle OB folds found in the four-OB-fold RPA1 of its relatives in the genus Methanosarcina (Fig. 1A). To test this hypothesis, the gene encoding the four-OB-fold-containing RPA (MacRPA1) was used as a template in a gene fusion method, based on PCR amplification, to generate a gene encoding an M. burtonii RPA1 (MbuRPA1)-like protein. The PCR method was described in our previous report (18). The initial experiment fused the two middle OB folds (B and C) at the exact position that matches the deletion in MbuRPA (Fig. 1A). Briefly, a forward primer MacRPA1F was combined with a reverse primer Chimera-2R to amplify a segment of the gene coding for MacRPA1. This segment encoded the amino acids from positions 1 to 195 of the MacRPA1 polypeptide. A second PCR amplification using the forward primer Chimera-2F and the reverse primer MacRPA1R amplified another segment of the gene. This second segment encoded the amino acids from positions 306 to 484 of the MacRPA1 polypeptide. The two primers Chimera-2R and Chimera-2F contained overlapping nucleotide sequences that are complementary. Thus, in a second PCR amplification, aliquots of the two PCR products are added as template; in the presence of all reagents except for primers, the complementary ends anneal to each other, and at the primer extension stage, the two products are fused into a single product. After five PCR cycles, the forward (MacRPA1F) and reverse (MacRPA1R) primers corresponding to the ends of the fused product are added to the PCR mixture, and amplification continued for another 20 cycles. The PCR amplifications of the individual segments were carried out with Vent DNA polymerase and reagents provided by the manufacturer (New England Biolabs), whereas the fusion was carried out using ExTaq DNA polymerase and reagents provided by the manufacturer (TakaRa Bio Inc.). The natural fusion leading to the formation of the M. burtonii RPA occurred through the fusion of OB folds B and C (Fig. 1B). However, we anticipated that a fusion between OB folds A and B and also between OB folds C and D could also yield functional three-OB-fold-containing RPAs. The gene fusion method was used to generate MacRPA1 derivatives that have the first two OB folds and the last two OB folds fused together. The gene products from the fused genes were named MacRPA1 chimera-1, MacRPA1 chimera-2 (MbuRPA-like protein), and MacRPA1 chimera-3 (Fig. 1B). The fusions of OB folds in MacRPA1 chimera-1 and MacRPA1 chimera-3 were carried out to mimic that of MacRPA1 chimera-2, i.e., the points of fusion were similar on the basis of the amino acid sequence alignment of the OB folds (Fig. 1E). Thus, for MacRPA1 chimera-1, the first (N-terminal) fragment was from amino acid residues 1 to 86 of the MacRPA1 polypeptide, and the second (C-terminal) fragment was from amino acid residues 196 to 484 of the MacRPA1 polypeptide. The MacRPA1 chimera-1 gene was amplified by using MacRPA1F/Chimera-1R and Chimera-1F/MacRPA1R primer pairs in the first PCR amplification to obtain the two fragments, and then the MacRPA1F/MacRPA1R primer pair was used in the second PCR to fuse the two products. In the case of MacRPA1 chimera-3, the N-terminal fragment consisted of amino acid residues 1 to 305 of the MacRPA1 polypeptide, and the C-terminal fragment consisted of amino acid residues 415 to 484 of the MacRPA1 polypeptide. To amplify the N- and C-terminal fragments, we used MacRPA1F/Chimera-3R and Chimera-3F/MacRPA1R as the primer pairs, respectively. All primers are shown in Table 1.All three chimeric genes were cloned into a TA-cloning vector, pGEM-T Easy vector (Promega), and sequenced (W. M. Keck Center for Functional and Comparative Genomics, University of Illinois at Urbana-Champaign) to confirm the correctness of the coding sequence. The correct fragment was released by digestion with NdeI and XhoI and then ligated (T4 DNA ligase; New England Biolabs) into an NdeI/XhoI-digested pET28a vector (Novagen) modified by replacing the kanamycin resistance gene with that for ampicillin resistance. The NdeI insertion resulted in each chimeric protein being produced with six histidines at the N terminus.
TABLE 1.
Oligonucleotide primers used in this study
| Expt | Oligonucleotide primera | Nucleotide sequenceb or description |
|---|---|---|
| MacRPA1 gene | MacRPA1F | 5′-AAAAACATATGACCGATATTGAGACTATTTATAAAAAG |
| MacRPA1R | 5′-TTTTTCTCGAGTCAGAGAATAATCACTCTACTCCTG | |
| MthRPA gene | MthRPA-R | 5′-CTCGAGTTAAAGTTCCTTCTTTACGACCTTTCT |
| MacRPA1 chimera-1 | Chimera-1F | 5′-GATGTCAAGGAGTTTACCCGCAAAGACGGAAGCGGTGGAAAA |
| Chimera-1R | 5′-ACCGCTTCCGTCTTTGCGGGTAAACTCCTTGACATCAAAAAC | |
| MacRPA1 chimera-2 | Chimera-2F | 5′-GAAATCAGAACTTTCCAGAAAAAGGATGGGACTGCCGGCAGG |
| Chimera-2R | 5′-GGCAGTCCCATCCTTTTTCTGGAAAGTTCTGATTTCAGAAAT | |
| MacRPA1 chimera-3 | Chimera-3F | 5′-TCAGAAGTTCGAACTTTCGAGAGGGAGGACGGAACCGAAAAC |
| Chimera-3R | 5′-GGTTCCGTCCTCCCTCTCGAAAGTTCGAACTTCTGAGATGTC | |
| MjaRPA-like protein | MjaRPA-like-F | 5′-AAAGTTTCCGAGATAGGAGAACTGAGGGAGTTCCAGAGGGGTGACGGC |
| MjaRPA-like-R | 5′-GCCGTCACCCCTCTGGAACTCCCTCAGTTCTCCTATCTCGGAAACTTT | |
| MthRPA-like protein | MthRPA-like-F | 5′-AAAGTTTCCGAGATAGGAGAACTGCGCGAATTTGAGAGGGAAGACGGG |
| MthRPA-like-R | 5′-CCCGTCTTCCCTCTCAAATTCGCGCAGTTCTCCTATCTCGGAAACTTT | |
| EMSA | MacMC-R | 5′-TTTTCTCGAGTTATGCCACGAGTTTTACATGCTCCTTGCCCC |
| dsDNA | A Thermoanaerobacterium polysaccharolyticum carbohydrate-binding module gene (375 bp) | |
| FPA | FL-22 | 5′-Fluorescein-(T/G)CCTCGCTGCCGTCGCCAGCGT-3′ |
| Flap structure | Oligonucleotide 1 | 5′-GATGTCAAGCAGTCCTAACTTTGAGGCAGAGTCC |
| Oligonucleotide 2 | 5′-CACGTTGACTACCGTC | |
| Oligonucleotide 3 | 5′-GGACTCTGCCTCAAGACGGTAGTCAACGTG | |
| Primer extension | (M13mp18 positions 6205 to 6234) | 5′-ATTCGTAATCATGGTCATAGCTGTTTCCTG |
The oligonucleotide primers used in the MacRPA1 gene, MthRPA gene, RPA1 chimera-1, RPA1 chimera-2 (MbuRPA1-like protein), RPA1 chimera-3, MjaRPA-like protein, and MthRPA-like protein experiments were used to create chimeric MacRPA1 genes. The EMSA oligonucleotide was the probe used for the electrophoretic mobility shift assay. The FPA oligonucleotide was used for fluorescence polarization anisotropy measurement. The flap structure oligonucleotides were used to create the substrate for an endonuclease assay. The primer extension oligonucleotide was used for primer extension analysis.
Restriction sites are underlined.
Gene fusion to create MjaRPA- and MthRPA-like genes.
The gene fusion method was also used to fuse segments of the genes encoding MacRPA1 and MthRPA to artificially construct chimeric genes that code for MjaRPA-like (Fig. 1C) and MthRPA-like (Fig. 1D) proteins. The fusion to create the MacRPA1/MthRPA chimeric genes followed the same procedure as described above for the MbuRPA1-like gene. The difference was that the segment containing the N-terminal region was amplified from MacRPA1 (positions 1 to 411 of the MacRPA1 polypeptide), and the C-terminus-encoding region was amplified from MthRPA (positions 551 to 793 of the MthRPA polypeptide). This fusion, as shown in Fig. 1C, created a gene that encodes four OB folds and a zinc finger domain (MjaRPA-like gene). The primer pairs used for the N- and C-terminal segments were MacRPA1F/MjaRPA-like-R and MjaRPA-like-F/MthRPA-R primers, respectively. In the second PCR that fused the two segments, the primer pair MacRPA1F/MthRPA-R was used with aliquots of the two PCR products as template. Using primers that targeted amino acid residue positions 1 to 411 of the MacRPA1 polypeptide and C-terminal amino acid residue positions 434 to 793 of the MthRPA polypeptide, two PCR products, which when fused and expressed, resulted in a polypeptide of five OB folds (MthRPA-like protein) were amplified (Fig. 1D). The primer pairs used to amplify the N- and C-terminal regions were MacRPA1F/MthRPA-like-R and MthRPA-like-F/MthRPA-R primers, respectively. The chimeric gene was obtained by a second PCR that used the two PCR products as template and primers MacRPA1F and MthRPA-R as the forward and reverse primers, respectively. The chimeric genes encoding the MjaRPA-like and MthRPA-like proteins were cloned into the TA-cloning vector and sequenced to confirm the correctness of the coding sequence. The correct fragment was ligated into the pET28a vector described above. Thus, when expressed, each gene contained a N-terminal six-histidine tag encoded by the plasmid.
Purification of recombinant proteins.
Wild-type MacRPA1 and its chimeras were expressed in E. coli BL21-CodonPlus (DE3) RIL cells (Stratagene) harboring the respective expression vectors introduced into the bacterium by heat shock transformation. A single colony from each cell line was grown at 37°C in LB broth containing ampicillin (100 μg/ml) and chloramphenicol (50 μg/ml). At an optical density of 0.3 at an absorbance of 600 nm, each culture was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM, and culturing was continued for 12 h at 16°C. The cells from each culture were harvested by centrifugation. The cell pellets were suspended in lysis buffer (50 mM sodium phosphate [pH 7.0] and 300 mM NaCl), and the cell contents were released by a French pressure cell (American Instruments Co.). After the cell debris was removed by centrifugation (10,000 × g for 20 min at 4°C), the supernatant was applied to a lysis buffer-equilibrated metal affinity resin (Talon cobalt affinity resin; Clontech). The resin was then washed with 10 column volumes of lysis buffer, and the bound protein was eluted with elution buffer (lysis buffer containing 150 mM imidazole). For further purification, the fractions were pooled and dialyzed against buffer A (50 mM Tris-HCl [pH 8.0] and 100 mM NaCl). The dialysate was applied to an anion-exchange column (HiTrap Q HP [5 ml]; GE Healthcare) equilibrated with buffer A, and the column was washed with five column volumes of the same buffer. The bound proteins were eluted with a linear gradient of buffer B (50 mM Tris-HCl [pH 8.0] and 1,000 mM NaCl). Aliquots of the eluted proteins were examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and all highly purified proteins were dialyzed against buffer C (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 10% glycerol, and 0.5 mM dithiothreitol [DTT]). The procedures used in expression and purification of recombinant MacFEN1 and MacPolBI were as previously described (13, 19), and those for recombinant wild-type MthRPA and M. thermautotrophicus DNA polymerase BI (MthPolBI) complex were as described elsewhere (9).
Oligomeric state analysis.
The estimation of subunit organization of each MacRPA1 derivative was carried out by gel filtration chromatography as described previously (19). In brief, 100 μg of wild-type MacRPA1 and each of its chimeric proteins was dialyzed against buffer D (50 mM sodium phosphate [pH 7.0] and 150 mM NaCl) and applied to a buffer D-equilibrated Superdex 200HR 10/30 column (GE Healthcare) fitted to a high-pressure liquid chromatography apparatus (AKTA explorer 10; GE Healthcare). The chromatograph was developed with the same buffer at a flow rate of 0.5 ml/min at 4°C, and the absorbance at 280 nm was monitored. Fractions with a volume of 500 μl were collected and analyzed by SDS-PAGE. To calibrate the column, ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), albumin (67 kDa), ovalbumin (43 kDa), and RNase A (13.7 kDa) (GE Healthcare) were used as molecular mass markers as described previously (19).
EMSA.
The wild-type MacRPA1 and its chimeras were tested for their ability to bind to ssDNA and dsDNA by electrophoretic mobility shift assay (EMSA). Five picomoles of wild-type MacRPA1 and its chimeras was incubated with 1 pmol of 32P-end-labeled 42-mer oligonucleotide (MacMC-R [Table 1]) in 20 μl of binding buffer (20 mM Tris-HCl [pH 8.8], 15 mM MgCl2, 2 mM DTT, and 0.05 mg/ml bovine serum albumin) at 25°C for 30 min. Binding specificity was verified by competition EMSA, using unlabeled ssDNA (MacMC-R) or dsDNA (Table 1) at 1-, 10-, and 50-fold excess concentration of the labeled probe. After 2 μl of loading buffer (250 mM Tris-HCl [pH 7.5], 0.2% bromophenol, and 40% glycerol) was added to the reaction mixtures, the mixtures were resolved by electrophoresis on 8% nondenaturing polyacrylamide gel using 1× Tris-borate-EDTA (TBE) buffer followed by autoradiography.
Fluorescence titrations.
In order to estimate the binding site size of each RPA, we followed the change in the intrinsic fluorescence of each protein during binding to ssDNA. Fluorescence titrations were performed in a fluorescence spectrophotometer (Cary Eclipse; Varian, Inc.). The binding reactions were carried out in 150 μl of reaction buffer containing 20 mM Tris-HCl (pH 8.8), 100 mM NaCl, and 2 mM DTT at 20°C. Small increments of φ174 phage ssDNA were added to a fixed amount (1 μM) of wild-type MacRPA1 and its chimeras until the fluorescence signal was saturated at a maximum value, which we attributed to 100% protein bound to DNA. The intrinsic protein fluorescence was measured using the excitation and emission wavelengths at 280 nm and 340 nm, respectively.
FPA.
Fluorescence polarization anisotropy (FPA) measurement was also used in this study to compare the interaction of wild-type MacRPA1 and its chimeras with ssDNA. The details of the FPA method were described in previous studies (13, 18, 19). In brief, 2 nM of high-performance liquid chromatography-purified ssDNA, FL-22 (Table 1), tagged with fluorescence at the 5′ end was used as the biomolecule in the experiment, which was performed in a fluorescence spectrophotometer (Cary Eclipse; Varian, Inc.) at 23°C ± 1°C. The reaction mixtures were excited at 490 nm, and emission data were collected at 518 nm. Each MacRPA1 chimera was titrated in a reaction mixture containing 20 mM Tris-HCl (pH 8.0), 1 M NaCl, and 2 mM DTT until anisotropy was saturated at the maximum value. The anisotropy curves were fitted with a nonlinear regression 1:1 binding equation to calculate the equilibrium dissociation constant Kd using Prism 4 (GraphPad Software, Inc.). The equation is as follows:
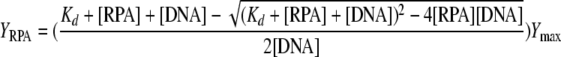 |
where [RPA] is the concentration of the RPA dimer, [DNA] is the concentration of the fluorescein-labeled oligonucleotide (in molecules), Ymax is the value of the FPA at saturation (the second variable obtained from fitting the experimental data), and YRPA is the FPA value at a given RPA concentration. The anisotropy measurements were repeated three times at each concentration, and the values were averaged.
MacFEN1 endonuclease assay.
In a previous study we discovered that wild-type MacRPA1 inhibits the endonuclease activity of MacFEN1 (13). Therefore, we tested the engineered RPAs for this property. The annealing reactions for creating the flap substrate were performed as reported previously (13). In brief, 4 pmol of 32P-end-labeled 34-mer oligonucleotide (oligonucleotide 1) were mixed with 4 pmol of unlabeled 16-mer oligonucleotide (oligonucleotide 2) and 30-mer oligonucleotide (oligonucleotide 3) in 20 μl of the reaction buffer containing 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl. The reaction mixture was incubated at 95°C for 5 min and then slowly cooled to room temperature. The FEN1 endonuclease assay was performed in 20 μl of reaction buffer containing 20 mM Tris-HCl (pH 8.8), 15 mM MgCl2, 2 mM DTT, 0.05 mg/ml bovine serum albumin, and 1 pmol of flap substrate. MacFEN1 was added at 25 pmol, and where the effects of either wild-type MacRPA1 or its engineered derivatives were tested, they were added in increasing amounts of 1.0, 2.5, 5.0, 7.5, and 10.0 pmol per reaction mixture. The reaction mixtures were incubated at 37°C for 5 min and terminated by adding 4 μl of stop solution (98% formamide, 1 mM EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). The samples were heated at 95°C for 5 min, and the products were analyzed on denaturing 15% polyacrylamide gels containing 7 M urea in 1× TBE buffer. The gels were dried, and the samples were visualized by autoradiography with X-ray film and quantified using a BAS-1800 II bioimaging analyzer (Fuji Photo Film Co., Ltd.).
Primer extension analysis.
In another functional analysis, wild-type MacRPA1 and its engineered derivatives were compared through their effect on the primer extension capacity of M. acetivorans DNA polymerase BI. One microgram of M13mp18 ssDNA template and 1 pmol of 5′-32P-end-labeled primer (Table 1), complementary to positions 6205 to 6234 of the template were annealed by heating at 95°C for 5 min and cooling slowly to room temperature. A standard primer extension reaction mixture (20 μl) contained the labeled substrate in 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM MgCl2, 2 mM β-mercaptoethanol, and 250 μM of each deoxynucleoside triphosphate. Wild-type MacRPA1 or each of its engineered derivatives was added at 15 or 30 pmol/reaction mixture, and to initiate primer extension, 0.5 μg of MacPolBI was added to the reaction mixture and incubated at 37°C for 30 min. Wild-type MacRPA1, wild-type MthRPA, and MthRPA-like proteins were also compared through their effects on the primer extension capacity of M. thermautotrophicus DNA polymerase BI. The standard primer extension reaction mixture (20 μl) was the same as described above. The RPAs were added at 15, 30, or 60 pmol/reaction mixture, and to initiate DNA synthesis, 1.5 μg of MthPolBI was added to the reaction mixture and incubated at 65°C or 37°C for 30 min. The reactions were terminated by adding 6 μl of stop solution (98% formamide, 1 mM EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue) and resolved on 1% alkali agarose gel in 50 mM sodium hydroxide and 1 mM EDTA.
Homology modeling of OB folds.
The homology models for the four native OB folds constituting the wild-type MacRPA1 protein (OB folds A, B, C, and D) and three fusion OB folds (fusion OB folds A/B, B/C, and C/D) were constructed using The Chemical Computing Group's Molecular Operating Environment (2008).
The high-resolution (1.26-Å) structure (PDB accession no. 1O7I.A) of the S. solfataricus SSB (SsoSSB) protein (10) was used as the template for modeling of each OB fold. Prior to building of the homology model, primary sequences for all OB folds were aligned with the sequence for the SsoSSB using the Blosum62 substitution matrix with the gap penalties at opening and extension selected as 7 and 1, respectively (Fig. 1E, panel ii). Ten intermediate homology models resulting from a permutational selection of different loop candidates and side chain rotamers were built for each OB fold. The intermediate models, which scored the best according to a packing evaluation function, were chosen as the final models. Each of the intermediate models was subjected to a degree of energy minimization using the forcefield Amber99, with a solvent continuum model (R-Field).
RESULTS
Engineering of functional replication protein A homologs.
Due to their modular organization and diversity, the archaeal RPAs represent an excellent model system for understanding how proteins evolve through shuffling of discrete domains. Each of the three Methanosarcina species (M. acetivorans, M. mazei, and M. barkeri) in the alignment (Fig. 1A) possesses a RPA with four OB folds. The two central OB folds (OB folds B and C in Fig. 1A and E) are very similar, sharing an identity of ∼60% at the amino acid sequence level (18). Interestingly, it appears that a deletion in the B and C OB folds has generated a unique form of RPA in Methanococcoides burtonii, a psychrophilic member of the order Methanosarcinales. To investigate the roles of individual domains, we made a deletion in MacRPA1 mimicking that in M. burtonii and called this derivative MbuRPA1-like or MacRPA1 chimera-2 protein (Fig. 1B). To investigate whether similar deletions occurring between other adjacent OB folds in MacRPA1 will result in a functional ssDNA-binding protein, we also made two similar deletions between the A and B OB folds and the C and D OB folds in MacRPA1 and called them MacRPA1 chimera-1 and MacRPA1 chimera-3, respectively (Fig. 1B).
The RPAs from two thermophilic methanogenic archaea, Methanocaldococcus jannaschii and Methanothermobacter thermautotrophicus, are composed of four and five OB folds, respectively, in the same polypeptide. In addition, each protein also contains a putative zinc finger module at the C terminus (7, 9) (Fig. 1C and D). Interestingly, the RPA from the mesophilic Methanosarcina species also possesses four OB folds. However, it lacks a zinc finger module. To investigate whether the four OB folds/one zinc finger and the five OB folds/one zinc finger proteins (Fig. 1C and D) could evolve from MacRPA1 and other RPAs with zinc finger modules at the C terminus, we created MjaRPA-like and MthRPA-like proteins by fusing MacRPA1 to the domains of interest from MthRPA. A schematic representation demonstrating the method is shown in Fig. 1C and D. Three OB folds (A, B, and C) and approximately one-third of OB fold D from MacRPA1 were fused with approximately two-thirds of OB fold E and the zinc finger module from MthRPA to create MjaRPA-like protein (Fig. 1C). Three OB folds (A, B, and C) and approximately one-third of OB fold D from MacRPA1 were fused with approximately two-thirds of OB fold D, OB fold E, and the zinc finger motif from MthRPA to create MthRPA-like protein (Fig. 1D). On the basis of the alignment of the last OB fold in MacRPA1 and MjaRPA and the last two OB folds (the D and E OB folds) of MthRPA (data not shown), the point of each fusion was at a conserved glutamate (MacRPA1 E412).
We also modeled each of the four OB folds in MacRPA1 after the single OB fold in Sulfolobus solfataricus RPA, which represents the only archaeal RPA with a known structure (10). In addition, each of the chimeric OB folds was also modeled after the SsoRPA. Pairwise comparison, at the amino acid level, among the MacRPA1 OB folds and the chimeric OB folds gave identities ranging from ∼30 to ∼85%, and homology modeling of the OB folds, based on the OB fold in the SsoRPA (10), gave root mean square deviation values (all atoms) of 1.06 to 1.67 (results not shown).
Gene expression, protein purification, and oligomeric state analysis.
All chimeric RPAs were successfully produced. The estimated molecular masses, based on polypeptide sequences, of wild-type MacRPA1, MacRPA1 chimera-1, MacRPA1 chimera-2 (MbuRPA1-like protein), MacRPA1 chimera-3, MjaRPA-like protein, MthRPA-like protein, wild-type MthRPA, and MthPolBI complex were 56.1, 44.4, 43.7, 43.4, 74.7, 88.1, 90.2, and 93.5 (68.0 plus 25.5) kDa, respectively. The molecular masses, as determined by SDS-PAGE, corresponded well with their estimated values (see Fig. S1 in the supplemental material). We also used gel filtration analysis to estimate the oligomeric state of each protein from the RPA constructs. The oligomerization state of wild-type MacRPA1 is dependent on the concentration of MacRPA1, and it exists as a homodimer and homotetramer (19). The results of the gel filtration analysis suggested that MacRPA1 chimera-1, MacRPA1 chimera-2, and MacRPA1 chimera-3 were able to oligomerize to form homotetramers. Single peaks could be observed for MacRPA1 chimera-1 and MacRPA1 chimera-3 (results not shown), and their elution volumes suggested relative molecular masses of 195.9 ± 5.6 and 197.7 ± 4.4 kDa, respectively, which suggested that they form homotetramers in solution. Interestingly, two peaks could be observed in the case of MacRPA1 chimera-2, but unlike wild-type MacRPA1, the ratio of the peaks was not concentration dependent (results not shown). The first and second peaks represented relative molecular masses of 158.3 ± 2.6 and 71.0 ± 1.2 kDa, respectively, which suggested that MacRPA1 chimera-2 could form homodimers or homotetramers in solution. We observed only single peaks for the MjaRPA-like and MthRPA-like proteins, and their elution volumes coincided with relative molecular masses of 183.1 ± 3.4 and 211.0 ± 4.0, respectively, suggesting that each of the two proteins exists as homodimers in solution (results not shown).
Chimeric RPAs are competent in ssDNA binding by EMSA.
In order to determine whether the chimeric RPAs can function as ssDNA-binding proteins, the recombinant wild-type MacRPA1 and its chimeras (MacRPA1 chimera-1, MacRPA1 chimera-2, MacRPA1 chimera-3, MjaRPA-like, and MthRPA-like proteins) were tested for their ability to bind to ssDNA by using EMSA. Each chimeric RPA was able to bind to ssDNA as shown in Fig. 2A, panels ii to vi. Using the EMSA method, the ssDNA-binding affinities of MacRPA1 chimera-1, MacRPA1 chimera-3, and MthRPA-like proteins (Fig. 2A, panels ii, iv, and vi) appeared to be somewhat reduced compared with that of wild-type MacRPA1 (Fig. 2A, panel i). The MjaRPA-like and MacRPA1 chimera-2 proteins seem to have higher ssDNA-binding activity than other chimeric RPAs (Fig. 2A, panels iii and v). In lane 3 (at a concentration of 2 pmol/reaction mixture), only these proteins, as also seen with the wild-type protein, seem to shift the entire probe. As the concentration of wild-type MacRPA1 and MacRPA1 chimera-2 (or MbuRPA1-like protein) increased, two stable binding states were observed (Fig. 2A, panels i and iii). For MacRPA1 chimera-1 and MacRPA1 chimera-3, we could observe only a single shifted band with the same ssDNA substrate and equimolar amounts of proteins as used for wild-type MacRPA1 (Fig. 2A, panels i, ii, and iv). The MjaRPA-like and MthRPA-like proteins also exhibited two binding states with the same substrate as the labeled ssDNA. Some of these proteins remained in the well, suggesting the presence of a very large molecular complex (Fig. 2A, panels v and vi). Each chimeric RPA was also tested for the ability to discriminate between ssDNA and dsDNA. Similar to results reported earlier (19), wild-type MacRPA1 exhibited selective binding to ssDNA (Fig. 2B, panel i). The ssDNA-binding activity of wild-type MacRPA1 to the labeled ssDNA was outcompeted by cold ssDNA until at 50-fold excess cold ssDNA (42 nucleotides), binding to the labeled ssDNA was totally outcompeted (Fig. 2B, panel i, lane 5). However, 1-fold, 10-fold, and 50-fold excess cold dsDNA of larger size (3 kb) failed to outcompete labeled ssDNA binding by wild-type MacRPA1 (Fig. 2B, panel i, lanes 6, 7, and 8). The discrimination of binding to ssDNA and dsDNA by the MjaRPA-like and MthRPA-like proteins was similar to that of the wild-type protein (Fig. 2B, panels v and vi), suggesting that the fusion of the zinc finger module to MacRPA1 did not drastically affect the ssDNA-binding properties of this chimeric RPA. In contrast, the binding of labeled ssDNA by the three-OB-fold-containing MacRPA1 derivatives (MacRPA1 chimera-1, MacRPA1 chimera-2, and MacRPA1 chimera-3) was each outcompeted in the presence of 50-fold excess dsDNA in the reaction mixture (Fig. 2B, panels ii to iv, lanes 8). This suggested that the creation or conversion of the protein to three-OB-fold RPA might lead to a lesser capacity to discriminate ssDNA from dsDNA. The decreased capacity to discriminate ssDNA from dsDNA may also be due to the presence of the chimeric OB fold, since the original OB folds might have evolved to precisely confer this property to MacRPA1.
FIG. 2.

(A) ssDNA-binding activity of wild-type MacRPA1 and its chimeras. A fixed amount (1 pmol) of 32P-labeled ssDNA (lane 1) was incubated with increasing amounts (1, 2, 3, 4, and 5 pmol [indicated by the height of the triangle above the gel) of wild-type MacRPA1 or each of its chimeras (lanes 2 to 6). The MacRPA1 chimera under investigation is indicated above each panel as follows: wild-type MacRPA1 (i), MacRPA1 chimera-1 (ii), MacRPA1 chimera-2 (MbuRPA1-like) (iii), MacRPA1 chimera-3 (iv), MjaRPA-like protein (v), and MthRPA-like protein (vi). The positions of free DNA and protein-DNA complex (protein-bound ssDNA) are shown by the black arrows labeled I and II to the left of the gels, respectively. (B) The ssDNA-binding and dsDNA-binding activities of wild-type MacRPA1 and its chimeras. A fixed amount (1 pmol) of 32P-labeled ssDNA (lane 1) was incubated with 5 pmol each of MacRPA1 or its derivative (lane 2) and challenged with 1 pmol of unlabeled ssDNA (lane 3), 10 pmol of unlabeled ssDNA (lane 4), 50 pmol of unlabeled ssDNA (lane 5), 1 pmol of unlabeled dsDNA (lane 6), 10 pmol of unlabeled dsDNA (lane 7), and 50 pmol of unlabeled dsDNA (lane 8). The MacRPA1 chimera under investigation is indicated under each panel as follows: wild-type MacRPA1(i), MacRPA1 chimera-1 (ii), MacRPA1 chimera-2 (MbuRPA1-like protein) (iii), MacRPA1 chimera-3 (iv), MjaRPA-like protein (v), and MthRPA-like protein (vi). Free DNA and protein-DNA complex were resolved by 8% polyacrylamide gel electrophoresis followed by visualization using autoradiography. The positions of free DNA and protein-DNA complex (protein-bound ssDNA) are shown by the black arrows labeled I and II to the left of the gels, respectively.
All chimeric RPAs have similar ssDNA-binding site sizes.
To further determine any differences resulting from the engineered proteins, we determined the binding site sizes of wild-type MacRPA1 and its chimeras. Differences in spectra in the presence and absence of φ174 phage ssDNA allowed us to monitor DNA binding by all RPAs by monitoring the change of the intrinsic fluorescence of the tryptophan in the protein. We carried out the fluorescence quenching experiments under stoichiometric binding conditions and determined the binding site size for each construct as described elsewhere (7, 11). When a fixed amount of RPAs (1 μM monomers) was titrated with φ174 phage ssDNA (Fig. 3), the quenching of RPA fluorescence increased linearly with ssDNA concentration until it saturated at a maximum value. The intercept between two phases corresponds to a binding site size, or the amount of DNA occluded by a monomer of the RPA under investigation. The estimated binding site sizes of wild-type MacRPA1, MacRPA1 chimera-1, MacRPA1 chimera-2, MacRPA1 chimera-3, MjaRPA-like, and MthRPA-like proteins were 11.1 ± 0.7, 9.0 ± 0.6, 11.4 ± 0.9, 9.1 ± 1.2, 11.4 ± 0.6, and 9.2 ± 1.0 nucleotides, respectively (Fig. 3). When twice the concentration (2 μM monomers) of each RPA was titrated with φ174 phage ssDNA, the intercept point was also shifted twofold (∼20 nucleotides per 2 μM RPA), confirming stoichiometric binding conditions (data not shown). These results showed that the binding site sizes for all RPA constructs were the same within experimental error, indicating that partial deletions and fusions of two OB folds did not affect the ssDNA-binding site size of RPAs. The dimer form of wild-type MacRPA1, MjaRPA-like, and MthRPA-like proteins would have a similar binding site size of ∼20 nucleotides, as reported earlier for MjaRPA wild-type (21 nucleotides) (7) and wild-type MacRPA1 (19). The tetramer forms of the chimeric RPAs (MacRPA1 chimera-1, MacRPA1 chimera-2, and MacRPA1 chimera-3) occlude ∼40 nucleotides. Depending on the structural arrangements of these proteins, however, the tetramer may have two independent binding sites, one being similar to that of the dimeric forms.
FIG. 3.

Fluorescence quenching of wild-type MacRPA1 and its chimeras. Reaction mixtures contain a fixed amount of wild-type (WT) MacRPA1 or its chimeras (1 μM). Increasing amounts of φ174 phage ssDNA were then added, and the fluorescence at 340 nm was measured at 20°C with an excitation wavelength of 280 nm. The percentage of free RPA was determined by the changes in fluorescence with MacRPA1 and its chimeras. The binding site sizes for wild-type MacRPA1, MacRPA1 chimera-1, MacRPA1 chimera-2 (MbuRPA1-like protein), MacRPA1 chimera-3, MjaRPA-like protein, and MthRPA-like protein were calculated to be 11.1 ± 0.8, 9.0 ± 0.6, 11.4 ± 0.9, 9.1 ± 1.2, 11.4 ± 0.6, and 9.2 ± 1.0 nucleotides (nt), respectively, per monomer.
Reduction in the number of OB folds did not impair RPA affinity for ssDNA.
We used fluorescence polarization anisotropy to study how the deletions and fusions of the OB fold affect ssDNA-binding affinity of the chimeric RPAs. Since all investigated proteins occluded approximately 10 nucleotides of ssDNA per monomer and all existed as either dimers (with expected binding site size of 20 nucleotides) or tetramers (with presumably two independent ssDNA-binding sites), we analyzed binding of these proteins to a fluorescently labeled 22-nucleotide ssDNA substrate, FL-22 (Table 1). Using this substrate allowed us to fit the binding isotherms to a simple equation that assumes 1:1 binding stoichiometry. To confirm that the latter is true, we first carried out the FPA experiments at the stoichiometric binding conditions. As more protein was added to the reaction mixture, the relative anisotropy increased until the DNA was completely saturated with protein. With no salt, all the chimeric proteins showed stoichiometric binding activity and strong binding affinity (data not shown). The binding curves confirmed that each DNA substrate could accommodate two monomers of each protein under this condition. To compare relative affinities of the chimeric proteins for ssDNA, we carried out the FPA titrations in the presence of 1 M NaCl (Fig. 4), since the proteins bound too tightly in the absence of salt. Therefore, the equilibrium dissociation constants were determined by fitting the experimental data obtained under nonstoichiometric conditions (high salt, 2 μM DNA) to the binding equation. Under this condition, wild- type MacRPA1, MacRPA1 chimera-1, MacRPA1 chimera-2, MacRPA1 chimera-3, MjaRPA-like, and MthRPA-like proteins displayed Kd values of 31.4, 42.7, 44.9, 82.7, 37.6, and 51.2 nM, respectively (Table 2). Thus, under these conditions, MacRPA1 chimera-3 bound to ssDNA less tightly than the wild-type MacRPA1 and the other two three-OB-fold RPAs (MacRPA1 chimera-1 and MacRPA1 chimera-2) did. Therefore, the creation of the chimeric OB folds in MacRPA1 and the introduction of a zinc finger module at the C terminus, as well as an increase in the number of OB folds beyond four in the polypeptide, had only a marginal effect on the ssDNA-binding affinity. This lack of difference in ssDNA-binding affinities of the wild-type protein and its chimeras was, however, less evident when we used the EMSA (Fig. 2A).
FIG. 4.

Anisotropy measurements reveal binding of ssDNA by wild-type MacRPA1 and its chimeras. The changes in anisotropy of a 22-base-long oligonucleotide (FL-22) were recorded as increasing amounts of wild-type (WT) MacRPA1 or its chimeras were added to the reaction mixture. The FPA curve of each RPA was then fitted to a binding model to calculate the dissociation constants (Table 2).
TABLE 2.
DNA-binding properties and oligomerization state of wild-type MacRPA1 and its chimerasa
| Protein | Kd (nM) (at 1 M NaCl)b | Oligomerization state | Binding site size (nt per monomer)c |
|---|---|---|---|
| Wild-type MacRPA1 | 31.5 ± 5.2 | Dimer or tetramer | 11.1 ± 0.8 |
| MacRPA1 chimera-1 | 42.7 ± 4.9 | Tetramer | 9.0 ± 0.6 |
| MacRPA1 chimera-2 | 44.9 ± 11.1 | Dimer or tetramer | 11.4 ± 0.9 |
| MacRPA1 chimera-3 | 82.7 ± 24.2 | Tetramer | 9.1 ± 1.2 |
| MjaRPA-like protein | 37.6 ± 8.1 | Dimer | 11.4 ± 0.6 |
| MthRPA-like protein | 51.2 ± 15.3 | Dimer | 9.2 ± 1.0 |
Wild-type MacRPA1 and its chimeras were titrated against a 22-base-long ssDNA (FL-22) to measure fluorescence polarization anisotropy (for details, see Materials and Methods). The subunit organizations were estimated by size exclusion chromatography. The concentration of each RPA protein for FPA experiments is based on monomeric proteins.
The Kd values are averages ± standard deviations for three measurements.
The binding site size (in nucleotides [nt] per monomer) was determined by a fluorescence quenching method (for details, see Materials and Methods). The values are averages ± standard deviations for three measurements.
Effects of wild-type MacRPA1 and its chimeras on cleavage of flap DNA by MacFEN1.
In a previous report (13), we demonstrated that MacRPA1 can inhibit the cleavage of a DNA flap by its cognate flap endonuclease 1 from M. acetivorans (MacFEN1). On the other hand, two other RPAs in this archaeon (MacRPA2 and MacRPA3, both composed of two OB folds and a zinc finger domain) failed to exhibit this inhibition. In order to determine whether the chimeric RPAs derived from MacRPA1 still have inhibitory activity on flap cleavage by MacFEN1, we tested each chimeric protein for the ability to inhibit cleavage of flap DNA by MacFEN1. Similar to our earlier report (13), wild-type MacRPA1 at a concentration of 2.5 pmol/reaction mixture was able to completely inhibit the cleavage of the DNA flap structure by MacFEN1 (Fig. 5A, lane 4). We also observed that all the chimeric RPAs were able to inhibit the endonuclease activity of MacFEN1 but to different degrees (Fig. 5B to F). The MjaRPA-like protein appeared to have the strongest inhibitory effect on MacFEN1 endonuclease activity (Fig. 5E). A concentration of 1.0 pmol/reaction mixture of this chimeric protein completely inhibited MacFEN1 endonuclease activity. Similar to the wild-type MacRPA1 protein, the MthRPA-like protein also had a strong inhibitory effect on MacFEN1 endonuclease activity (Fig. 5F). In contrast, the chimeric RPAs (MacRPA1 chimera-1, MacRPA1 chimera-2, and MacRPA1 chimera-3) with only three OB folds showed weaker inhibitory effects on MacFEN1 endonuclease activity (Fig. 5B to D). MacRPA1 chimera-1 showed complete inhibition of MacFEN1 activity at about 5 pmol/reaction mixture (Fig. 5B). At 10 pmol/reaction mixture, there were about 12.9% and 22.1% of cleaved product in the reaction with MacRPA1 chimera-2 and MacRPA1 chimera-3, respectively (Fig. 5C and D, lane 7). This demonstrated that although the artificially synthesized RPAs with three OB folds still maintain the inhibitory property of the parental protein, their effects were quite reduced. It is anticipated that future research that unravels the molecular basis of MacRPA1 inhibition of MacFEN1 will aid in explaining the behavior of the three-OB-fold-containing RPA chimeras.
FIG. 5.

Effects of wild-type MacRPA1 and its chimeras on flap endonuclease activity by MacFEN1. A fixed amount (1 pmol) of 32P-labeled flap structure DNA (lane 1) was incubated in the presence (+) of MacFEN1 alone (lane 2), and in the presence (+) of MacFEN1 and increasing amounts (1.0, 2.5, 5.0, 7.5, and 10.0 pmol) of either wild-type (WT) MacRPA1 or its artificially synthesized derivative under investigation (lanes 3 to 7). The products were resolved by 15% denaturing polyacrylamide gel electrophoresis, followed by visualization using autoradiography. The results are presented as wild-type MacRPA1 (A), MacRPA1 chimera-1 (B), MacRPA1 chimera-2 (C), MacRPA1 chimera-3 (D), MjaRPA-like (E), and MthRPA-like (F) proteins. The positions of cleaved product (DNA flap) and uncut DNA are shown by the black arrows labeled I and II to the left of the gels, respectively.
Effects of wild-type MacRPA1, wild-type MthRPA, and their chimeras on DNA synthesis by MacPolBI and MthPolBI complex.
The mesophilic RPAs from M. acetivorans have been shown to stimulate DNA synthesis by a cognate DNA polymerase BI (19). However, in the thermophilic archaeon M. thermautotrophicus, the RPA homolog was shown to inhibit DNA synthesis by its cognate DNA polymerase (MthPolBI complex), which is an ortholog of MacPolBI (9). Each chimeric RPA and wild-type MacRPA1 (positive control) was tested for the ability to stimulate or inhibit DNA synthesis activity of the mesophilic DNA polymerase (MacPolBI). Similar to our previous report (19), MacPolBI by itself was able to synthesize a product that is approximately 500 nucleotides long (Fig. 6A and B, lanes 2). Increasing amounts (15 and 30 pmol) of wild-type MacRPA1, MacRPA1 chimera-1, MacRPA1 chimera-2 (MbuRPA1-like protein), and MacRPA1 chimera-3 added to the reaction mixture stimulated primer extension by MacPolBI, and in some cases (MacRPA1 chimera-2 and chimera-3), completely replicated products could be observed as with the wild-type MacRPA1 (Fig. 6A, lanes 3 to 10). However, the artificially synthesized RPAs that mimic the thermophilic RPA (MjaRPA-like and MthRPA-like proteins) showed either no stimulation or very weak capacity to stimulate primer extension by MacPolBI (Fig. 6B, lanes 5 to 8) compared with the wild-type protein (Fig. 6B, lanes 3 and 4). No DNA synthesis inhibition was detected with either the MjaRPA-like or MthRPA-like proteins (Fig. 6B, lanes 5 to 8). In addition, we also tested the effects of wild-type MthRPA, MacRPA1, and MthRPA-like protein on DNA synthesis activity by the thermostable DNA polymerase MthPolBI complex at 65°C and 37°C. Note that the MthRPA-like protein is stable at 65°C (result not shown). The results showed that the MthPolBI complex alone was able to synthesize a product approximately 200 nucleotides long (Fig. 6C and D, lanes 2) at either 65°C or 37°C incubation. With 65°C incubation, increasing amounts (15, 30, and 60 pmol) of wild-type MthRPA added to the reaction mixture inhibited primer extension by the MthPolBI complex (Fig. 6C, lanes 3 to 5) as reported elsewhere (8). Increasing amounts (15, 30, and 60 pmol) of MthRPA-like protein in the reaction mixture stimulated primer extension by the MthPolBI complex (Fig. 6C, lanes 6 to 8). However, the stimulation did not yield full-length products (∼7.2 kb) under this condition. With 37°C incubation, increasing amounts (15, 30, and 60 pmol) of wild-type MthRPA also inhibited primer extension by the MthPolBI complex (Fig. 6D, lanes 3 to 5). Interestingly, unlike at 65°C, the MthRPA-like protein did not impact DNA synthesis by the MthPolBI complex at 37°C.
FIG. 6.

Effects of wild-type MacRPA1, its chimeras, and wild-type MthRPA on the primer extension capacities of two DNA polymerases (MacPolBI and MthPolBI complex). (A and B) Primer extension by MacPolBI was compared in the presence of wild-type MacRPA1 (15 and 30 pmol), MacRPA1 chimera-1 (15 and 30 pmol), MacRPA1 chimera-2 (MbuRPA1-like protein) (15 and 30 pmol), and MacRPA1 chimera-3 (15 and 30 pmol) (A) and wild-type MacRPA1 (15 and 30 pmol), MjaRPA-like protein (15 and 30 pmol), and MthRPA-like protein (15 and 30 pmol) (B) at 37°C. (C and D) Primer extension by MthPolBI complex was compared in the presence of wild-type MthRPA (15, 30, and 60 pmol) and MthRPA-like protein (15, 30, and 60 pmol) at 65°C (C) and wild-type MthRPA (15, 30, and 60 pmol) and wild-type MacRPA1 (15, 30, and 60 pmol) at 37°C (D). The products were resolved by 1% alkaline agarose gel electrophoresis followed by visualization using autoradiography.
DISCUSSION
The third domain of life, Archaea, displays an unusual complexity and diversity of organization of ssDNA-binding proteins (18). Not only do individual organisms contain single SSB or RPAs as in bacteria and eukaryotes, but certain organisms also have multiple RPAs in their cells, presumably functioning in different pathways or cellular processes (7, 9, 12, 18, 19). This fascinating observation prompted us to explore how this diversity of RPAs might have evolved.
In an earlier report, we demonstrated that domain shuffling could lead to functional RPAs (18). Furthermore, we reported that the two central OB folds of the four-OB-fold-containing M. acetivorans RPA1 were very similar at both the amino acid sequence (19) and nucleotide sequence levels (similarity of 64%). Surprisingly, while analyzing the genome of M. burtonii, a relative of the Methanosarcina spp., we found that there is a deletion that removed parts of the two central OB folds to create a single OB fold or a chimeric OB fold of the two original ones. Thus, a RPA that was most likely originally made up of four OB folds is currently a three-OB-fold protein in M. burtonii. We reasoned that this creation of a chimeric OB fold was likely one of the means by which nature has generated the diversity of OB folds found in different proteins (22). We hypothesized that the chimeric OB fold in M. burtonii evolved through intramolecular homologous recombination as illustrated in Fig. 7A. Similarly, we hypothesized that intermolecular homologous recombination, especially in organisms with multiple RPA homologs that exhibit high nucleotide sequence similarity, could also occur to generate new forms of RPAs with increased numbers of OB folds (Fig. 7B). To test whether the formation of chimeric OB folds would lead to functional RPAs, we constructed three different chimeric OB folds in the four-OB-fold-containing MacRPA1. Each of these proteins represented a possible intramolecular homologous recombination event, with that of MacRPA1 chimera-2 likely to have occurred in nature as seen in M. burtonii RPA1 (Fig. 1A and B, MbuRPA1). We also fused OB folds and zinc finger modules to MacRPA1 to create two polypeptides that mimic two naturally occurring RPAs in the two archaea M. jannaschii and M. thermautotrophicus. The two fusions illustrated intermolecular homologous recombination that could occur, for example, in an organism such as M. acetivorans (21), which has a four-OB-fold RPA (MacRPA1) and two RPAs with two OB folds/one zinc finger module (MacRPA2 and MacRPA3) (19).
FIG. 7.

Proposed evolutionary model showing deletions and fusions in modules occurring in genes coding for RPAs in archaea. (A) Model showing an OB fold deletion event from a four-OB-fold RPA (MacRPA1) to a three-OB-fold RPA (MbuRPA1) protein through intramolecular homologous recombination process. (B) Model showing a fusion event between a multiple-OB-fold RPA (MacRPA1) and an RPA containing a zinc finger module and multiple OB folds (MjaRPA or MthRPA) through homologous recombination process. The OB folds are shown by differently shaded boxes. The small black boxes labeled Zn represent zinc-binding modules. The black lines indicate genomic DNA. The motifs are not drawn to scale. (C) The mechanism of domain reshuffling may involve recombinational repair of double-strand damage occurring during DNA replication.
The subunit organization analysis suggested that MacRPA1 chimera-1, chimera-2, and chimera-3 mostly oligomerize in solution to form homotetramers, although a homodimer was also seen from the MbuRPA1-like protein. Thus, it is fascinating that the mimic of the naturally occurring protein exhibited the two states also seen in the wild-type MacRPA1 (Table 2) (19). In contrast, the MjaRPA-like and MthRPA-like proteins existed as homodimers only in solution, suggesting that the attachment of the zinc finger module to these proteins can change their subunit organization in solution. Note, however, that the recombinant form of the naturally occurring MjaRPA was reported as existing as a monomer (7).
Our EMSA results suggested that the RPAs with reduced numbers of OB folds have a lower capacity to discriminate ssDNA from dsDNA (Fig. 2B). On the other hand, the chimeras that were constructed by fusing the zinc finger modules to MacRPA1 resulted in proteins that were also very competent in discriminating ssDNA from dsDNA. Thus, the results were in agreement with our previous report that suggested that mutants harboring deletions of OB folds in MacRPA1 are less able to bind specifically to ssDNA (13).
Further biochemical analysis showed different effects of each chimeric RPA on ssDNA flap processing. All of the RPAs with the chimeric OB fold (derivatives containing three OB folds) showed weaker inhibition of flap cleavage activity by MacFEN1 than by wild-type MacRPA1. In contrast, increasing the number of OB folds or attaching a C-terminal zinc finger module resulted in strong inhibition of flap cleavage by MacFEN1. Each of the one-OB-fold deletion chimeras maintained stimulation of DNA synthesis activity by MacPolBI. Interestingly, although no complete inhibition was observed, the MjaRPA-like and MthRPA-like proteins showed weak to no stimulation of DNA synthesis activity by MacPolBI (Fig. 6B). Previous results showed that the MthRPA, which the MthRPA-like protein in the present experiment mimics, inhibited DNA synthesis by its cognate DNA polymerase (MthPolBI) (9). This finding was confirmed in the present experiment as shown in Fig. 6C and D. In contrast, neither the MthRPA-like protein nor the wild-type MacRPA1 inhibited DNA synthesis by MthPolBI, suggesting that the RPA inhibition of DNA synthesis by MthPolBI is specific to its cognate RPA.
In the present report, we artificially synthesized RPAs based on a natural occurrence in the cells of M. burtonii, a psychrophilic member of the Methanosarcinales. Interestingly, we found that an ortholog of this protein is also present in Methanosaeta thermophila, a thermophilic member of this group of archaea. The results from our functional studies on these engineered RPAs suggest that the deletions (MacRPA1 chimera-1, chimera-2, and chimera-3) and insertions (MjaRPA-like and MthRPA-like proteins) hypothesized to have taken place to generate some of the diversity seen in extant archaeal RPAs would have likely led to proteins that were endowed with or maintained the properties of RPAs. This would have been essential for the organisms harboring these newly generated proteins to survive in order to transfer the gene to subsequent generations.
Extant RPAs in archaea were hypothesized to have originated from an ancestral protein that was composed of only a single OB fold (3), and in fact although yet to be biochemically characterized, genes encoding such a protein remain in the genomes of several archaea and eukaryotes (18). The ancestral OB fold is presumed to have undergone several gene duplications or fission and recombination events to arrive at its current complexity (3, 18). Our data, based on the MbuRPA1-like, MthRPA-like, and MjaRPA-like proteins, suggest how homologous recombination events leading to chimeric or duplicated OB folds might have contributed to the diversity of RPAs in archaea. M. burtonii is a psychrophilic organism, and it is assumed that after the three-OB-fold RPA was invented and acquired by this organism, the protein went through other processes to adapt to a cold environment. Likewise, M. thermophila might have evolved a thermophilic version of the three-OB-fold RPA (Fig. 1A). This makes one wonder if this excision or the recombination event leading to the formation of the three-OB-fold protein facilitated its further evolution for adaptation to other environments. A search of the genomes of several haloarchaea suggests that Halobacterium sp. strain NRC-1, Haloarcula marismortui, and Natronomonas pharaonis also possess the three-OB-fold RPA with similar fusion of OB folds B and C (based on MacRPA1 architecture). However, the haloarchaeal protein seems to have acquired more genetic information through a fusion at the extreme C-terminal region of the three original OB folds (Fig. 1A). The acquired amino acid sequence could be a noncanonical OB fold, which will lead to a four-OB-fold RPA as in the Methanosarcinales. In contrast, this could also be a modification that was essential for adaptation to high-salt environments.
Multi-OB-fold RPAs provide a unique case to study domain deletion and fusion. A high nucleic acid sequence homology between individual OB folds will make the sequences of the genes encoding RPAs prone to expansion and deletion. The mechanism for domain reshuffling likely involves recombinational repair of a two-strand damage that may occur during replication (Fig. 7C) or restoration of a collapsed replication fork. Consecutive OB folds can be looked upon as direct repeats. Therefore, if a sequence corresponding to a highly homologous fold (either preceding or following the cognate fold) is used as the template for the homology-directed DNA repair, the repaired daughter chromosome would contain a different number of repeats than the parental DNA molecule. This mechanism may explain the deletion and introduction of complete OB folds and fusion of two OB folds leading to a chimeric OB fold.
Supplementary Material
Acknowledgments
We thank William W. Metcalf (University of Illinois at Urbana-Champaign) for providing M. acetivorans genomic DNA and Zvi Kelman (University of Maryland at College Park) for providing M. thermautotrophicus RPA and PolBI genes. We also thank Roderick I. Mackie, Satish K. Nair, H. Rex Gaskins, and Bryan A. White (University of Illinois at Urbana-Champaign) for insightful scientific discussions.
This research was supported by a National Science Foundation grant, MCB-0238451, to I.K.O.C. Also, Y.L., L.-J.L., and P.S. were supported by National Science Foundation grant MCB-0238451.
Footnotes
Published ahead of print on 27 June 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alexandrescu, A. T., V. A. Jaravine, S. A. Dames, and F. P. Lamour. 1999. NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J. Mol. Biol. 2891041-1054. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, D. A., J. M. Eggington, M. P. Killoran, A. M. Misic, M. M. Cox, and J. L. Keck. 2004. Crystal structure of the Deinococcus radiodurans single-stranded DNA-binding protein suggests a mechanism for coping with DNA damage. Proc. Natl. Acad. Sci. USA 1018575-8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chedin, F., E. M. Seitz, and S. C. Kowalczykowski. 1998. Novel homologs of replication protein A in archaea: implications for the evolution of ssDNA-binding proteins. Trends Biochem. Sci. 23273-277. [DOI] [PubMed] [Google Scholar]
- 4.Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1610881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerstein, M., and M. Levitt. 1997. A structural census of the current population of protein sequences. Proc. Natl. Acad. Sci. USA 9411911-11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haseltine, C. A., and S. C. Kowalczykowski. 2002. A distinctive single-strand DNA-binding protein from the archaeon Sulfolobus solfataricus. Mol. Microbiol. 431505-1515. [DOI] [PubMed] [Google Scholar]
- 7.Kelly, T. J., P. Simancek, and G. S. Brush. 1998. Identification and characterization of a single-stranded DNA-binding protein from the archaeon Methanococcus jannaschii. Proc. Natl. Acad. Sci. USA 9514634-14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelman, Z., and J. Hurwitz. 2000. A unique organization of the protein subunits of the DNA polymerase clamp loader in the archaeon Methanobacterium thermoautotrophicum deltaH. J. Biol. Chem. 2757327-7336. [DOI] [PubMed] [Google Scholar]
- 9.Kelman, Z., S. Pietrokovski, and J. Hurwitz. 1999. Isolation and characterization of a split B-type DNA polymerase from the archaeon Methanobacterium thermoautotrophicum deltaH. J. Biol. Chem. 27428751-28761. [DOI] [PubMed] [Google Scholar]
- 10.Kerr, I. D., R. I. Wadsworth, L. Cubeddu, W. Blankenfeldt, J. H. Naismith, and M. F. White. 2003. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J. 222561-2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim, C., B. F. Paulus, and M. S. Wold. 1994. Interactions of human replication protein A with oligonucleotides. Biochemistry 3314197-14206. [DOI] [PubMed] [Google Scholar]
- 12.Komori, K., and Y. Ishino. 2001. Replication protein A in Pyrococcus furiosus is involved in homologous DNA recombination. J. Biol. Chem. 27625654-25660. [DOI] [PubMed] [Google Scholar]
- 13.Lin, Y., C. E. Guzman, M. C. McKinney, S. K. Nair, T. Ha, and I. K. O. Cann. 2006. Methanosarcina acetivorans flap endonuclease 1 activity is inhibited by a cognate single-stranded-DNA-binding protein. J. Bacteriol. 1886153-6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lohman, T. M., and M. E. Ferrari. 1994. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 63527-570. [DOI] [PubMed] [Google Scholar]
- 15.Long, M., E. Betran, K. Thornton, and W. Wang. 2003. The origin of new genes: glimpses from the young and old. Nat. Rev. Genet. 4865-875. [DOI] [PubMed] [Google Scholar]
- 16.Murzin, A. G. 1993. OB (oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 12861-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patthy, L. 1999. Protein evolution. Blackwell Science, Oxford, United Kingdom.
- 18.Robbins, J. B., M. C. McKinney, C. E. Guzman, B. Sriratana, S. Fitz-Gibbon, T. Ha, and I. K. O. Cann. 2005. The Euryarchaeota: nature's medium for engineering of single-stranded DNA binding proteins. J. Biol. Chem. 28015325-15339. [DOI] [PubMed] [Google Scholar]
- 19.Robbins, J. B., M. C. Murphy, B. A. White, R. I. Mackie, T. Ha, and I. K. O. Cann. 2004. Functional analysis of multiple single-stranded DNA-binding proteins from Methanosarcina acetivorans and their effects on DNA synthesis by DNA polymerase BI. J. Biol. Chem. 2796315-6326. [DOI] [PubMed] [Google Scholar]
- 20.Schindelin, H., M. A. Marahiel, and U. Heinemann. 1993. Universal nucleic acid-binding domain revealed by crystal structure of the B. subtilis major cold-shock protein. Nature 364164-168. [DOI] [PubMed] [Google Scholar]
- 21.Sowers, K. R., S. F. Baron, and J. G. Ferry. 1984. Methanosarcina acetivorans sp. nov., an acetotrophic methane-producing bacterium isolated from marine sediments. Appl. Environ. Microbiol. 47971-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theobald, D. L., R. M. Mitton-Fry, and D. S. Wuttke. 2003. Nucleic acid recognition OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 32115-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wadsworth, R. I., and M. F. White. 2001. Identification and properties of the crenarchaeal single-stranded DNA binding protein from Sulfolobus solfataricus. Nucleic Acids Res. 29914-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiner, J., F. Beaussart, and E. Bornberg-Bauer. 2006. Domain deletions and substitutions in the modular protein evolution. FEBS J. 2732037-2047. [DOI] [PubMed] [Google Scholar]
- 25.Wold, M. S. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 6661-92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.