

Abstract
Understanding immunodominance, the phenomenon of epitope-specific T cells expanding in an often distinctly hierarchical fashion, is important for the design of T-cell-based intervention strategies. Several recent studies have investigated immunodominance of H-2Db-restricted CD8+ T cells specific for the nucleoprotein NP366 and acid polymerase PA224 epitopes during influenza A virus infection of C57BL/6 mice. CD8+ T cells specific for these two epitopes are codominant during primary infection; NP366 dominates during secondary infection. While a number of explanations for this observation have been proposed, none of them can fully account for all the observed data. In this article, we use a simple mathematical model to explain the seemingly inconsistent data. We show that the dynamic interactions between CD8+ T cells and antigen presentation lead to a situation where CD8+ T cells are limiting during the initial response whereas antigen is limiting in the secondary response. This “numbers game” between antigen and CD8+ T cells can reproduce the observed immunodominance of the NP336- and PA224-specific CD8+ T cells, thereby explaining the reported experimental data.
Successful clearance of viral infections often relies on CD8+ T cells (11, 23, 24). CD8+ T cells (hereinafter also called cytotoxic T lymphocytes [CTL]) can recognize viral epitopes that are presented by other cells on major histocompatibility complex (MHC) class I (27, 29, 32). In many infections, CTL specific for only a few viral epitopes become activated. The activated CTL expand to reach different peak numbers. This hierarchy in numbers of epitope-specific CTL is called immunodominance (20, 26). Since immunodominance is an important aspect of the dynamics of CTL responses, it is necessary to properly understand it, not only to gain fundamental insights into the functioning of immune responses but also for the design of T-cell-based vaccines and cancer treatments (6, 12, 15, 22).
Despite all we do know about immunodominance, much remains to be understood (13, 25, 26). Because the processes involved in immunodominance are so complex, studies combining experiments with mathematical models might be useful (28), and a few mathematical studies have shed light on some aspects of immunodominance (18, 19). Nevertheless, we are still far from a full understanding. Indeed, it is likely that there is no universal mechanism causing immunodominance, but instead, in different pathogen-host systems, different mechanisms are responsible for the observed immunodominance hierarchies (25).
One pathogen-host system that has been used in a number of recent studies on immunodominance is influenza A virus infections in C57BL/6 mice. During a primary influenza virus infection, H-2Db-restricted CTL specific for the nucleoprotein NP366 and acid polymerase PA224 epitopes are codominant: the CTL responses to these two epitopes are of similar magnitudes. Following rechallenge with a heterologous influenza virus strain, the NP366 response is several times larger than the PA224 response (3, 8). Different explanations for this finding have been proposed, which we describe briefly below.
Different numbers of NP366- and PA224-specific memory CTL (Fig. 1A).
FIG. 1.

Possible explanations for observed NP366 (NP) and PA224 (PA) responses. (A) Differences in secondary CTL numbers due to differences in memory cells. (B) Availability of higher levels of NP366 to memory CTL during secondary response leads to higher levels of NP366-specific CTL. (C) Different levels of NP366 and PA224 antigen presentation in both primary and secondary responses. Equal CTL peaks in primary response due to fewer naive NP366 CD8 T cells. (D) Different levels of NP366 and PA224 antigen presentation in both primary and secondary responses. Equal numbers of NP366 and PA224 naive and memory CTL. Differences in peak sizes due to nonlinear dynamical interactions between limiting CTL in the primary and limiting antigen in the secondary responses. For a more-detailed explanation, see the text. AgP, antigen presentation).
The simplest explanation for a larger NP366-specific CTL response during secondary infections is to assume that after primary infection, NP366 CTL contract less, leading to a higher number of NP366 memory CTL. However, measurement of the memory cells found no difference between the two epitope-specific CTL (8).
Differences in antigen presentation between primary and secondary responses (Fig. 1B).
A study by Crowe et al. suggested that only dendritic cells (DC) are efficient presenters of PA224 while NP366 was expressed by a broader range of cells (8). Under the assumption that naive CTL need activation by DC while memory CTL can also be activated by other antigen-presenting cells, a higher level of NP366 presentation available for CTL activation during the secondary response could explain the preferential expansion of NP366 over PA224 CTL. However, other studies have since suggested that memory cells also need DC to be activated (31) and might in fact have more-stringent activation requirements (2). Additionally, it was shown that non-DC can also process and present PA224, albeit with lower efficiency (7).
Unequal antigen presentation and CTL numbers between NP366 and PA224 (Fig. 1C).
Using a technique that allowed disabling of the NP366 and PA224 epitopes in their native location and their reexpression in the neuraminidase stalk of the virus, La Gruta et al. showed that DC present NP366 at higher levels than those of PA224 and that changing peptide-MHC (pMHC) levels on DC affected the CTL immunodominance hierarchy (16). Based on these findings, the authors proposed a model whereby high NP366 antigen presentation by DC, combined with low numbers of NP366-specific naive CTL, results in a primary response equal to the PA244 response, with lower epitope-specific antigen presentation but more PA224-specific naive CTL. In the secondary response, equal numbers of NP366 and PA224 memory cells, combined with higher NP366 presentation, lead to the observed dominance of the NP366 CTL. However, a recent study found no appreciable differences in the numbers of NP366- and PA224-specific naive CTL (14).
Dynamic interactions between CTL and antigen (Fig. 1D).
None of the just-described verbal models can fully explain the experimental data. In this article, we show that even if the numbers of naive and memory cells do not differ between NP366- and PA224-specific CTL and additionally NP366 antigen is expressed at higher levels than PA224 during both the primary and secondary responses, we can recover the observed codominance of CTL during the primary response and dominance of NP366 CTL during the secondary response, thus reconciling current experimental studies (7, 8, 14, 16). This perhaps surprising result comes about due to dynamic interactions between T-cell numbers and levels of antigen presentation. In the initial response, the CTL are limiting. All epitope-specific CTL become activated, and the equal levels of naive NP366 and NP224 cells lead to equal peaks. However, during the secondary response, antigen presentation is limiting, and the higher level of NP366 presentation leads to a dominant NP366 response.
MATERIALS AND METHODS
To understand the observed immunodominance hierarchies of NP366- and PA224-specific CTL during primary and secondary influenza virus infections, we use a simple mathematical model that describes the dynamics of virus, antigen presentation, and CTL activation and expansion. This model is similar to one used previously (18). The model (Fig. 2) considers virus load (V), epitope-specific pMHC complexes on DC (Pi), unactivated epitope-specific CTL (Ti), and activated epitope-specific CTL ( ). The index i is a label for NP366 (i = 1) or PA224 (i = 2) specificity.
). The index i is a label for NP366 (i = 1) or PA224 (i = 2) specificity.
FIG. 2.

Schematic of the model. Virus grows initially at rate r and is later reduced through killing of infected cells by activated CTL. Epitope-specific pMHC on activated DC, Pi, are created in proportion to the virus load at rates f1 and f2 for the NP366 and PA224 epitopes, respectively. Unactivated CTL, Ti, are activated by their cognate epitope at rates ai. Activated CD8 T cells, 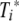 , proliferate at rates gi and reduce virus at rates ki. Equations and more details are given in the text.
, proliferate at rates gi and reduce virus at rates ki. Equations and more details are given in the text.
In line with experimental data, we assume that virus initially grows exponentially at rate r. It is then reduced by the activated CTL at rate ki. This happens through killing of infected cells, which we do not model explicitly. Presentation of pMHC on activated DC increases proportionally to the amount of virus at rate fi and decays at a fixed rate, di. The pMHC on DC can then activate epitope-specific CTL, Ti, according to a mass-action term with rate ai. The activated CTL, 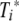 , proliferate by clonal expansion at rate gi. To keep the model simple, we study only the dynamics during the expansion phase of the CTL, up to the peak of the response, which occurs around days 8 to 10 for both primary and secondary infections (3, 4). It would be possible to include the CTL contraction phase (9), but the model would become more complicated, and for this study, we are interested only in the CTL dynamics up to the peak.
, proliferate by clonal expansion at rate gi. To keep the model simple, we study only the dynamics during the expansion phase of the CTL, up to the peak of the response, which occurs around days 8 to 10 for both primary and secondary infections (3, 4). It would be possible to include the CTL contraction phase (9), but the model would become more complicated, and for this study, we are interested only in the CTL dynamics up to the peak.
There are two ways in which activated CTL can reduce antigen presentation on DC. The first mechanism, which we just described, is through killing of infected cells, which reduces virus load and thereby antigen uptake and presentation on DC. An alternative mechanism involves direct reduction of antigen presentation through killing or otherwise disabling of DC by CTL. Some evidence suggests that antigen presentation on DC during influenza virus infections can be regulated directly by CTL in a perforin-dependent manner, at least during secondary infections (5). However, it is not clear to what extent this also occurs during primary responses. Additionally, one study did not find any indication that knockout of either the pfp or gld gene (both of which are thought to be involved in killing by CTL) changed the immunodominance hierarchy between primary and secondary responses, arguing that elimination of DC is not a mechanism by which CTL of different epitope specificities compete (7).
We investigated both possibilities, one in which pMHC levels are indirectly regulated through CTL reducing the viral load and one in which CTL directly reduce pMHC levels by inactivating DC. The two models produced very similar results. For reasons of simplicity, we present only the model where CTL reduce the virus load. The other model is identical to the one shown below, apart from a CTL-dependent killing term analogous to the second term in the virus equation, added to the equation describing pMHC dynamics. The model is shown schematically in Fig. 2; the equations are given by
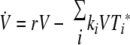 |
(1) |
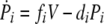 |
(2) |
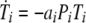 |
(3) |
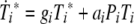 |
(4) |
where variables are as defined above and the dot indicates differentiation with respect to time.
A few of the model parameters can be obtained from the literature. In line with recent findings, we set the number of unactivated, epitope-specific CTL to 500 and 10,000 for the naive and memory responses, respectively (8, 14). The initial growth rate of virus can be obtained from studies reporting viral load data. There is limited evidence that initial virus growth is similar during primary infection and secondary infection with a heterologous challenge strain (30). We used these data to estimate an approximate value for the initial growth of the virus at around r = 3 per day. Different values for r also lead to the results described below if the other parameters of the model are adjusted accordingly. Since virus load in our model is given in arbitrary units, we set the inoculum size to 1 at the beginning of the infection. No antigen-presenting DC or activated CTL exist initially. The rates of pMHC expression, fi, rate of activation and clonal expansion of CTL, ai and gi, and rate of virus reduction, ki, are not known and were chosen by us to match reported data (see Results). The rate of pMHC decline is set at d = 1 per day, equal for the two different epitopes, in line with recent data (5). The parameter values are listed in Table 1. Note that, crucially, all parameters apart from the number of unactivated CTL are identical between primary and secondary responses. Additionally, the only difference between the parameters describing the NP366 and PA224 responses are a higher rate of pMHC expression for the NP366 epitopes (5, 16).
TABLE 1.
Model parametersa
| Symbol | Meaning | Value for NP366/PA224 (reference) |
|---|---|---|
| r | Initial virus growth rate | 3 (30) |
| di | pMHC deactivation rate | 1 (5) |
| fi | Increase of pMHC on DC | 1,000/100 |
| ai | CTL activation rate | 5 × 10−4 |
| gi | CTL expansion rate | 0.45 |
| ki | Rate of virus removal by CTL | 3 × 10−3 |
All parameters are in units of 1/day. Note that only the rate of increase of pMHC is chosen differently between the two epitopes. See the equations, Fig. 2, and the text for more details.
RESULTS
We use the model described in Methods to show that it is possible to reproduce the immunodominance hierarchy observed during primary and secondary influenza A virus infections given the known experimental data. Unfortunately, the currently available data are somewhat limited. To our knowledge, direct measurements of the number of pMHC complexes on DC specific for either epitope and the rate at which pMHC/DC activate CTL are not available. However, a recent study reported the ability of pMHC/DC to activate NP366- or PA224-specific CD8 T cells during primary and secondary responses of influenza A virus infection (5). Here we make the assumption that these data are proportional to the number of pMHC on activated DC. For simplicity, we assume a direct correspondence to Pi in our model. In addition, from a different study, we obtained data for the NP366- and PA224-specific CTL responses in vivo after primary and secondary influenza virus infection (4). We used these data (4, 5) to help us guide our choice for the parameters gi, fi, ki, and ai. The parameter values of the model were chosen “by hand” to match the data. This should not be confused with a more rigorous approach of fitting models to data. Due to a lack of data that directly correspond to the variables of the model and because we combined data from different studies, the fitting approach is not feasible here. We therefore want to stress that the simultaneous plotting of data and model, as shown in the figures below, is done to illustrate overall agreement between our model and the experimental findings and should not be confused with data fitting.
The model reproduces the primary and secondary responses of NP366 and PA224 CTL in wild-type infections.
With the parameter choices given in Table 1, our model produced results that match the data (Fig. 3). We found that for the primary response, the NP366- and PA224-specific CTL reached almost identical levels despite much higher NP366-specific presentation than that for PA224 and equal numbers of naive NP366 and PA224 T cells (Fig. 3, top). This is due to the fact that all epitope-specific, unactivated CTL get activated and expand, independently of the amount of antigen presentation. Even for the less-expressed PA224 epitope, there is plenty of stimulus to rapidly activate virtually all precursor CTL, leading to equal responses.
FIG. 3.

Dynamics of pMHC/DC and CTL during primary and secondary infections with influenza A virus. Top, primary infection. Number of initially unactivated, naive CTL, T0 = 500 for both epitopes (14). Bottom, secondary infection. Same as top but T0 = 10,000 epitope-specific, unactivated memory cells (8). The only difference between the NP366 and PA224 epitopes is that NP366-specific pMHC (black) are created at a higher rate than the PA224 pMHC (gray). The symbols in the left-column figures denote data for epitope-specific CTL activated by antigen-presenting cells ex vivo (see Fig. 1 and 2 in reference 5), which is assumed to be proportional to pMHC levels (see the text). The data in the right column figures are numbers of CTL obtained from the mediastinal lymph nodes (MLN) (see Fig. 5 in reference 4). The model starts 1 day p.i. to incorporate the time for DC migration to the MLN.
For a secondary response with a heterologous challenge strain, the immunodominance hierarchy changes, favoring NP366, the level of which is about five times higher than that of PA224 (4, 8). The mathematical model with the same parameter values as used for the primary response, only with different numbers of initially unactivated memory cells, can also reproduce the CTL dynamics during secondary responses (Fig. 3, bottom). The number of unactivated CTL is much higher than that in the primary response, and these CTL, once activated, quickly reduce the viral load (through killing of infected cells). This leads to much lower levels of pMHC expression, and antigen presentation now becomes the limiting factor in the secondary response. Since NP366 is presented at a higher level, CTL specific for this epitope dominate the response.
While the rapid increase in activated CTL between days 1 and 2 shown in the figure might seem surprising, we think that if it were possible to measure numbers of endogenous, activated CTL this early (which to our knowledge has not been possible due to limits in assay sensitivity), one would see such a rapid increase. Recent findings for the dynamics of CTL activation, using two-photon intravital microscopy, seem to back this (10, 17). Several hours after infection, CTL start to form long-lasting conjugates with DC, which then leads to activation. This activation, which we model with a mass-action term (the aiPiTi term), leads to the rapid increase in activated CTL, followed by clonal expansion.
The model reproduces the primary and secondary responses of NP366 and PA224 CTL in virus epitope knockout experiments.
We wanted to further test our model using additional data. A few recent studies selectively knocked out the PA224 or NP366 epitope and studied how this influenced the immunodominance hierarchies during primary and secondary responses (1, 21). It was found that during the primary response, knocking out either the PA224 or NP366 epitope did not lead to a significant increase in the CTL response of the other epitope, while during the secondary response, knocking out the NP366 epitope led to PA224 compensation but not vice versa (1, 21). We checked if our model could reproduce these experimental findings. As Fig. 4 shows, this was indeed the case. The results can again be explained by the fact that in the primary response, naive CTL are limiting, not antigen. Therefore, removing one epitope (and thereby CTL which reduce virus/pMHC levels) does not matter. For the secondary response, the high presentation of NP366 on DC provides the NP366-specific T cells with enough stimulation, independently of the presence or absence of the PA224 CTL. However, removing the NP366 CTL population leads to a higher viral load, which in turn provides the increase in pMHC needed by the PA224 CTL to expand more.
FIG. 4.

Dynamics of CTL during primary and secondary infections with wild-type (wt), PA224 knockout (PA k.o.), or NP366 knockout (NP k.o.) virus. The symbols denote data for CTL in mediastinal lymph nodes (MLN) after infection with the wild-type or NP366 or PA224 knockout virus from reference 1. No data are shown for the primary response, since MLN data are not available. However, spleen data agree with the finding that neither NP366 nor PA224 epitope knockout changes the CTL responses (1).
DISCUSSION
Understanding immunodominance is important, not only to gain better insights into the function of immune responses but also for practical purposes, such as the development of T-cell-based vaccines. The immunodominance hierarchies during influenza A virus infections in H57BL/6 mice have been studied in detail (7, 8, 16). While the results from these studies have led to important insights, none of the proposed verbal models was completely satisfactory in explaining the data. Here we used a simple mathematical model to explain the seemingly inconsistent data. Maybe surprisingly, our results show that even when NP366 antigen presentation is higher than PA224 presentation during both primary and secondary responses and the number of nonactivated CTL are the same for NP366 and PA224 epitopes, the outcome is equal NP366 and PA224 responses during the primary response, while the NP366 CTL dominate the secondary response. This result can be explained by virtue of a “numbers game” between antigen presentation levels and CTL numbers. During the primary response, CTL are limiting; during the secondary response, antigen presentation is limiting. This alone, combined with the dynamic interactions between pMHC/DC and CTL, can lead to the observed results.
We were able to further test our model by comparing it to results from experimental studies with NP366 or PA224 knockout virus. We found that our model was also able to reproduce these experimentally observed data. The explanation for the knockout studies is again that if antigen is limiting (which is the case for PA224 during secondary responses), knocking out a competing epitope leads to an increase in CTL for the remaining epitope, while epitope knockouts for situations where antigen is not limiting (all other cases) do not influence the CTL responses.
We have focused on the NP366 and PA224 epitopes, the two most prominent H-2Db-restricted CD8+ T-cell responses during primary influenza A virus infection of C57BL/6 mice. We did so because most data were available for these two epitopes, especially data on epitope-specific CTL activation by DC reported in reference 5, which we used as a proxy for pMHC levels. If more data were available for other epitopes, it would be straightforward to extend our model and test if it will also properly predict the immunodominance dynamics for these additional epitopes.
Overall, our results show that the dynamic interactions between populations of cells can lead to different outcomes solely due to changes in the quantitative levels of the involved cell populations. It is not necessary to invoke qualitatively different mechanisms. In our opinion, this is an often-overlooked point. We argue that while uncovering novel mechanisms is important, so are quantitative analyses of the complicated dynamics governing infections and immune responses. In focusing on a purely mechanistic, qualitative approach, valuable insights might be missed. We hope that this study illustrates the usefulness of a quantitative, dynamic approach. There is strong evidence that no unifying, simple model can account for the complex, multifactorial processes that lead to immunodominance in different host-pathogen systems (25). Nevertheless, we think that studies combining mechanisms with dynamics and mathematical models with experimental data can be very useful in furthering our understanding of this complicated and important aspect of immunology.
Acknowledgments
We acknowledge support from the NIH.
Footnotes
Published ahead of print on 11 June 2008.
REFERENCES
- 1.Andreansky, S. S., J. Stambas, P. G. Thomas, W. Xie, R. J. Webby, and P. C. Doherty. 2005. Consequences of immunodominant epitope deletion for minor influenza virus-specific CD8+-T-cell responses. J. Virol. 794329-4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belz, G. T., S. Bedoui, F. Kupresanin, F. R. Carbone, and W. R. Heath. 2007. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat. Immunol. 81060-1066. [DOI] [PubMed] [Google Scholar]
- 3.Belz, G. T., W. Xie, J. D. Altman, and P. C. Doherty. 2000. A previously unrecognized H-2Db-restricted peptide prominent in the primary influenza A virus-specific CD8+ T-cell response is much less apparent following secondary challenge. J. Virol. 743486-3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belz, G. T., W. Xie, and P. C. Doherty. 2001. Diversity of epitope and cytokine profiles for primary and secondary influenza A virus-specific CD8+ T cell responses. J. Immunol. 1664627-4633. [DOI] [PubMed] [Google Scholar]
- 5.Belz, G. T., L. Zhang, M. D. H. Lay, F. Kupresanin, and M. P. Davenport. 2007. Killer T cells regulate antigen presentation for early expansion of memory, but not naive, CD8+ T cells. Proc. Natl. Acad. Sci. USA 1046341-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, W., and J. McCluskey. 2006. Immunodominance and immunodomination: critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv. Cancer Res. 95203-247. [DOI] [PubMed] [Google Scholar]
- 7.Chen, W., K. Pang, K.-A. Masterman, G. Kennedy, S. Basta, N. Dimopoulos, F. Hornung, M. Smyth, J. R. Bennink, and J. W. Yewdell. 2004. Reversal in the immunodominance hierarchy in secondary CD8+ T cell responses to influenza A virus: roles for cross-presentation and lysis-independent immunodomination. J. Immunol. 1735021-5027. [DOI] [PubMed] [Google Scholar]
- 8.Crowe, S. R., S. J. Turner, S. C. Miller, A. D. Roberts, R. A. Rappolo, P. C. Doherty, K. H. Ely, and D. L. Woodland. 2003. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J. Exp. Med. 198399-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Boer, R., M. Oprea, R. Antia, K. Murali-Krishna, R. Ahmed, and A. Perelson. 2001. Recruitment times, proliferation, and apoptosis rates during the CD8+ T-cell response to lymphocytic choriomeningitis virus. J. Virol. 7510663-10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germain, R. N., M. Bajnoff, F. Castellino, M. Chieppa, J. G. Egen, A. Y. C. Huang, M. Ishii, L. Y. Koo, and H. Qi. 2008. Making friends in out-of-the-way places: how cells of the immune system get together and how they conduct their business as revealed by intravital imaging. Immunol. Rev. 221163-181. [DOI] [PubMed] [Google Scholar]
- 11.Harty, J. T., A. R. Tvinnereim, and D. W. White. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 18275-308. [DOI] [PubMed] [Google Scholar]
- 12.Kaech, S. M., E. J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2251-262. [DOI] [PubMed] [Google Scholar]
- 13.Kedl, R. M., J. W. Kappler, and P. Marrack. 2003. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 15120-127. [DOI] [PubMed] [Google Scholar]
- 14.Kedzierska, K., E. B. Day, J. Pi, S. B. Heard, P. C. Doherty, S. J. Turner, and S. Perlman. 2006. Quantification of repertoire diversity of influenza-specific epitopes with predominant public or private TCR usage. J. Immunol. 1776705-6712. [DOI] [PubMed] [Google Scholar]
- 15.Kim, C. J., D. R. Parkinson, and F. Marincola. 1998. Immunodominance across HLA polymorphism: implications for cancer immunotherapy. J. Immunother. 211-16. [PubMed] [Google Scholar]
- 16.La Gruta, N. L., K. Kedzierska, K. Pang, R. Webby, M. Davenport, W. Chen, S. J. Turner, and P. C. Doherty. 2006. A virus-specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc. Natl. Acad. Sci. USA 103994-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mempel, T. R., S. E. Henrickson, and U. H. V. Andrian. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427154-159. [DOI] [PubMed] [Google Scholar]
- 18.Scherer, A., and S. Bonhoeffer. 2005. Epitope down-modulation as a mechanism for the coexistence of competing T-cells. J. Theor. Biol. 233379-390. [DOI] [PubMed] [Google Scholar]
- 19.Scherer, A., M. Salath, and S. Bonhoeffer. 2006. High epitope expression levels increase competition between T cells. PLoS Comput. Biol. 2e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sercarz, E. E., P. V. Lehmann, A. Ametani, G. Benichou, A. Miller, and K. Moudgil. 1993. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 11729-766. [DOI] [PubMed] [Google Scholar]
- 21.Webby, R. J., S. Andreansky, J. Stambas, J. E. Rehg, R. G. Webster, P. C. Doherty, and S. J. Turner. 2003. Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc. Natl. Acad. Sci. USA 1007235-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welsh, R. M., L. K. Selin, and E. Szomolanyi-Tsuda. 2004. Immunological memory to viral infections. Annu. Rev. Immunol. 22711-743. [DOI] [PubMed] [Google Scholar]
- 23.Wherry, E. J., and R. Ahmed. 2004. Memory CD8 T-cell differentiation during viral infection. J. Virol. 785535-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong, P., and E. G. Pamer. 2003. CD8 T cell responses to infectious pathogens. Annu. Rev. Immunol. 2129-70. [DOI] [PubMed] [Google Scholar]
- 25.Yewdell, J. W. 2006. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25533-543. [DOI] [PubMed] [Google Scholar]
- 26.Yewdell, J. W., and J. R. Bennink. 1999. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1751-88. [DOI] [PubMed] [Google Scholar]
- 27.Yewdell, J. W., and S. M. M. Haeryfar. 2005. Understanding presentation of viral antigens to CD8+ T cells in vivo: the key to rational vaccine design. Annu. Rev. Immunol. 23651-682. [DOI] [PubMed] [Google Scholar]
- 28.Yewdell, J. W., and M. D. Val. 2004. Immunodominance in TCD8+ responses to viruses: cell biology, cellular immunology, and mathematical models. Immunity 21149-153. [DOI] [PubMed] [Google Scholar]
- 29.York, I. A., and K. L. Rock. 1996. Antigen processing and presentation by the class I major histocompatibility complex. Annu. Rev. Immunol. 14369-396. [DOI] [PubMed] [Google Scholar]
- 30.Yoshikawa, T., K. Matsuo, K. Matsuo, Y. Suzuki, A. Nomoto, S.-I. Tamura, T. Kurata, and T. Sata. 2004. Total viral genome copies and virus-Ig complexes after infection with influenza virus in the nasal secretions of immunized mice. J. Gen. Virol. 852339-2346. [DOI] [PubMed] [Google Scholar]
- 31.Zammit, D. J., L. S. Cauley, Q.-M. Pham, and L. Lefrancois. 2005. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity 22561-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zinkernagel, R. M., and P. C. Doherty. 1974. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 248701-702. [DOI] [PubMed] [Google Scholar]