

Abstract
An operationally simple and efficient work-up method for TBAF-mediated TBS-deprotection has been developed. The procedure includes addition of a sulfonic acid resin and calcium carbonate, followed by filtration and evaporation. This method eliminates the tedious aqueous-phase extraction process to remove excess TBAF and materials derived from TBAF, thereby making the protocol highly amenable to multiple TBS-deprotection. Its efficiency and usefulness were demonstrated by using the transformation of 1a to 3a in the halichondrin synthesis.
t-Butyldimethylsilyl (TBS) group is one of the most widely used protecting groups for alcohols, phenols, carboxylic acids, and amines, because it is: (1) easy to introduce, (2) stable under various conditions, and (3) readily and selectively cleaved under the fluoride-mediated, typically tetrabutylammonium fluoride (TBAF), conditions.1,2 However, deprotection with TBAF often requires an excess amount of the reagent, and an aqueous-phase extraction protocol is commonly used to remove excess TBAF and materials derived from TBAF. For the cases where deprotected products have a high water-solubility, the protocol of aqueous-phase extraction is not ideal. Despite this concern, very limited TBAF work-up methods are known to avoid an aqueous-phase extraction. Thus, a TBAF work-up with no use of aqueous-phase extraction is potentially useful.
This need came to our attention for a slightly different reason. In conjunction with the synthesis of marine natural products halichondrins,3,4,5,6,7 we recently reported an ion-exchange resin based device to achieve a complete conversion of the enone 2a into the polycyclic ketal 3a in an excellent chemical yield (Scheme 1).4d The enone 2a was in turn obtained via TBAF-deprotection of the penta-TBS enone 1a; this deprotection was accomplished in the presence of 7.5–10.0 equiv of TBAF (1.5–2.0 equiv TBAF per TBS ether) and 2a8 was isolated in a high yield by the standard aqueous-phase extraction work-up. However, tediously extensive extraction was required, to obtain 2a free from excess TBAF and materials derived from TBAF. Thus, we recognized the need of developing a non-aqueous work-up method of TBAF deprotection, not only because such a method should eliminate the labor-intensive step, but also because such a method might allow us to achieve the conversion of the enone 1a to the polycyclic ketal 3a without isolation of the intermediate 2a. Herein, we report an operationally simple, efficient, and versatile work-up protocol for TBAF-mediated desilylation.
Scheme 1.
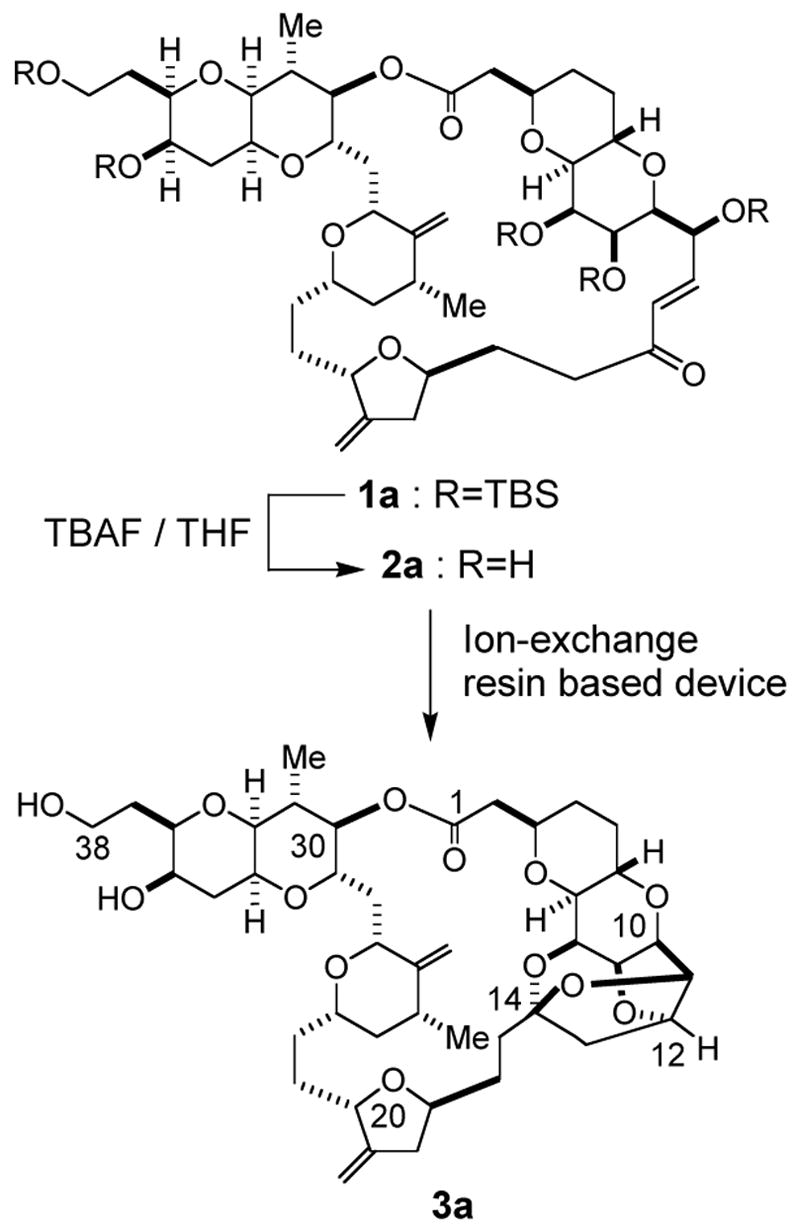
Transformation of 1a to 3a.
Related to the current work, we noticed two relevant methods known in the literature. First, Craig and Everhart used sodium perchlorate, to remove excess TBAF as the insoluble tetra-n-butylammonium perchlorate salt.9 Second, Parlow, Vazquez, and Flynn reported the simultaneous use of a calcium sulfonate resin with a sulfonic acid resin.10 We appreciated an appealing future for both methods but, at the same time, recognized some room for improvement.11
At the outset of our work, we envisioned the possibility of removing tetrabutylammonium cation with the use of acidic ion-exchange resin. In order to test this possibility, we treated TBAF in THF with excess sulfonic acid resin at room temperature, removed the resin by filtration, and evaporated the filtrate. A 1H NMR analysis of the residue showed that only a small portion of TBAF was removed by this operation, thereby suggesting that sulfonic acid resin alone cannot drive eq. 1 toward the right side (Scheme 2). Based on this observation, we focused on a method to remove liberated HF from THF solution. Calcium carbonate (insoluble in THF) seemed to be an attractive HF scavenger for the following reasons: (1) the equilibrium should shift toward the right side, because formed CaF2 precipitates out from the system (CaF2 is insoluble in THF) and (2) the products, i.e., CaF2 (insoluble in THF), water, and CO2, can be removed by filtration and evaporation (see eq. 2 in Scheme 2). To test this possibility experimentally, we treated TBAF in THF in the presence of calcium carbonate at room temperature, removed the insoluble materials by filtration, and evaporated the filtrate to dryness under reduced pressure. A 1H NMR analysis of the residue showed that more than 98% of TBAF was removed via this simple operation. Among the sulfonic acid resins tested,12 DOWEX 50WX8-400 was found to be the most effective for this purpose.
Scheme 2.
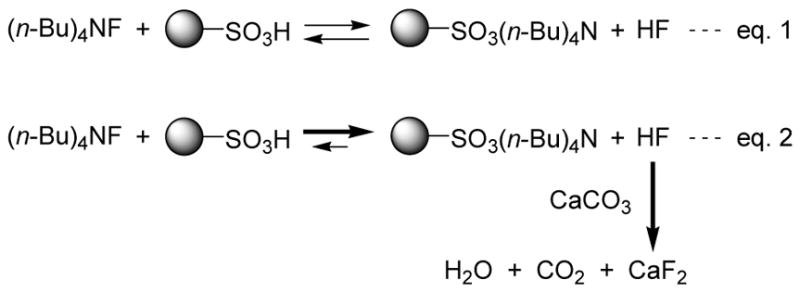
Reaction of TBAF and sulfonic acid resin in the absence (eq. 1) or presence (eq. 2) of calcium carbonate.
Having demonstrated that a combination of sulfonic acid resin and calcium carbonate can effectively remove TBAF from THF solution, we then studied the effectiveness of this work-up method of TBAF-promoted TBS-deprotection. The anticipated overall transformation is depicted in Scheme 3. TBS ether is treated with excess TBAF, followed by addition of DOWEX 50WX8-400 (H+-form; excess) and calcium carbonate (powder, excess). The products formed in this transformation are the desired alcohol, DOWEX 50WX8-400 (n-Bu4N+-form), calcium fluoride, water, CO2, and t-butyldimethylsilyl fluoride (TBSF). DOWEX 50WX8-400 (n-Bu4N+-form) and calcium fluoride, as well as excess DOWEX 50WX8-400 (H+-form) and calcium carbonate, can be removed by filtration, whereas water and TBSF can be removed by evaporation. Thus, only the desired alcohol should be left after filtration and evaporation.
Scheme 3.
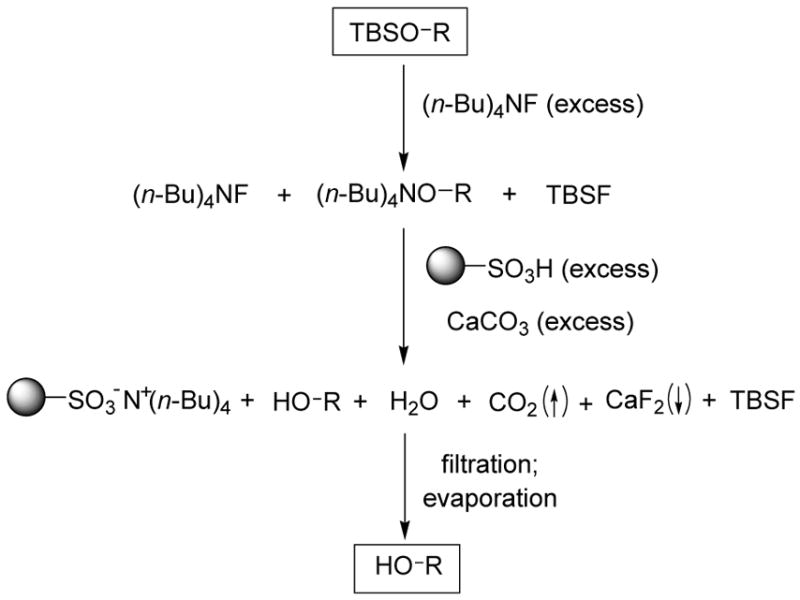
Overall transformation.
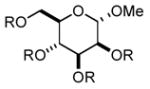
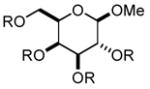
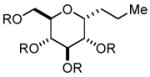
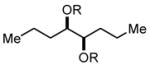
In order to test the feasibility of this method, we selected seven substrates that should furnish the deprotected products with a broad range of polarity (Table 1). According to the general procedure given below,13 deprotection and work-up were carried out. 1H NMR analysis revealed that the crude product thus obtained was the expected deprotected alcohol only contaminated with a small amount of the TBAF-derived materials. Based on the signal intensity in the 1H NMR spectrum of the crude product, at least 99% of the TBAF-derived materials were removed through this procedure for all the cases. Interestingly, practically no TBS-derived material was contaminated in the crude product. This procedure thus proved effective for all the seven substrates tested. In our view, this method offers an attractive option to deal with water-soluble products for which a conventional aqueous phase work-up is difficult to apply.
Table 1.
Substrates used to test the feasibility and efficiency of the TBAF work-up protocol.a
| entry | substrate (R = TBS)/
product (R = H) |
equiv of TBAF | crude yield (%)b | removed TBAF (%)c |
|---|---|---|---|---|
| 1 |
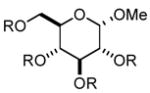
|
8 | 111 | 99.3 |
| 2 |
|
8 | 110 | 99.5 |
| 3 |
|
8 | 107 | 99.5 |
| 4 |
|
8 | 106 | 99.5 |
| 5 |
|
4 | 95d | 99.5 |
| 6 |
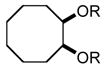
|
8 | 110 | 99.6 |
| 7 |
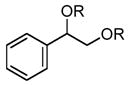
|
3 | 103 | 99.8 |
Reaction conditions employed for desilylation: substrate (1 equiv), TBAF (3–8 equiv), THF, rt or heat, 4-28 h; CaCO3, DOWEX 50WX8-400 (used as supplied), MeOH, rt, 1 h.
Based on the weight obtained after filtration and evaporation.
Estimated from 1H NMR spectra of crude compound.
Product was volatile.
As mentioned earlier, we had one specific goal for this developmental work. Thus, the penta-TBS enone 1a in the halichondrin B series was subjected to TBAF-promoted TBS removal in THF (TBAF: total 10 equiv or 2.0 equiv per TBS, RT, 49 h), followed by this work-up method, to furnish the crude desilylated enone 2a (126% of the theoretical weight). Then, the crude product was directly applied to the ion-exchange resin device,4d to give the crude ketal 3a (107% of the theoretical weight), which was passed through a short plug of silica gel to furnish the pure ketal 3a in 96% overall yield from 1a (Scheme 4). This protocol was also successfully applied to the transformation of the penta-TBS enone 1b to the polycyclic ketal 3b in the E7389 series.14
Scheme 4.
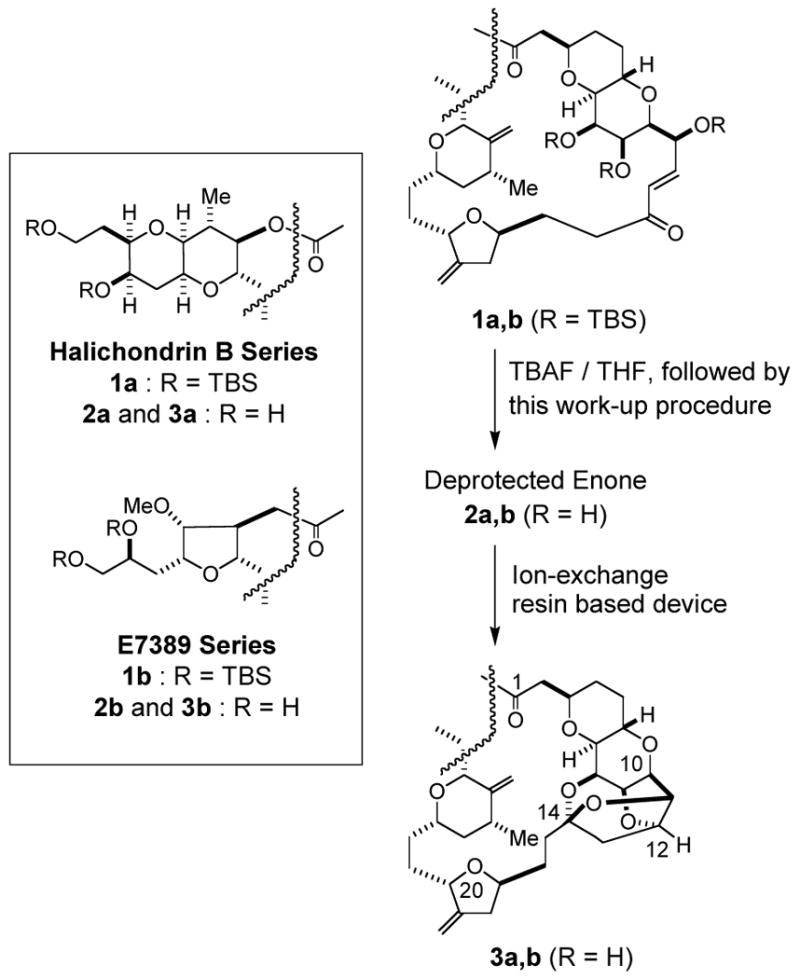
Application of this TBAF work-up method to the advanced synthetic stage in both halichondrin B and E7389 series.
In summary, we developed an operationally simple and efficient work-up method for TBAF-promoted desilylations, with the use of commercially available sulfonic acid resin (used as supplied) and calcium carbonate. This method eliminates the tedious aqueous-phase extraction protocol, to remove excess TBAF and TBAF-derived materials. The effectiveness and versatility of this method was demonstrated for the seven examples shown in Table 1. The usefulness of this method was further demonstrated by using the transformation of 1a,b to 3a,b in the halichondrin synthesis. We believe that this method offers an attractive option to deal with water-soluble products for which a conventional aqueous phase work-up is difficult to apply.
Supplementary Material
Experimental details and 1H NMR spectra (19 pages). This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
We are grateful to the National Institutes of Health and Eisai Research Institute for generous financial support.
References
- 1.Corey EJ, Venkateswarlu AJ. Am Chem Soc. 1972;94:6190–6191. [Google Scholar]
- 2.For example, see: Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. John Wiley & Sons; New York: 1999.
- 3.For the first isolation of the halichondrins from a marine sponge Halichondria okadai Kadota, see: Uemura D, Takahashi K, Yamamoto T, Katayama C, Tanaka J, Okumura Y, Hirata YJ. Am Chem Soc. 1985;107:4796–4798.Hirata Y, Uemura D. Pure Appl Chem. 1986;58:701–710. For isolation of the halichondrins from different species of sponges, see: Pettit GR, Herald CL, Boyd MR, Leet JE, Dufresne C, Doubek DL, Schmidt JM, Cerny RL, Hooper JNA, Rutzler KCJ. Med Chem. 1991;34:3339–3340. doi: 10.1021/jm00115a027.Pettit GR, Tan R, Gao F, Williams MD, Doubek DL, Boyd MR, Schmidt JM, Chapuis JC, Hamel E, Bai R, Hooper JNA, Tackett LPJ. Org Chem. 1993;58:2538–2543.Litaudon M, Hart JB, Blunt JW, Lake RJ, Munro MHG. Tetrahedron Lett. 1994;35:9435–9438.Litaudon M, Hickford SJH, Lill RE, Lake RJ, Blunt JW, Munro MHGJ. Org Chem. 1997;62:1868–1871.
- 4.For the synthetic work from this laboratory, see: Aicher TD, Kishi Y. Tetrahedron Lett. 1987;28:3463–3466.Aicher TD, Buszek KR, Fang FG, Forsyth CJ, Jung SH, Matelich MC, Scola PM, Spero DM, Yoon SK, Kishi YJ. Am Chem Soc. 1992;114:3162–3164.Choi H, Demeke D, Kang FA, Kishi Y, Nakajima K, Nowak P, Wan ZK, Xie C. Pure Appl Chem. 2003;75:1–17. and the references cited therein. Namba K, Kishi YJ. Am Chem Soc. 2004;126:7770–7771. doi: 10.1021/ja047826b.Namba K, Kishi YJ. Am Chem Soc. 2005;127:15382–15383. doi: 10.1021/ja055966v. and references cited therein.
- 5.For synthetic work by Salomon, Burke, and Yonemitsu, see: Kim S, Salomon RG. Tetrahedron Lett. 1989;30:6279–6782.Cooper AJ, Pan W, Salomon RG. Tetrahedron Lett. 1993;34:8193–8196. and the references cited therein. Lambert WT, Hanson GH, Benayoud F, Burke SDJ. Org Chem. 2005;70:9382–9398. doi: 10.1021/jo051479m. and the references cited therein. Horita K, Hachiya S, Nagasawa M, Hikota M, Yonemitsu O. Synlett. 1994:38–39.Horita K, Nagasawa M, Sakurai Y, Yonemitsu O. Chem Pharm Bull. 1998;46:1199–1216.Horita K, Nishibe S, Yonemitsu O. Phytochem Phytopharm. 2000:386–397. and the references cited therein.
- 6.For mechanism of action, see: Bai R, Paull KD, Herald CL, Malspeis L, Pettit GR, Hamel EJ. Biol Chem. 1991;266:15882–15889.Hamel E. Pharmacol Ther. 1992;55:31–51. doi: 10.1016/0163-7258(92)90028-x.Dabydeen DA, Burnett JC, Bai R, Verdier-Pinard P, Hickford SJH, Pettit GR, Blunt JW, Munro MHG, Gussio R, Hamel E. Mol Pharmacol. 2006;70:1866–1875. doi: 10.1124/mol.106.026641. and the references cited therein. Luduena RF, Roach MC, Prasad V, Pettit GR. Biochem Pharmacol. 1993;45:421–427. doi: 10.1016/0006-2952(93)90079-c.
- 7.Recently Jordan and co-workers reported that the primary antimitotic mechanism of action of halichondrin analog E7389 is suppression of microtuble growth. For details, see: Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L. Mol Cancer Ther. 2005;4:1086–1095. doi: 10.1158/1535-7163.MCT-04-0345.
-
8.Compound 2 exists primarily as the intramolecular oxy-Michael adducts shown below.
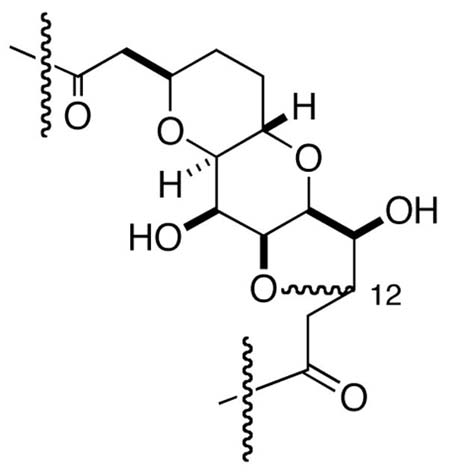
- 9.Craig JC, Everhart ET. Synth Commun. 1990;20:2147–2150. [Google Scholar]
- 10.Parlow JJ, Vazquez ML, Flynn DL. Bioorg Med Chem Lett. 1999;8:2391–2394. doi: 10.1016/s0960-894x(98)00432-6. [DOI] [PubMed] [Google Scholar]
- 11.For instance, perchlorates are potentially hazardous, whereas calcium sulfonate resin is not commercially available.
- 12.Other sulfonic acid resins tested included Rexyn 101 and Amberlyst 15.
- 13.General work-up procedure for removal of TBAF residue (Table 1. Entry 1): A 10-mL flask containing a stirring bar was charged with substrate (110 mg, 0.169 mmol). TBAF (1.0 M in THF, 1.35 mL, 1.35 mmol) was added via syringe. After the solution was stirred at rt for 4 h (no SM nor partially deprotected intermediate detected by TLC), CaCO3 (280 mg), DOWEX 50WX8-400 (840 mg, used as supplied), and MeOH (2.0 mL) were added. The suspension was stirred at rt for 1 h. All insoluble materials were removed by filtration through a pad of Celite, and the filter cake was washed with MeOH thoroughly. The combined filtrates were evaporated to dryness under reduced pressure to give crude product (36.3 mg, 111%) as a white solid. 1H NMR spectra of the crude product showed that 99.3% of tetrabutylammonium salt were removed by these operations.
- 14.Zheng W, Seletsky BM, Palme MH, Lydon PJ, Singer LA, Chase CE, Lemelin CA, Shen Y, Davis H, Tremblay L, Towle MJ, Salvato KA, Wels BR, Aalfs KK, Kishi Y, Littlefield BA, Yu MJ. Bioorg Med Chem Lett. 2004;14:5551–5554. doi: 10.1016/j.bmcl.2004.08.069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and 1H NMR spectra (19 pages). This material is available free of charge via the Internet at http://pubs.acs.org