

Abstract
The cytotoxic, anti-proliferative and apoptotic effects of 3-Bromoacetoxy Calcidiol (B3CD), a derivative of vitamin D3 precursor calcidiol, on human neuroblastoma (NB) cells were examined. NB, predominantly a tumor of early childhood, is the most common extracranial solid tumor. Despite aggressive treatments, survival for advanced stages remains low and novel treatment strategies are needed. B3CD-induced apoptosis in various neuroblastic cells via caspases-3 and -9 activation. B3CD upregulated mitochondrial pro-apoptotic Bax and anti-apoptotic Bcl-2 expression, caused cytochrome c release, downregulated N-Myc expression and activated pro-survival marker Akt. Accordingly, B3CD treatment dose dependently reduced the viability of NB cells with IC50 values between 1 and 3 μM. The cytotoxicity of B3CD was significantly higher than for the calcemic parent-compound vitamin D3 (IC50 between 10 and 30 μM). Further studies revealed that B3CD treatment inhibits the proliferation of NB cells at low concentrations (IC50 between 30 and 100 nM). Cell cycle analysis showed a dramatic increase in the apoptotic sub-diploidal population along with a cell cycle block. In summary, the present study shows that B3CD is toxic to NB cells via suppression of cell proliferation and cell viability by caspase activation and regulation of survival signals. These results suggest that B3CD could be developed as a treatment for NB.
Keywords: chemical biology, drug design, kinases, signal transduction
Neuroblastoma (NB), predominantly a tumor of early childhood, is the most common extracranial solid tumor. Two-thirds of the cases occur in children below the age of five. NB account for 7–10% of all childhood cancers; in the majority of patients older than 1 year of age, the disease is fatal (1). There are approximately 500–1000 new cases of NB in the USA each year (2). Treatment methods currently available include surgery, radiation therapy, chemotherapy, and autologous stem cell transplantation (3–5) either alone or in combination, depending on the location and biological characteristics of the cancer cells, stage and the risk group to which the patient belongs. However, despite intensive multimodality treatment, more than 50% of children with high-risk disease relapse, because of drug-resistant residual disease (6–8). Eradication of refractory microscopic disease remains one of the most significant challenges in the treatment of the high-risk NB and innovative treatments need to be designed.
Calcitriol/vitamin D3 (1,25-dihydroxy-vitamin D3) (Figure 1), an endocrine hormone responsible for calcium and mineral homeostasis, inhibits cell proliferation and induces differentiation, via binding to the vitamin D receptor (VDR) (9). Its clinical use in cancer therapy is limited by its hypercalcemic side-effects (10–12). Synthetic analogs of calcitriol/vitamin D3 have been developed to overcome these side-effects. However, secondary to hypercalcemia their therapeutic efficacy remains limited (13). The natural precursor to calcitriol/vitamin D3 is calcidiol, the precursor of the latter is cholecalciferol (Figure 1). Calcidiol, found abundantly in serum, is a substrate to 1,α-vitamin D hydroxylase and biologically inactive both in terms of binding to the VDR and transcription regulation (14). Because the production of calcitriol/vitamin D3 by renal 1α-vitamin D hydroxylase is under tight transcriptional control, an elevation in serum calcidiol level does not necessarily translate into higher serum calcitriol/vitamin D3 or calcium levels (15). Consequently, calcidiol does not suffer from lethal calcemic side-effects, even at supra-physiological doses (16). Past studies led to the development of bromoacetoxy-calcidiol (B3CD; Figure 1), a bromoacetoxy-ester derivative of calcidiol, which exerted potent selective anti-proliferative effects on prostate cancer cells (17–19). Bromoacetoxy analogs generally display an improved pharmacologic profile, exert less toxicity and greater stability compared with their parent compounds (20,21). For B3CD, a systemic study in CD-1 mice showed that this calcidiol analog did not raise serum-calcium nor exhibit toxicity (166 μg/kg, repeated intraperitoneal administration) unlike other synthetic vitamin D3 derivatives (17, and references therein). Moreover, bromoacetic acid, a possible metabolite after potential B3CD administration in clinically relevant doses, is unlikely to reach toxic concentrations; the LD50 of bromoacetic acid in male rats is several hundred-fold higher (87.8 mg/kg)a. The objective of the present study was to investigate the therapeutic potential of B3CD to treat NB by analyzing the cytotoxic, anti-proliferative, and apoptotic effects of B3CD, on human NB cell lines.
Figure 1.
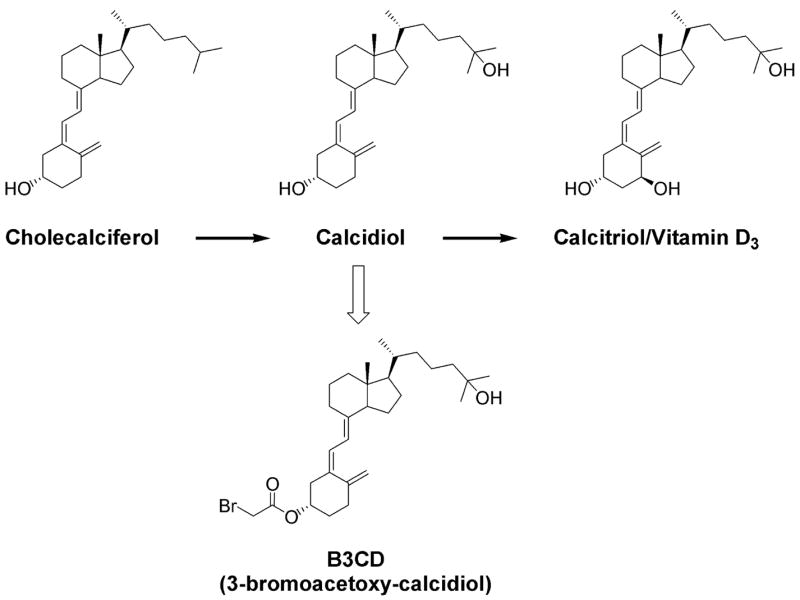
Structure of cholecaliferol, calcidiol, calcitriol/vitamin D3 and derivative B3CD.
Materials and Methods
Synthesis of B3CD
A procedure described earlier, with suitable modifications was used to synthesize B3CD (22,23). Briefly, equimolar amounts of calcidiol and bromoacetic acid were stirred with excess of dicyclohexylcarbodiimide and dry pryridine in dichloromethane in an ice bath for 2–4 h. Our modifications entail preparative high performance liquid chromatography (HPLC; Waters, Milford, MA, USA) using a C18 Luna column (4.6 ×150 mm, 5 μm; Phenomenex (Torrance, CA, USA) of B3CD followed by 1H NMR and Mass spectroscopy characterizationb.
Cell culture
SH-SY5Y (human NB) and IMR-32 (human NB) cells were obtained from American Type Culture Collection (Manassas, VA, USA). SMS-KCNR (human NB) cells were provided by John Maris (CHOP, Philadelphia, PA, USA). All cells were seeded at 5 × 105/T75 flask (Corning, Inc., Corning, NY, USA) and cultured to ~80% confluency according to the suppliers recommendations at 37 °C, 5% CO2, in a humidified incubator.
Cell viability assay
Viability of various NB cell lines was determined by the CellTiter 96® AQueous One Solution Assay (Promega Corp., Madison, WI, USA) following the manufacturer’s recommendations. An ELISA plate reader (Thermo Labsystems, Waltham, MA, USA) allowed quantification of this colorimetric assay [conversion of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium ‘MTS’ reagent in the presence of phenazine methosulfate into a soluble formazan product] at 490 nM. Briefly, cells (5 × 103 per well) were plated into 96-well flat-bottom plates (Corning, Inc.) and allowed to attach overnight before treatment with various drugs as indicated (see ‘Result’ section) in FCS-free medium. Stock solutions of drugs dissolved in DMSO as well as these vehicles alone (control) were serially diluted in serum-free medium and added to the wells. Following incubation at 37 °C in a cell-culture incubator for 20 h, MTS reagent was added at a 1:10 dilution to the medium. The samples were incubated for an additional 4 h before absorbance was measured at 490 nM. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X ± SD) of a representative experiment in % of absorbance in samples with untreated cells [100%].
Western blot analysis
Cells were seeded into 100-mm2 tissue culture dishes (5 × 105 cells per dish), cultured to ~80% confluency, treated in serum-free medium with B3CD as indicated (see ‘Result’ section), rinsed in PBS, pH 7.4, scraped off, spun down in a microcentrifuge (10 000 × g, 5 min) and pellets resuspended in lysis buffer (1% NP-40, 20 mM Tris pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM activated sodium orthovanadate, 10 μg/mL Aprotinin, and 10 μg/mL Leupeptin, Inhibitor Cocktail P-2714; Sigma–Aldrich, St Louis, MO, USA). Lysates were rocked at 4 °C for 5 min, sonicated (10 pulses 5 seconds), centrifuged at 140 000 ×g for 10 min, and the protein concentration of the supernatant quantitated (Bio-Rad protein estimation kit; Bio-Rad, Hercules, CA, USA). The samples were boiled in the presence of 5 ×SDS-PAGE sample buffer and 50 μg total protein per lane were separated on 12% SDS-polyacrylamide gels and blotted onto PVDF membranes. The blots were blocked with 5% non-fat dry milk in PBST for 1 h at room temperature and incubated overnight at 4 °C with the antibodies against various caspases (Cell Signaling Technology, Beverly, MA, USA), Bcl-2 or Bax (BD Pharmigen, San Jose, CA, USA), N-Myc (Stratagen, La Jolla, CA, USA) or cleaved PARP-1, beta-actin, phosphorylated Akt, Akt, cytochrome c (Cell Signaling Technology) at a 1:1000 dilution in 5% BSA in PBST on a rotating platform. After washing in PBST, the blots were incubated with secondary antibody (peroxidase-conjugated antibodies; Amersham-Pharmacia Biotech, Piscataway, NJ, USA). The bands were visualized by enhanced chemiluminescence (Upstate, Waltham, MA, USA) and documented using the ChemiDocTM XRS System (Bio-Rad).
For cytochrome c determination, NB cells were suspended in 0.5 mL of ice-cold buffer, containing 20 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.1 mM phenylmethylsulfonyl fluoride, 10 μg/mL Aprotinin, 10 μg/mL Leupeptin, and 20 mM sucrose. After standing on ice for 30 min, the cells were disrupted by stroking 40 times in a glass homogenizer. The nonlysed cells and nuclei were spun down at 1000 ×g for 10 min at 4 °C. To pellet mitochondria, the resulting supernatant was centrifuged at 10 000 ×g for 30 min and was resuspended in HEPES buffer. The supernatant was spun at 100 000 ×g for 1 h. The supernatant from this final centrifugation represents the cytosolic fraction.
Cell proliferation assay
Proliferation of various cell lines was determined by a BrdU (5-bromo-2′-deoxyuridine) incorporation assay (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s recommendations. Briefly, cells (5 ×103 per well) were plated into 96-well flat-bottom plates (Corning, Inc.) and allowed to attach overnight before treatment with B3CD (see ‘Result’ section) for 18 h in FCS-free medium. BrdU (10 μM final concentration) was added to the cells grown for a further 6 h. After washing, the cells were fixed and incubated for 2 h at 37 °C with an anti-BrdU antibody-peroxidase conjugate. Immune complexes were detected by addition of a tetramethyl-benzidine (TMB) substrate solution according to the manufacturer’s recommendations. The reaction was stopped by adding 50 μL of 1 M sulfuric acid, and the absorbance was measured with an ELISA plate reader (Thermo Labsystems) at 450 nM. In this assay, the color intensity correlates directly to the amount of BrdU incorporated into the DNA, which in turn represents proliferation. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X SD) of a representative experiment in % of absorbance of samples with untreated cells [100%].
DNA fragmentation analysis
Nuclear DNA fragments were isolated by using Apoptotic DNA Ladder Kit (Roche Molecular Biochemicals, Indianapolis, IN, USA). Cells were seeded into 100-mm2 tissue culture dishes (5 ×105 cells per dish), cultured to ~80% confluency, and treated in serum-free medium with 1 μM B3CD for 24 h. Floating and attached cells were collected, lysed in 0.5 mL of lysis buffer and fragmented DNA was extracted using a glass fiber spin column provided by the manufacturer. DNA was analyzed using 1.8% agarose gel in the presence of 0.5 μg/mL ethidium bromide, alongside a DNA size standard (Bio-Rad). Apoptotic DNA fragment ladders were detected on a UV transilluminator and photographed on a Bio-Rad, Gel Document System, GDS 8000 (Bio-Rad). All experiments were performed in triplicate.
Cell cycle analysis (by FACS)
Cell cycle analysis and quantification of apoptosis was carried out by flow cytometry. Cells were seeded into 100-mm2 tissue culture dishes (7.5 ×105 cells per dish), allowed to attach overnight, and treated for 48 h with B3CD (300 nM or 3.0 μM). At the end of the incubation period, detached cells were collected in 15 mL polypropylene centrifuge tubes along with the medium; culture dishes were washed once with PBS, adherent cells scraped off and combined in the same tube. After centrifugation (250 g, 5 min), cells were fixed and permeabilized with 70% ice-cold ethanol for 30 min, followed by incubation with 50 μg/mL of propidium iodide and 100 μg/mL of RNase for 30 min at 37 °C in the dark. Data were acquired on a BD FACSort flow cytometer using CELLQUEST software (BD Immunocytometry Systems) and analyzed using MODFIT LT software (Verity Software House, Inc., Topsham, ME, USA). Ten thousand events were analyzed for each sample. Appropriate gating was used to select the single cell population NB cells. The same gate was used on all samples, ensuring that the measurements were made on a standardized cell population.
Results and Discussion
The present study investigates the cytotoxic, anti-proliferative, and apoptotic effect of B3CD, a derivative of the natural vitamin D3 precursor calcidiol on various human NB cell lines. Previous work showed that B3CD at concentrations as low as 1.0 μM displayed strong growth-inhibitory effects both with respect to proliferation and viability in prostate cancer cell lines while other cancer cells such as MCF-7 (breast cancer) or primary keratinocytes were significantly less affected (17,18). Other investigators have confirmed that, similarly, the calcemic parent compound calcitriol/vitamin D3 acts differentially on distinct cell, tissue, and tumor types (24–27).
Viability of human NB cell lines after B3CD or calcitriol/vitamin D3 treatment
In an initial approach to analyze the cytotoxicity of B3CD on several NB cell lines (stromal S-type SMS-KCNR, neuronal N-type SH-SY5Y, and IMR-32), some of which are representative of the type-3 childhood NB, we performed a colorimetric viability assay. The assay is based on the ability of a cell’s mitochondria to reduce the substrate (MTS) and the absorbance of the resulting product is directly proportional to the number of living cells in culture (see ‘Materials and Methods’ section). B3CD treatment for 24 h dose dependently reduced the viability of SMS-KCNR, SH-SY5Y, and IMR-32 NB cells (Figure 2, left panel). Viability was slightly reduced at concentrations of B3CD as low as 100 nM, reduced by 20–25% at 300 nM with IC50 values between 1 and 3 μM for all NB cell lines. B3CD exerted a 10-fold stronger effect on NB cell viability than the calcemic parent compound calcitriol/vitamin D3 (Figure 2, right panel; IC50 between 10 and 30 μM). Similarly, calcitriol/vitamin D3 in previous studies at μM concentrations did not induce growth-inhibitory effects in prostate cancer cell lines (17,18).
Figure 2.
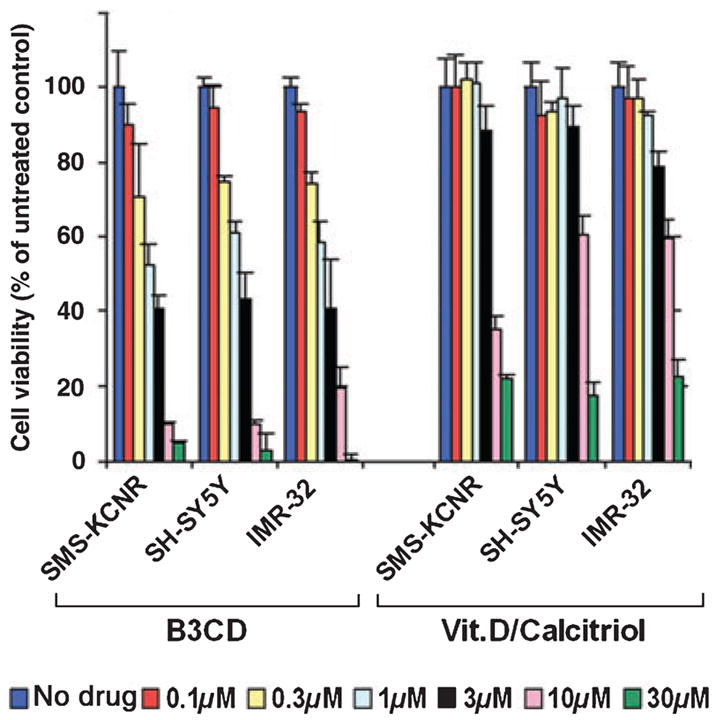
Comparative analysis of the cytotoxic effect of B3CD and calcitriol/vitamin D3 on various human NB cell lines. NB cells (SMS-KCNR, SH-SY5Y, and IMR-32) were treated with various concentrations (0.1–30 μM) of B3CD and calcitriol/vitamin D3 for 24 h. The MTS viability assay was carried out as described in ‘Materials and Methods’ section. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X SD) of a representative experiment in % cell viability of untreated cells [100%].
B3CD induces apoptosis in NB cells via the intrinsic pathway
To define the cellular response upon B3CD treatment of NB cells, we next analyzed the expression and/or activation of cellular markers that are characteristics for induction of apoptosis by immunblotting (see ‘Materials and Methods’ section). Apoptosis is executed by caspases: initiator caspases (such as caspase-2, -8, -9, and -10) function mainly as upstream apoptotic signals. Once activated, the initiator caspases cleave and activate downstream effector caspases (such as caspase-3, -6, and -7), which are responsible for the cleavage of many intracellular proteins, leading to the morphological and biochemical changes associated with apoptosis (28,29).
As shown in Figure 3A, the treatment of NB cells with 100 nM to 2 μM B3CD resulted in a dose-dependent activation of procaspase-3 (17 and 19 kD intermediates). Similarly, B3CD activated caspase-9 (35 kD intermediate) in all the three NB cell lines. In contrast to caspase-9, the caspase-8 pathway was not activated, although pro-caspase-8 was expressed in SMS-KCNR cells (Figure 3A). Two major signaling pathways have been described for activation of initiator caspases in mammalian cells. The intrinsic pathway mediates apoptotic responses to various stress signals such as DNA damage, hypoxia, and growth factor deprivation. These signals lead to the activation of pro-apoptotic members of the Bcl-2 family, resulting in mitochondrial release of cytochrome c and other pro-apoptotic proteins (30,31). The extrinsic pathway is initiated by interaction of specific ligands with their corresponding ‘death’ receptors such as Fas, TNF receptor-1, and TRAIL resulting in receptor oligomerization and caspase-8 activation. Activated caspase-8 can process downstream effector caspases directly, leading to apoptosis (26,30,32). It is interesting to note that for certain cell types, however, the death-receptor induced apoptosis requires a mitochondrial pathway controlled by Bcl-2 family proteins (33).
Figure 3.
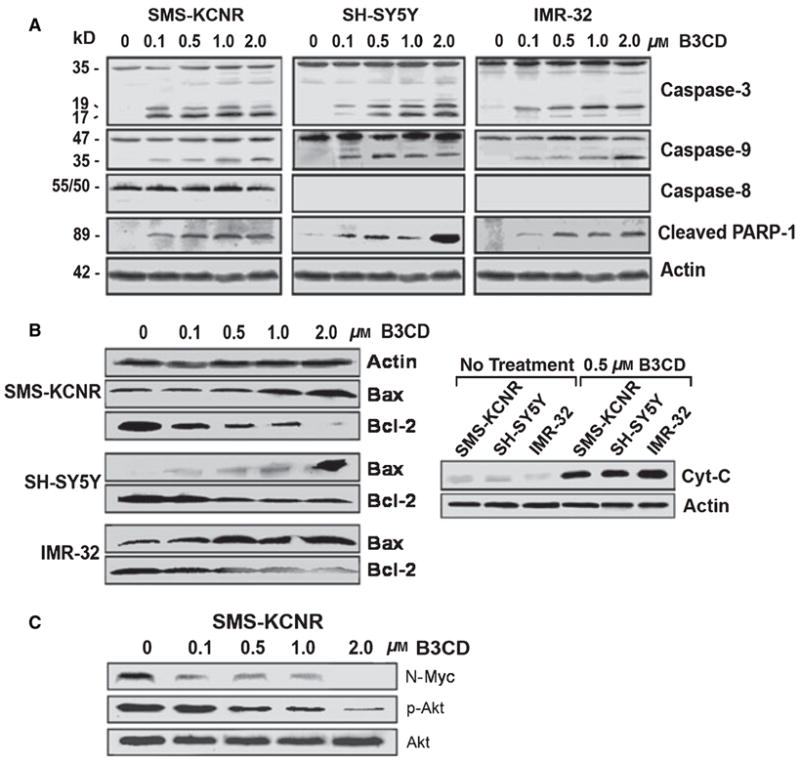
Expression of apoptotic and pro-survival markers in NB cells after B3CD treatment. NB cells (SMS-KCNR, SH-SY5Y, and IMR-32) were treated with increasing concentrations (0.1–2.0 μM) of B3CD for 48 h. Expression of proteins in the lysates of treated and untreated cells by PAGE and Western blot analysis was carried out as described in ‘Materials and Methods’ section. (A) Caspase activation; primary antibodies against pro- and activated caspase-3, -8, -9, and inactivated/cleaved PARP-1. As an internal standard for equal loading, blots were probed with an anti-beta-Actin antibody. (B) Mitochondria-mediated apoptosis; primary antibodies against cytochrome c, Bax, and Bcl-2. (C) Inhibition of N-MYC expression and Akt activation; primary antibodies against N-MYC, Akt, and phosphorylated Akt.
Our observations suggest that the extrinsic pathway is not involved in the B3CD-induced apoptosis of SH-SY5Y, SMS-KCNR, and IMR-32 NB cells and B3CD likely induces apoptosis via activation of the intrinsic pathway. Thus, treatment with B3CD can bypass the reported resistance of certain NB cell lines to apoptosis because of the lack of caspase-8 activation or expression (34). In addition to analysis of caspase activation, a commonly used method for detection of apoptosis is the presence of cleaved Poly-ADP Ribose Poly-merase-1 (PARP-1) in many cell types (35). Accordingly, after B3CD treatment, we observed a dose-dependent increase of cleaved PARP-1 (89 kD) in all the three NB cell lines (Figure 3A) because of caspase action (36).
As the intrinsic pathway of apoptosis leads to the activation of members of the Bcl-2 family and mitochondrial release of cytochrome C (33.39), we analyzed the expression of Bax and Bcl-2 in B3CD-treated NB cells. Bcl-2 promotes cell survival, whereas Bax is a pro-apoptotic factor (37). Immunblotting of the lysates of SMS-KCNR, SH-SY5Y, and IMR-32 revealed an increase in Bax expression at concentrations of B3CD as low as 100 nM (48 h treatment), reaching a maximum level at 2.0 μM (Figure 3B). In contrast, the levels of Bcl-2 protein were significantly reduced following treatment with B3CD (Figure 3B). These studies indicate that B3CD enhances expression of the pro-apoptotic factor Bax, while simultaneously suppressing anti-apoptotic factor, Bcl-2, thereby efficiently inducing apoptosis in NB cells. Cancer cells with augmented Bcl-2 are resistant to cytotoxic drugs and, thus, Bcl-2 proteins are increasingly targets for new anticancer drugs (38). The balance between the anti-apoptotic Bcl-2 and death-promoting Bax controls mitochondrial permeability changes and the release of cytochrome c, which is an early event in the apoptotic process, preceding morphological signs of apoptosis (39,40). Treatment with 500 nM B3CD for 48 h also resulted in an increase in cytosolic cytochrome C in all the three NB cell lines tested (Figure 3B).
Suppression of pro-survival markers in NB cells by B3CD treatment
To elucidate the effect of B3CD on pro-survival signaling in NB, the activation/phosphorylation of a hallmark survival factor, Akt, as well as expression of N-Myc was investigated. Akt is a proteine–serine/threonine kinase and when activated targets the phosphorylation of regulators of cell survival, transcription factors, and other kinases cells (41,42). N-Myc is a transcription factor that is expressed in the brain and peripheral nervous system and plays an important role in survival of aggressive NB in which caspase-8, component of the death receptor pathways, is inactivated (43).
Immunblotting of lysates of SMS-KCNR was carried out using antibodies that specifically recognize N-Myc or the phosphorylated or inactive form of Akt. We did not observe a regulation in the expression of inactive Akt by B3CD concentrations up to 2.0 μM (Figure 3C). In contrast, activated/phosphorylated Akt, which is in untreated cells was highly expressed after B3CD treatment (100 nM to 2.0 μM, 48 h) dose dependently decreased to background levels (Figure 3C). B3CD also inhibited the expression of N-Myc. No expression was seen at 2.0 μM B3CD (Figure 3C). Together, these studies clearly show that B3CD suppressed the oncogenic transcription factor N-Myc as well as Akt-mediated pro-survival signaling in SMS-KCNR NB cells. These observations are of particular interest for the potential therapeutic use of B3CD as Akt activation has previously been linked to drug resistance in NB cells (42). Similarly, N-Myc is a potential target for drug development, as N-Myc has been reported to be overexpressed in more than 65% of human NB (44).
B3CD effect on cell proliferation and cell cycle progression
As described in the previous section, B3CD acts as a cytotoxic drug and leads to expression and/or activation of apoptotic markers linked to the intrinsic pathway of apoptosis. To investigate if B3CD affects the proliferation of NB cells (particularly at drug concentrations at or below 300 nM when viability is not affected or partially reduced; Figure 2), we performed BrdU incorporation assays and cell cycle analysis. Cell cycle analysis of SMS-KCNR NB cells after B3CD treatment revealed an increase in the sub-diploidal apoptotic population at 300 nM (23.7% apoptotic) with 55% of the population in an apoptotic state at 3.0 μM B3CD (Figure 4A). This observation directly correlates with the reduction of SMS-KCNR viability by B3CD at the same concentrations (Figure 2). The sub-diploidal population represents cells with significant DNA damage. DNA fragmentation analysis (Figure 4B) proved that DNA in chromatin of cells treated with B3CD degraded into oligonucleosome-length fragments (DNA laddering, n ×123 bp) characteristic for apoptosis, whereas DNA smear indicating necrotic events was not observed (45).
Figure 4.
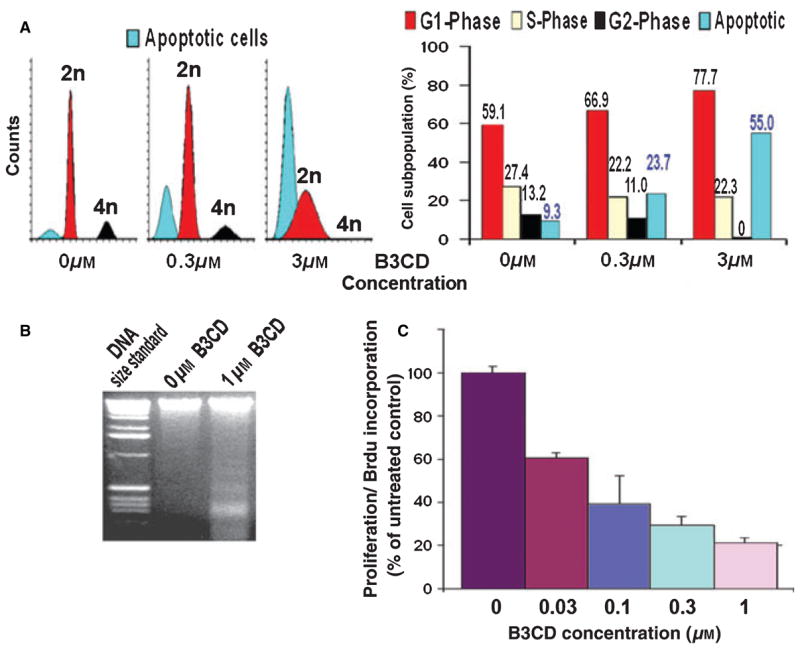
B3CD effect on SMS-KCNR NB cell proliferation and cell cycle progression. (A) Cell cycle analysis by FACS: NB cells were treated with 300 nM or 3.0 μM B3CD for 48 h. Cell cycle analysis by FACS of treated and untreated cells followed was carried out as described in ‘Materials and Methods’ section. Data are presented as the relative fluorescence intensity of cell sub-populations in the two-dimensional FACS profile (left panel) or bar diagram (right panel). (B) DNA fragmentation: NB cells were treated with 1 μM B3CD for 24 h. DNA fragmentation assay was carried out and apoptotic DNA fragment ladder (n ×123 bp) detected as described in ‘Materials and Methods’ section. (C) BrdU incorporation: NB cells were treated with various concentrations (30 nM to 3.0 μM) of B3CD for 24 h. The proliferation assay was carried out as described in ‘Materials and Methods’ section. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X SD) in % cell proliferation of untreated cells [100%].
With respect to the cycling cells, B3CD at 300 nM causes a prominent increase of G0/G1 population along with slight decrease of cells in S-phase or G2/M phase (Figure 4A). Treatment of SMS-KCNR with 3.0 μM B3CD for 48 h fully blocked cell cycle progression with no cells left in the G2/M phase. Accordingly, the majority of the cycling cells were arrested in G0/G1 phase (Figure 4A) while the S-phase sub-population, compared with untreated NB cells, was decreased (to 22%; Figure 4A). Apparently, in this asynchronous cell culture, B3CD treatment affected cell cycle checkpoints in G0/G1 and S-phase. Although not the objective of the present report, further studies emphasizing cancer-related cell cycle features (46,47) could focus on the specific checkpoints in G0/G1 or S-phase affected by B3CD treatment. This would require the study of expression pattern of cell cycle regulators (cyclin-dependent kinases and cyclins) and replication-start and -progression signals of the S-phase (48,49) in synchronized NB cultures. In summary, cell cycle analysis revealed a dramatic increase in the apoptotic sub-diploidal population (severe DNA damage) after B3CD treatment along with a full block of cell cycle progression (G2/M sub-population = 0%).
The observation that B3CD treatment at concentrations of 300 nM significantly increased the apoptotic sub-population and reduced S-phase progression of NB cells (Figure 4A) correlates with the effect of B3CD on BrdU incorporation/DNA replication as analyzed in our proliferation assay (Figure 4B). B3CD dose dependently reduced SMS-KCNR proliferation. Even at low drug concentrations (30 and 100 nM), BrdU incorporation was dramatically suppressed (by 39% and 60%, respectively) compared with untreated cells (Figure 4C). Previous work showed that 1.0 μM B3CD similarly displayed anti-proliferative effects in prostate cancer while proliferation of other cell lines such as MCF-7 (breast cancer) and primary keratinocytes was less affected (17,18). More importantly, the authors also proved in an SCID mouse model that administration of B3CD did not cause any apparent systemic toxicity, weight loss, or increased serum calcium levels (17). The present report suggests that B3CD is a potent and growth-suppressing agent to cell lines derived from NB and a potential therapeutic drug to treat such tumors in vivo.
Conclusion
In summary, the present study shows that B3CD is toxic to NB cells via suppression of cell proliferation and cell viability by caspase activation, N-Myc downregulation, mitochondria-mediated apoptosis, and de-activation of survival marker Akt. Apoptosis was mediated by cas-pase-3 and -9 activation, and not by the extrinsic (caspase-8 mediated) pathway. This wide effect of the bromoacetoxy derivative of calcidiol, natural precursor to calcitriol/vitamin D3 on NB cell signaling and survival suggests that B3CD could be developed as a therapeutic agent against NB.
Acknowledgments
This work was supported by an NINDS/NIH 1R21NS051408-01A2 grant to Dr Brard. The authors also thank Dr Sunil K. Shaw for advice and technical guidance and NIH COBRE Grant 1-P20RR018728 for providing instrumentation support.
Footnotes
Notes: United Sates Environmental Protection Agency (USEPA) report (2005) Bromoacetic acid – identification, toxicity, use, water pollution potential, ecological toxicity and regulatory information. CAS number 79-08-3. http://yosemite.epa.gov
For preparative HPLC, gradient elution with two solvents at a flow rate of 1.5 mL/min was used. Solvent A consisted of 10% MeOH in H2O adjusted to pH 3.5 with formic acid. Solvent B consisted of 20% H2O (pH 3.5), 20% MeOH, and 60% acetonitrile. The gradient consisted of 0 min-90% A, 10% B followed by 15 min-0% A,100% B and held at 100% B for 20 min. Five minutes of equilibration at 100% A was performed before and after each injection. Retention time for B3CD: 15.178 min. NMR spectra were recorded on ‘Bruker Advance NMR spectrometers’ operating at 300 MHz or 400 MHz 1H frequency. Chemical shifts are referenced to CDCl3 at room temperature. Mass spectra data were obtained using either JEOL JMS600 under FAB mode or API 150 MCA under electro spray mode. 1H NMR δ6.27–6.26 (d, 1H, J = 3.0 Hz), 6.08–6.07 (d, 1H, J = 3.0 Hz), 5.41 (s, 2H, CH2Br), 5.21 (s, 1H), 4.88 (s, 1H), 2.98 (d, 2H, J = 4.2 Hz), 2.76 (d, 2H, J = 3.6 Hz), 2.67–2.31 (m, 3H), 2.19–1.76 (m, 4H), 1.65–1.21 (m, 15H), 0.61–0.28 (m, 10H), 0.23 (s, 3H); MS (ES): m/z 474 [M + H]+.
References
- 1.Brodeur GM, Maris JM. Neuroblastoma. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. 4. Philadelphia: Lippincott-Raven; 2001. pp. 895–937. [Google Scholar]
- 2.Weinstein JL, Katzenstein HM, Cohn SL. Advances in the diagnosis and treatment of neuroblastoma. Oncologist. 2003;8:278–292. doi: 10.1634/theoncologist.8-3-278. [DOI] [PubMed] [Google Scholar]
- 3.Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, Black CT, Gerbing R, Haase GM, Stram DO, Swift P, Lukens JN. Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children’s Cancer Group study. J Clin Oncol. 1998;16:1256–1264. doi: 10.1200/JCO.1998.16.4.1256. [DOI] [PubMed] [Google Scholar]
- 4.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cisretinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 5.Perez CA, Matthay KK, Atkinson JB, Seeger RC, Shimada H, Haase GM, Stram DO, Gerbing RB, Lukens JN. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: a Children’s Cancer Group study. J Clin Oncol. 2000;18:18–26. doi: 10.1200/JCO.2000.18.1.18. [DOI] [PubMed] [Google Scholar]
- 6.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 7.Goldsby RE, Matthay KK. Neuroblastoma: evolving therapies for a disease with many faces. Paediatr Drugs. 2004;6:107–122. doi: 10.2165/00148581-200406020-00004. [DOI] [PubMed] [Google Scholar]
- 8.Matthay KK, Atkinson JB, Stram DO, Selch M, Reynolds CP, Seeger RC. Patterns of relapse after autologous purged bone marrow transplantation for neuroblastoma: a Childrens Cancer Group pilot study. J Clin Oncol. 1993;11:2226–2233. doi: 10.1200/JCO.1993.11.11.2226. [DOI] [PubMed] [Google Scholar]
- 9.Kumar R. 1Alpha,25-dihydroxyvitamin D(3) – not just a calciotropic hormone. Nephron. 2002;91:576–581. doi: 10.1159/000065015. [DOI] [PubMed] [Google Scholar]
- 10.DeLuca HF, Zierold C. Mechanisms and functions of vitamin D. Nutr Rev. 1998;56:S4–S10. S54–S75. doi: 10.1111/j.1753-4887.1998.tb01686.x. [DOI] [PubMed] [Google Scholar]
- 11.Dusso AS, Brown AJ. Mechanism of vitamin D action and its regulation. Am J Kidney Dis. 1998;32(Suppl 2):S13–S24. doi: 10.1053/ajkd.1998.v32.pm9808140. [DOI] [PubMed] [Google Scholar]
- 12.Olick MF. Vitamin D: the underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr Opin Endocrinol Diabetes. 2002;9:87–98. [Google Scholar]
- 13.Reichrath J. Will analogs of 1,25-dihydroxyvitamin D(3) (calcitriol/vitamin D3) open a new era in cancer therapy? Onkologie. 1997;24:128–133. doi: 10.1159/000050299. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt-Gayk H, Bouillon R, Roth HJ. Measurement of vitamin D and its metabolites (calcidiol and calcitriol/vitamin D3) and their clinical significance. Scand J Clin Lab Invest Suppl. 1997;227:35–45. [PubMed] [Google Scholar]
- 15.Brown AJ. Mechanisms for the selective actions of vitamin D analogues. Curr Pharm Des. 2000;6:701–716. doi: 10.2174/1381612003400416. [DOI] [PubMed] [Google Scholar]
- 16.Addad JG, Abrams J, Walgate J. Affinity chromatography with 25-hydroxycholecalciferol ester in the isolation of the binding protein for vitamin D and its metabolites from human serum. Metab Bone Dis Relat Res. 1981;3:43–46. doi: 10.1016/s0221-8747(81)80022-7. [DOI] [PubMed] [Google Scholar]
- 17.Swamy N, Chen TC, Peleg S, Dhawan P, Christakos S, Stewart LV, Weigel NL, Mehta RG, Holick MF, Ray R. Inhibition of proliferation and induction of apoptosis by 25-hydroxyvitamin D3-3beta-(2)-Bromoacetate, a nontoxic and vitamin D receptor-alkylating analog of 25-hydroxyvitamin D3 in prostate cancer cells. Clin Cancer Res. 2004;10:8018–8027. doi: 10.1158/1078-0432.CCR-04-0881. [DOI] [PubMed] [Google Scholar]
- 18.Swamy N, Persons KS, Chen TC, Ray R. 1α,25-Dihydroxyvitamin D3-3beta-(2)-bromoacetate, an affinity labeling derivative of 1α,25-dihydroxyvitamin D3 displays strong antiproliferative and cytotoxic behavior in prostate cancer cells. J Cell Biochem. 2003;89:909–916. doi: 10.1002/jcb.10585. [DOI] [PubMed] [Google Scholar]
- 19.Lambert JR, Young CD, Persons KS, Ray R. Mechanistic and pharmacodynamic studies of a 25-hydroxyvitamin D(3) derivative in prostate cancer cells. Biochem Biophys Res Commun. 2007;361:189–195. doi: 10.1016/j.bbrc.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Cottens S, Sedrani R. O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants. 6440990 United States Patent. 2002
- 21.Driedger PE, Quick J. Protein kinase C modulators, E. 5886017 United States Patent. 1999
- 22.Swamy N, Ray R. Affinity labeling of rat serum vitamin D binding protein. Arch Biochem Biophys. 1996;333:139–144. doi: 10.1006/abbi.1996.0374. [DOI] [PubMed] [Google Scholar]
- 23.Warren JC, Sweet F. Synthesis and use of affinity labeling steroids for analysis of macromolecular steroid-binding sites. Methods Enzymol. 1975;36:374–410. doi: 10.1016/s0076-6879(75)36036-9. [DOI] [PubMed] [Google Scholar]
- 24.Carlberg C. Molecular basis of the selective activity of vitamin D analogues. J Cell Biochem. 2003;88:274–281. doi: 10.1002/jcb.10337. [DOI] [PubMed] [Google Scholar]
- 25.Gruber BM, Anuszewska EL. Studies on the influence of vitamin D3 metabolites on apoptosis induction in human neoplastic cells. Acta Pol Pharm. 2003;60:363–366. [PubMed] [Google Scholar]
- 26.Peleg S, Ismail A, Uskokovic MR, Avnur Z. Evidence for tissue- and cell-type selective activation of the vitamin D receptor by Ro-26-9228, a noncalcemic analog of vitamin D3. J Cell Biochem. 2003;88:267–273. doi: 10.1002/jcb.10344. [DOI] [PubMed] [Google Scholar]
- 27.Van den Bemd GJ, Pols HA, Van Leeuwen JP. Anti-tumor effects of 1,25-dihydroxyvitamin D3 and vitamin D analogs. Curr Pharm Des. 2000;6:717–732. doi: 10.2174/1381612003400498. [DOI] [PubMed] [Google Scholar]
- 28.Salvesen GS, Abrams JM. Caspase activation – stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene. 2004;23:2774–2784. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- 29.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 30.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 31.Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–453. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulda S, Friesen C, Los M, Scaffidi C, Mier W, Benedict M, NuÇez G, Krammer PH, Peter ME, Debatin KM. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulaki V, Mitsiades N, Romero ME, Tsokos M. Fas-mediated apoptosis in neuroblastoma requires mitochondrial activation and is inhibited by FLICE inhibitor protein and Bcl-2. Cancer Res. 2001;61:4864–4872. [PubMed] [Google Scholar]
- 34.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. 2001;79:428–436. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 35.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- 36.Garrido C, Kroemer G. Life’s smile, death’s grin: vital functions of apoptosis-executing proteins. Curr Opin Cell Biol. 2004;16:639–646. doi: 10.1016/j.ceb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 38.O’Neill J, Manion M, Schwartz P, Hockenbery DM. Promises and challenges of targeting Bcl-2 anti-apoptotic proteins for cancer therapy. Biochim Biophys Acta. 2004;1705:43–51. doi: 10.1016/j.bbcan.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 40.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emran MA, Rebbaa A, Mirkin BL. Doxorubicin resistant neuroblastoma cells secrete factors that activate AKT and attenuate cytotoxicity in drug-sensitive cells. Cancer Lett. 2002;182:53–59. doi: 10.1016/s0304-3835(02)00062-9. [DOI] [PubMed] [Google Scholar]
- 43.Cui H, Li T, Ding HF. Linking of N-Myc to death receptor machinery in neuroblastoma cells. J Biol Chem. 2005;280:9474–9481. doi: 10.1074/jbc.M410450200. [DOI] [PubMed] [Google Scholar]
- 44.Pession A, Tonelli R. The MYCN oncogene as a specific and selective drug target for peripheral and central nervous system tumors. Curr Cancer Drug Targets. 2005;5:273–283. doi: 10.2174/1568009054064606. [DOI] [PubMed] [Google Scholar]
- 45.Zhivotovsky B, Orrenius S. Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria. In: Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. Current Protocols in Cell Biology. New York: John Wiley and Sons, Inc; 2001. pp. 18.3.1–18.3.23. [DOI] [PubMed] [Google Scholar]
- 46.Mazumder S, DuPree EL, Almasan A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr Cancer Drug Targets. 2004;4:65–75. doi: 10.2174/1568009043481669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gladden AB, Diehl JA. Cell cycle progression without cyclin E/CDK2: breaking down the walls of dogma. Cancer Cell. 2003;4:160–162. doi: 10.1016/s1535-6108(03)00217-4. [DOI] [PubMed] [Google Scholar]
- 48.Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol. 1999;1:73–79. doi: 10.1038/11041. [DOI] [PubMed] [Google Scholar]
- 49.Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]