

Abstract
Monoamine oxidases A and B (MAO A and B) catalyze neurotransmitters degradation and represent drug targets for the treatment of neurodegenerative disorders. Rasagiline is an irreversible, MAO B-selective inhibitor that has been approved as a novel anti-Parkinson’s drug. In this study we investigate the inhibition of recombinant human MAO A and MAO B by several rasagiline analogues. Different substituents added onto the rasagiline scaffold alter the binding affinity depending on the position on the aminoindan ring and on the size of the substituent. Compounds with a hydroxyl group on either the C4 or the C6 atom inhibit both isozymes, whereas a bulkier substituent such as a carbamate is tolerated only at the C4 position. The 1.7 Å crystal structure of MAO B in complex with 4-(N-methyl-N-ethyl-carbamoyloxy)-N-methyl-N-propargyl-1(R)-aminoindan shows that the binding mode is similar to that of rasagiline with the carbamate moiety occupying the entrance cavity space. 1(R)-aminoindan, the major metabolic product of rasagiline, and its analogues reversibly inhibit both MAO A and MAO B. The crystal structure of N-methyl-1(R)-aminoindan bound to MAO B shows that its aminoindan ring adopts a different orientation compared to that of rasagiline.
Keywords: human MAO B structure, inhibitor binding, rasagiline, flavin
Introduction
Human monoamine oxidases A and B (MAO A and MAO B) modulate the intracellular levels of arylalkylamines such as dopamine and serotonin by catalyzing their oxidative deamination with the concomitant production of hydrogen peroxide1. Impairment in neurotransmitters catabolism and oxidative damage are important factors in the physiology of neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases. For this reason, MAO B inhibition represents one of the strategies to alleviate symptoms of patients suffering from these pathological conditions and there have been considerable efforts in the last years to develop new MAO inhibitors to be used as neuroprotective agents.2
Rasagiline [N-propargyl-1(R)-aminoindan] (Figure 1a) is a novel drug for Parkinson’s disease treatment both as monotherapy in early stages and as adjunct to L-dopa.3 It is a selective and irreversible MAO B inhibitor that prevents the degradation of dopamine. Rasagiline has been demonstrated to reduce motor fluctuations and exhibits superior efficacy with respect to the therapy based on dopamine agonists. Moreover, rasagiline is well tolerated and in contrast to other anti-parkinsonians can be given once-daily with no need for dose titration.3 Pharmacological studies are currently ongoing on rasagiline and related compounds (Figures 1a and 1b) as potential neuroprotective agents in Alzheimer’s disease.4 The idea is that a molecule with two pharmacophores that confer both MAO and acetylcholine esterase inhibition as well as neuroprotective activity may represent a comprehensive approach to deal with the complexity of Alzheimer’s disease.4,5
Figure 1.

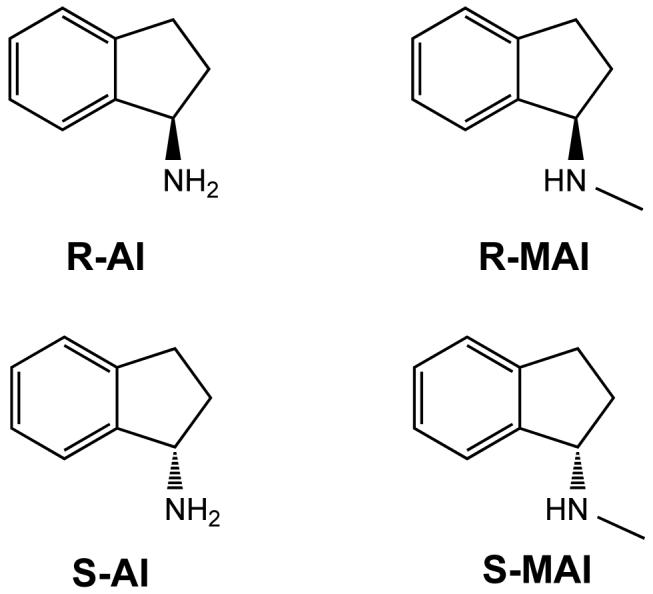
Chemical formula of rasagiline-related inhibitors. (a) Irreversible inhibitors that form a covalent adduct with the flavin: 6-(N-methyl-N-ethyl-carbamoyloxy)-N-methyl-N-propargyl-1(R)-aminoindan (R-M6CPAI); 4-(N-methyl-N-ethyl-carbamoyloxy)-N-propargyl-1(R)-aminoindan (R-4CPAI); 4-(N-methyl-N-ethyl-carbamoyloxy)-N-methyl-N-propargyl-1(R)-aminoindan (R-M4CPAI); 4-hydroxy-N-propargyl-1(R)-aminoindan (R-4HPAI); 6-(N-methyl-N-ethyl-carbamoyloxy)-N-propargyl-1(R)-aminoindan (Ladostigil); N-propargyl-1(R)-aminoindan (Rasagiline). The rasagiline flavocyanine adduct with atom numbering is shown. (b) Reversible inhibitors of the aminoindan class: 1(R)-aminoindan (R-AI); 1(S)-aminoindan (S-AI); N-methyl-1(R)-aminoindan (R-MAI); N-methyl-1(R)-aminoindan (S-MAI).
MAO inhibition by rasagiline, ladostigil (6-(N-methyl-N-ethyl-carbamoyloxy)-N-propargyl-1(R)-aminoindan (Figure 1a), an inhibitor of MAOs and acetylcholine esterase) and some of their analogues has been extensively studied in our laboratories.6,7 Both MAO A and MAO B are flavin-dependent enzymes monotopically bound to the outer mitochondrial membrane. The crystal structure of human MAO B revealed that its active site consists of two cavities, named substrate and entrance cavity respectively, which can exist as a single entity when bulky ligands are bound8 (Figure 2a). Rasagiline and its analogues occupy the active site cavity and react with the flavin forming an irreversible covalent adduct with the N5 atom of the cofactor (Figure 1a). Our studies have shown that the addition of substituents in different positions of the rasagiline structure can considerably affect the inhibition properties with respect to MAO A and B, which has provided clues for drug optimization. This analysis is now extended to rasagiline analogues that carry substituents on different positions of the aminoindan ring (R-M6CPAI, R-4CPAI, R-M4CPAI and R-4HPAI; Figure 1a) and were shown before to inhibit MAO’s4. In addition, we study compounds that lack the acetylenic moiety required for the covalent bond to the flavin (Figure1b). These covalent and non-covalent inhibitors are biochemically and crystallographically characterized with respect to MAO A and MAO B inhibition. The aim of this study is to give insights into two questions:
How do the different substituents influence MAO inhibition and how is this effect related to the mode of binding?
What is the role of the covalent bond that rasagiline and its analogues form with the flavin in their inhibitory activity towards MAO?
Here we report on these experiments that are comparatively interpreted on the basis of the previous data.
Figure 2.
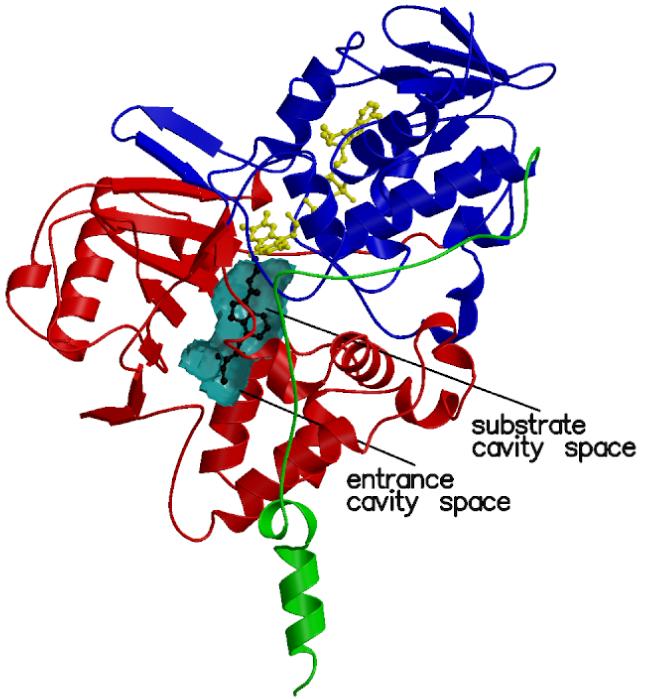
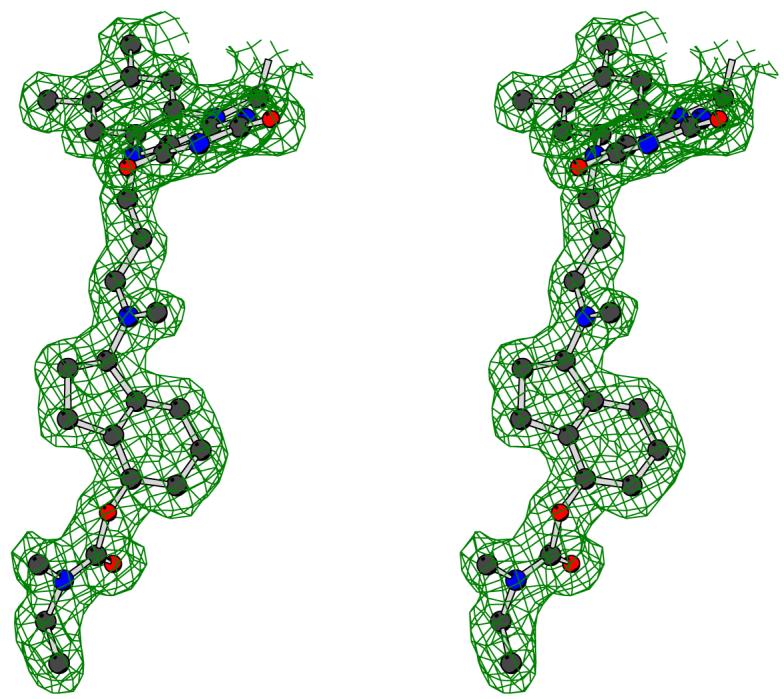
Three-dimensional structure of human MAO B in complex with R-M4CPAI. (a) Ribbon representation of the MAO B subunit. The FAD-binding domain is in blue, the substrate-binding domain in red, and the membrane-binding C-terminal region in green. The FAD cofactor and R-M4CPAI are shown as yellow and black ball-and-stick, respectively. The active site cavity is outlined by a cyan semitransparent surface. (b) Stereoview of the unbiased 2Fo-Fc electron density map for the covalent adduct of R-M4CPAI with the flavin (contoured at 1 σ level). Carbon atoms are in black, oxygen atoms in red, and nitrogen atoms in blue.
Results
Irreversible inhibitors
The first part of this work focuses on four N-propargylaminoindan derivatives that carry a substituent on either position C4 or C6 of the aminoindan ring (R-M6CPAI, R-4CPAI, R-M4CPAI and R-4HPAI; Figure 1a). Similarly to rasagiline and other acetylenic inhibitors, these molecules irreversibly inactivate both MAO A and MAO B by forming a covalent adduct with the flavin cofactor (Figure 1a).6 Enzyme inactivation produces a typical perturbation of the UV/Vis spectrum characterized by the bleaching of the 450 nm peak with the concomitant appearance of another peak at a wavelength ranging from 411 to 418 nm. Because the inactivation process is relatively slow, it has been possible to determine the inhibition constants for these compounds (Table 1). The crystal structures of human MAO B in complex with R-M6CPAI, R-M4CPAI and R-4HPAI have been solved at a resolution better than 2.5 Å (Table 2) and in all cases the inhibitor could be unambiguously modeled in the electron density (Figure 2b). The root-mean-square deviations calculated from the superposition of these complexes onto the MAO B-rasagiline structure7 fall in the 0.20-0.34 Å range for 986 Cα atoms of the dimeric enzyme, which allows a comparative analysis of these compounds bound in the MAO B active site. In all these complexes the substrate and entrance cavities are fused and Ile199 is in the “open” conformation as found in the complex with rasagiline (Figure 2a).
Table 1.
Inhibition Data of Human Recombinant MAO A and MAO B by the Investigated Inhibitors
| Ki (μM)b | kinact (10-3min-1)c | kinact/Ki (103M-1min-1) | MAO A massd | MAO B massd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MAO A | MAO B | MAO A | MAO B | MAO A | MAO B | Observed | Calculated | Observed | Calculated | |
| Rasagilinea | 9.7 | 0.7 | 6.7±1.5 | 53.3±2.2 | 0.69 | 76.2 | 60,674±6 | 60,683 | 59,621±23 | 59,646 |
| R-M6CPAI | 295 | 137 | 19±1.8 | ND e,f | 0.06 | ND e,f | 60,728±15 | 60,798 | 59,673±23 | 59,761 |
| R-M4CPAI | 6 | 130 | 700±13.6 | 170±7.4 | 18.3 | 1.31 | 60,803±6 | 60,798 | 59,739±6 | 59,761 |
| R-4CPAI | 7 | 154 | 6±0.1 | 29±0.8 | 0.86 | 0.19 | 60,784±6 | 60,784 | 59,731±6 | 59,747 |
| R-4HPAI | 2 | 31 | 1±0.1 | 20±0.9 | 0.50 | 0.65 | ND,f | ND,f | ND,f | ND,f |
| R-MAI | 56 | 17 | - | - | - | - | - | - | - | - |
| S-MAI | 50 | 270 | - | - | - | - | - | - | - | - |
| R-AI | 67 | 32 | - | - | - | - | - | - | - | - |
| S-AI | 160 | 1,080 | - | - | - | - | - | - | - | - |
Data on rasagiline6 have been included as comparison.
MAO A and MAO B activities were measured at 25 °C by kynuramine and benzylamine assays, respectively, in 50 mM potassium phosphate buffer pH 7.5, 0.5 % (w/v) reduced Triton X-100. The errors in determinations of Ki constants are within 5% of their values.
Apparent rates of inactivation were measured following the increase in absorbance at λ=415 nm at 15 °C using 10 μM enzyme and 1 mM inhibitor in 50 mM potassium phosphate pH 7.5, 0.8% w/v β-octylglucoside.
MAO A and MAO B masses were calculated assuming the presence of only one flavocyanine per enzyme monomer.
No rates constants were obtained due to high turbidity of the samples.
ND: not determined.
Table 2.
Data collection and refinement statistics
| R-M6CPAI | R-M4CPAI | R-4HPAI | R-MAI | |
|---|---|---|---|---|
| space group | C222 | C222 | C222 | C222 |
| unit cell (Å) | a = 140.9 | a = 131.7 | a = 132.5 | a = 131.3 |
| b = 221.1 | b = 222.4 | b = 223.2 | b = 222.4 | |
| c = 86.3 | c = 86.1 | c = 86.3 | c = 86.2 | |
| resolution (Å) | 2.2 | 1.7 | 2.5 | 1.7 |
| Rsyma,b (%) | 9.2 (59.8) | 8.8 (48.3) | 14.2 (49.3) | 6.8 (28.9) |
| completenessb (%) | 99.0 (99.9) | 98.2 (99.1) | 93.6 (94.7) | 98.2 (99.4) |
| unique reflections | 65,730 | 131,876 | 40,147 | 131,493 |
| redundancy | 2.6 | 3.1 | 3.0 | 3.1 |
| I/σ b | 9.3 (2.4) | 11.0 (2.2) | 9.0 (1.7) | 12.0 (3.5) |
| n° of atoms protein/ligand/waterc average B value for | 8017/1×14/293 | 8017/2×21/839 | 8017/2×14/189 | 8017/2×11/627 |
| ligand atoms (Å2) | 43.5 | 18.2 | 23.8 | 13.0 |
| Rcrystd (%) | 22.2 | 19.3 | 21.5 | 19.4 |
| Rfreed (%) | 26.0 | 21.0 | 28.5 | 21.2 |
| rms bond length (Å) | 0.016 | 0.007 | 0.019 | 0.007 |
| rms bond angles (°) | 1.53 | 1.13 | 1.69 | 1.14 |
Rsym=Σ|Ii-<I>|/ΣIi, where Ii is the intensity of ith observation and <I> is the mean intensity of the reflection.
Values in parentheses are for reflections in the highest resolution shell.
In the complex with R-M6CPAI, the hydrolyzed inhibitor lacking the carbamate substituent was modeled only in monomer B. In the other crystallographically independent monomer (monomer A), the electron density was not sufficiently well defined to allow modeling of the inhibitor atoms.
Rcryst=Σ|Fobs-Fcalc|/Σ|Fobs| where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. Rcryst and Rfree were calculated using the working and test set, respectively.
R-4HPAI
Inspection of the crystal structure of MAO B in complex with rasagiline predicted the C4 position of the aminoindan ring to be a promising site for introducing a substituent, which was expected to bind in the entrance cavity space (Figure 2a).7 We first studied R-4HPAI that bears a small hydroxyl substituent on position 4. This compound shows a higher affinity for MAO A (Ki=2 μM) than MAO B (Ki=31 μM), although the inhibition potencies calculated as kinact/Ki are essentially identical (Table 1). These values indicate that the 4-hydroxyl group does not greatly alter the binding to MAO A whereas it causes a 45-fold reduction in affinity towards MAO B. The crystal structure of MAO B in complex with R-4HPAI reveals that the mode of binding is identical to that of rasagiline with the 4-hydroxyl group H-bonded to an ordered water molecule located in the entrance cavity space (Figure 3).
Figure 3.

Stereo representations illustrating the binding modes from top to bottom of R-4HPAI, R-M4CPAI, and R-M6CPAI. The carbamate group of the enzyme-bound R-M6CPAI is hydrolyzed. Carbon atoms are in black, oxygen atoms in red, nitrogen atoms in blue, and sulfur atoms in yellow. The inhibitor molecule is highlighted in black. Water molecules are shown as cyan spheres. H-bonds are outlined by dotted lines. With respect to Figure 2a, the model has been rotated by about 120° around an axis perpendicular to the plane of the paper.
R-4CPAI and R-M4CPAI
R-4CPAI and R-M4CPAI carry a bulky carbamate substituent at the C4 position (Figure 1a). They have been developed as dual function compounds able to inhibit both MAO B and acetylcholine esterase.4 Our analysis with the recombinant proteins confirms that these two molecules are moderately effective as MAO inhibitors and shows that the affinity for MAO A is 20-fold higher than MAO B although the inactivation rates are higher for MAO B than MAO A (Table 1). These data indicate that compared to R-4HPAI the presence of the bulky carbamate substituent on position 4 reduces the affinity (especially for MAO B, Table 1) but it does not abolish binding. This feature is confirmed by Electron Spray Ionisation Mass Spectrometry and crystallographic analysis of its covalent complex with MAO B. In particular, the three-dimensional structure of the complex between MAO B and R-M4CPAI (Figures 2a and 3) shows that the inhibitor binding mode is identical to that of rasagiline and R-4HPAI. The structure nicely confirms that the carbamate moiety fills the entrance cavity space and establishes a number of van der Waals contacts with the surrounding amino acids, although no H-bonds are formed.
R-M6CPAI
R-M6CPAI is the 6-N-methyl-N-ethyl-carbamoyloxy derivative of N-methylated rasagiline. Although this compound has weak inhibitory properties towards both MAO B (Ki=137 μM) and MAO A (Ki=295 μM), the UV/Vis spectra of the inhibited enzymes show the perturbation typical of the flavocyanine adduct formation. A surprising finding was that the molecular masses of the inhibited A and B isozymes as measured by mass spectrometry are lower than the values calculated assuming a 1:1 molar stoichiometry between enzyme and inhibitor (Table 1). These findings are corroborated by the crystal structure of MAO B in complex with R-M6CPAI (Figure 3). The inhibitor is covalently bound to the flavin in a way essentially identical to that of rasagiline but the electron density contiguous with the C6 position of the aminoindan ring is consistent with a hydroxyl group rather than a N-methyl-N-ethyl-carbamoyloxy substituent. Taken together, the mass-spectrometry and crystallographic data can be explained by the hypothesis that the carbamoyloxy moiety of R-M6CPAI undergoes a slow hydrolytic process, which results in the formation of a compound corresponding to the 6-hydroxyl derivative of N-methyl rasagiline. This is the compound that is ultimately bound to the protein and its binding appears to be virtually identical to that of 6-hydroxy-N-propargyl-1(R)-aminoindan.7 The hydroxyl group in position 6 does not create any steric hindrance and is engaged in a H-bond with the Sγ atom of Cys172 (Figure 3) as observed in the 6-hydroxy-N-propargyl-1(R)-aminoindan complex.7 The hydrolysis of the carbamoyloxy group might be a non-enzymatic process with the enzyme preferentially binding the hydrolyzed form of the inhibitor.
Reversible inhibitors
A second group of rasagiline analogues has been studied which comprises four compounds that lack the acetylenic moiety required for the covalent bond to the flavin (Figure 1b). They include the S and R enantiomers of aminoindan (R-AI and S-AI) and the corresponding N-methylated derivatives (R-MAI and S-MAI). These molecules bear either a primary or secondary amino group but they are not substrates for either MAO A or MAO B. All four compounds, except S-AI for MAO B, have been found to competitively inhibit both enzymes although this inhibition is weak compared to that of rasagiline (Table 1). In general, the R-isomers show higher binding affinities than the corresponding compounds in the S configuration and this difference is more pronounced for MAO B than for MAO A. They are all weaker inhibitors relative to rasagiline, although R-MAI, S-MAI and R-AI are all better inhibitors than the S-enantiomer of rasagiline.6 Most importantly, no perturbation of the flavin absorption spectrum in either MAO A or B is detected with any of these aminoindan molecules, suggesting that they do not covalently bind to the flavin cofactor.
The crystal structure of MAO B in complex with R-MAI has been determined at 1.7 Å resolution (Figure 4). The inhibitor binds in front of the flavin cofactor and occupies the substrate cavity space. The inhibitor amino group establishes H-bonds with two water molecules and the N-methyl group lies in the aromatic cage formed by Tyr398 and Tyr435, displacing a water molecule that is present in all MAO B structures. The distances of the amino group and the N-methyl group from the flavin ring are 4.6 Å and 4.1 Å, respectively. The superposition of the structures of the R-MAI and rasagiline MAO B complexes highlights the substantially different binding modes between the two inhibitors (Figure 4). Compared to rasagiline, the indan ring of R-MAI is shifted by about 3 Å towards the flavin. This movement is associated with a 45° rotation of the R-MAI ring around an axis approximately perpendicular to the indan plane. Thus, R-MAI differs from rasagiline in its binding mode to MAO B not only in lacking a covalent interaction, but also in the orientation and position of its ring moiety. This feature is coupled to the different conformation of Ile199 that in the R-MAI complex adopts the closed conformation so that the entrance and substrate cavities are not fused. This binding mode resembles that of isatin, a reversible non-covalent MAO B inhibitor.8 Isatin also binds in the substrate cavity without forcing Ile199 to adopt the open conformation and its dioxoindole ring binds in a position similar to that of the indan ring of R-MAI (Figure 4). This structural similarity is reflected in the similar Ki values of these two inhibitors (3 μM for isatin and 17 μM for R-MAI).
Figure 4.

Stereoview of R-MAI bound in the MAO B active site. Color code for atoms is the same as in Figure 3. For binding mode comparison, the position of the superposed (top) rasagiline7 and (bottom) isatin8 has been highlighted in white dashed ball-and-stick representation. The orientation is the same as in Figure 3. For the sake of clarity, Q206 has been omitted.
Discussion
Rasagiline can be used as a molecular scaffold to introduce functional groups that confer additional pharmacological properties in a strategy of developing multi-pharmacophore drugs. This idea has led to the development of carbamate derivatives of rasagiline which can inhibit both MAO B and acetylcholine esterase; these dual function inhibitors are also of potential value as neuroprotective agents for the treatment of Alzheimer’s disease.4 The MAO B active site consists of a large cavity that originates on the surface of the protein and reaches the core of the structure in front of the flavin cofactor (Figure 2a). When small ligands bind to MAO B a conformational change at Ile199 occurs, which causes the cavity to be divided into two parts, the substrate cavity and the entrance cavity.8 Rasagiline was shown to occupy the substrate cavity space with the aminoindan ring extending far enough to make Ile199 adopt the “open” conformation.7 We have investigated several rasagiline analogues carrying different substituents on the C4 and C6 positions of their aminoidan ring (Figure 1a). Small substituents such as a hydroxyl group can be borne on both the C6 and C4 positions of the aminoindan ring7 (Figure 3). This is not possible for bulkier substituents which are not tolerated on the C6 atom because of the tight packing of the surrounding amino acid residues. Thus, R-M6CPAI can bind only when the carbamate moiety is hydrolyzed (Figure 3). Conversely, the mass spectrometry analysis clearly indicates that R-M4CPAI binds to both MAO A and MAO B in its intact form and in the case of MAO B the crystal structure shows that this is enabled by the fact that the carbamate group is accommodated in the entrance cavity space (Figure 2a). The binding mode of the rasagiline scaffold in all these analogues appears to be highly conserved with respect to that of the parent compound and the different substituents do not produce any conformational change in the active site structure. These observations lead to the conclusion that in the design of novel rasagiline derivatives the active site of MAO B can be considered as a quite rigid cavity that allows the introduction of bulky substituents only if they can bind in the entrance cavity space.
We have investigated four rasagiline analogues that lack the propargyl moiety (Figure 1b) and are not expected to form the covalent adduct with the flavin. In addition, one of these compounds (R-AI) is a major metabolite of rasagiline and therefore it was of interest to investigate the effects of these products with respect to MAO A and MAO B. Although they contain either a primary or secondary amino group, they are not MAO substrates and the structure of MAO B in complex with R-MAI explains this observation. The ligand binds closer to the flavin compared to rasagiline but the distance between the inhibitor -NH-CH3 group and the N5 atom of the cofactor is too long (4.6 Å) to allow oxidation. All the investigated aminoindan molecules have been found to non-covalently inhibit MAO A and/or MAO B although to different extents and they are all weaker than rasagiline (Table 1). However, from a structural standpoint, these compounds should not be considered as “non-covalent” analogues of rasagiline. The complex with R-MAI indicates that its binding mode is substantially different, not only because it does not covalently attack the flavin but also because its aminoindan ring has a different orientation compared to the ring of rasagiline and it does not induce the opening of Ile199 and the associated cavity fusion. This notion is supported by the observation that the binding mode of R-MAI is similar to that of the non-covalent MAO inhibitor isatin. A key conclusion outlined by these studies is that the same chemical group (i.e. the indan ring) can bind in different ways depending on the nature of the substituents added to the ring, an observation often made in medicinal chemistry studies. The structure with rasagiline and its S-enantiomer revealed that the indan ring can bind in two “flipped” orientations7; the complex with R-MAI now shows that the indan ring can have a third mode of binding that is closer to the flavin and does not involve the fusion of the entrance and substrate cavities. Attempts to co-crystallize MAO B in complex with S-AI and S-MAI failed probably due to their low binding affinity. Therefore we cannot compare the binding mode of R-MAI with its S-enantiomers, but the lower inhibitory potency that is observed for the S-series of compounds with respect to their corresponding R-analogs is likely to reflect the worse fitting of the aminoindan ring when it is in the S-configuration, independently on the covalent/non-covalent inhibition or on the presence of substituents.
Conclusions
Rasagiline is a novel anti-Parkison’s drug that has shown significant advantages in clinical studies of Parkinson’s disease. It is not converted to amphetamine and the present work shows that its metabolic product (R-AI) is not a substrate for MAO oxidation but, conversely, is a weak reversible inhibitor (the relatively high Ki value suggests that it does not effectively function as a MAO inhibitor at pharmacological and clinical doses). A number of rasagiline derivatives are subject of pharmacological investigation as compounds that exert effects on multiple targets. In particular those that contain a carbamate moiety would combine the neuroprotective effect by MAO inactivation with the inhibitory activity on acetylcholine esterase, which is known to benefit patients suffering from Alzheimer’s disease. The structural details of human MAO B in complex with such analogues as well as the data obtained from the kinetic analysis provide clues that may be important for the optimization of neuroprotective drugs.
Experimental Section
Rasagiline analogues were synthesized at TEVA Pharmaceuticals as previously reported4. All other reagents used were purchased from Sigma-Aldrich.
Biochemical characterization
Human recombinant MAO A and MAO B were expressed in Pichia pastoris and purified as described previously.9,10 Both enzymes (1-2 mg) were desalted from a glycerol stock solution using a G-25 (fine) Sephadex column (1 × 20 cm, Sigma) in 50 mM potassium phosphate buffer, pH=7.5 containing 0.8% (w/v) β-octylglucoside before use. Enzymatic activity measurements, kinetics parameters determination and electrospray mass spectrometry analysis (Table 1) have been carried out as previously described.6 Briefly, MAO A and MAO B activities were determined spectrophotometrically using kynuramine (316 nm) and benzylamine (250 nm) as substrates, respectively, in 50 mM potassium phosphate buffer pH 7.5, 0.5 % (w/v) reduced Triton X-100 at 25 °C. Apparent rates of inactivation were measured following the increase in absorbance at λ=415 nm (flavocyanine adduct formation) at 15 °C using 10 μM enzyme and 1 mM inhibitor in 50 mM potassium phosphate pH 7.5, 0.8% w/v β-octylglucoside.
X-ray crystallography
Crystals of MAO B in complex with the rasagiline analogues have been prepared following published protocols.7 X-ray diffraction data were collected at the Swiss Light Source in Villigen and at the beam-line ID14-EH1 of European Synchrotron Radiation Facility in Grenoble. For data collection, crystals were transferred into a mother liquor solution containing 18% (v/v) glycerol and flash-cooled in a stream of gaseous nitrogen at 100 K. Data processing and scaling (Table 1) were carried out using MOSFLM11 and programs of the CCP4 package.12 The structure of MAO B in complex with rasagiline7 after removal of all water and inhibitor atoms provided the initial model for refinement. Unbiased 2Fo-Fc and Fo-Fc maps were used to model the inhibitors (Figure 2b) by means of the program O. 13 Crystallographic refinements were performed with the programs REFMAC514 and WARP.15 Tight non-crystallographic symmetry restraints were applied throughout the refinement calculations. Refinement statistics are listed in Table 1. Cavities were identified with the program Voidoo.16 Pictures were produced by using Bobscript17, Molscript18 and Raster3d19.
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences (GM-29433), the MIUR (FIRB and COFIN04). C.B. is supported by an Investigator Fellowship from Collegio Ghislieri, Pavia, Italy. We thank SLS and ESRF beam line groups whose outstanding efforts have made these experiments possible. Ms. M. Aldeco provided excellent technical assistance with this project. We thank Dr. M.B. Youdim for his interest in our work.
Data deposition: Coordinates have been deposited with the Protein Data Bank with ID codes XXXX, XXXX, XXXX and XXXX.
Abbreviations
- MAO
monoamine oxidase
- R-M6CPAI
6-(N-methyl-N-ethyl-carbamoyloxy)-N-methyl-N-propargyl-1(R)-aminoindan
- R-4CPAI
4-(N-methyl-N-ethyl-carbamoyloxy)-N-propargyl-1(R)-aminoindan
- R-M4CPAI
4-(N-methyl-N-ethyl-carbamoyloxy)-N-methyl-N-propargyl-1(R)-aminoindan
- R-4HPAI
4-hydroxy-N-propargyl-1(R)-aminoindan
- R-AI
1(R)-aminoindan
- S-AI
1(S)-aminoindan
- R-MAI
N-methyl-1(R)-aminoindan
- S-MAI
N-methyl-1(R)-aminoindan.
References
- 1.Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu. Rev. Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesura AM, Pletscher A. The new generation of monoamine oxidase inhibitors. Prog. Drug Res. 1992;38:171–297. doi: 10.1007/978-3-0348-7141-9_3. [DOI] [PubMed] [Google Scholar]
- 3.Rascol O, Brooks DJ, Melamed E, Oertel W, Poewe W, Stocchi F, Tolosa E, LARGO study group Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet. 2005;365:947–954. doi: 10.1016/S0140-6736(05)71083-7. [DOI] [PubMed] [Google Scholar]
- 4.Sterling J, Herzig Y, Goren T, Finkelstein N, Lerner D, Goldenberg W, Miskolczi I, Molnar S, Rantal F, Tamas T, Toth G, Zagyva A, Zekany A, Lavian G, Gross A, Friedman R, Razin M, Huang W, Krais B, Chorev M, Youdim MB, Weinstock M. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J. Med. Chem. 2002;45:5260–5279. doi: 10.1021/jm020120c. [DOI] [PubMed] [Google Scholar]
- 5.Mandel S, Weinreb O, Amit T, Youdim MB. Mechanism of neuroprotective action of the anti-Parkinson drug rasagiline and its derivatives. Brain Res. Rev. 2005;48:379–387. doi: 10.1016/j.brainresrev.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 6.Hubalek F, Binda C, Li M, Herzig Y, Sterling J, Youdim MBH, Mattevi A, Edmondson DE. Inactivation of purified human recombinant monoamine oxidases A and B by rasagiline and its analogues. J. Med. Chem. 2004;47:1760–1766. doi: 10.1021/jm0310885. [DOI] [PubMed] [Google Scholar]
- 7.Binda C, Hubalek F, Li M, Herzig Y, Sterling J, Edmondson DE, Mattevi A. Crystal structures of monoamine oxidase B in complex with four inhibitors of the N-propargylaminoindan class. J. Med. Chem. 2004;47:1767–1774. doi: 10.1021/jm031087c. [DOI] [PubMed] [Google Scholar]
- 8.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. U S A. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, Hubálek F, Newton-Vinson P, Edmondson DE. High-Level Expression of Human Liver Monoamine Oxidase A in Pichia pastoris: Comparison with the Enzyme Expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 2002;24:152–162. doi: 10.1006/prep.2001.1546. [DOI] [PubMed] [Google Scholar]
- 10.Newton-Vinson P, Hubálek F, Edmondson DE. High-level expression of human liver monoamine oxidase B in Pichia pastoris. Protein Expr. Purif. 2000;20:334–345. doi: 10.1006/prep.2000.1309. [DOI] [PubMed] [Google Scholar]
- 11.Leslie AGW. Integration of macromolecular diffraction data. Acta Crystallogr. 1999;D55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 12.Collaborative Computational Project The CCP4 Suite: Programs for protein Crystallography. Acta Crystallogr. 1994;D50:760–767. doi: 10.1107/S0907444994003112. Number 4. [DOI] [PubMed] [Google Scholar]
- 13.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 14.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 15.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP’s model-building algorithms. I. The main chain. Acta Crystallogr. 2002;D58:968–75. doi: 10.1107/s0907444902005462. [DOI] [PubMed] [Google Scholar]
- 16.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. 1994;D50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 17.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. 1999;D55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 18.Kraulis PJJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 19.Merritt EA, Bacon DJ. Raster3D: Photorealistic Molecular Graphics. Methods in Enzymology. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]