

Abstract
Several laboratories have demonstrated that activation of drug metabolism by P450s may occur via a mechanism that resembles allosterism from an enzyme kinetic standpoint. Because the effector drug binding site may be located in the same P450 binding pocket where the drug substrate is located, the ability to find and characterize novel effectors (a.k.a. heteroactivators) will prove to be important in probing the mechanism of activation. We have used analogs of the prototypical CYP2C9 heteroactivator dapsone to validate a simple docking method that can be used to predict heteroactivators based on ligand binding location in a P450 crystal structure. As proof of concept for the described docking method, a protocol was developed to discover potential heteroactivators from a virtual chemical library through efficient sorting of >40,000 compounds. One of the top-scoring compounds identified was verified to be a CYP2C9 heteroactivator in vitro and it possessed activity similar to dapsone.
Keywords: cytochrome P450, heteroactivation, effector, virtual screen, high-throughput, docking
As the most abundant enzymes involved in the oxidative metabolism of xenobiotics in the liver, the cytochromes P450 aid in drug clearance1. Therefore, the rate of P450-mediated drug metabolism will affect the dosing regimen of a drug, and possibly, efficacy and/or toxicity. Upon dosing of multiple drugs whose metabolism is carried out by the same P450 isoform, a drug-drug interaction may result via inhibition of metabolism2;3. More recently, evidence of drug-drug interactions that result in an increase in P450 metabolism in vitro have been described4;5. The enzyme kinetic profiles in these cases are most analogous to allosterism; however, it has been shown that the steric interactions that lead to this heteroactivation can occur via the simultaneous binding of a drug substrate and a second drug that acts as an effector, to the same P450 enzyme active site6–8 (Scheme 1). Yet, the precise mechanisms by which multiple ligand binding alters the P450 catalytic cycle to increase the oxidation rate of a drug are still under investigation. The discovery of novel heteroactivators of P450 metabolism could be used to study the mechanisms of heteroactivation and to further dissect distinct steps in the P450 catalytic cycle9. Unfortunately, finding new classes of activators is challenging due to the highly substrate-dependent nature of P450 heteroactivation.
Opportunities might also exist in drug development to characterize heteroactivation. While one study demonstrated that the magnitude of drug-drug interactions resulting from CYP2C9 activation appears to be more modest in vivo10, in principle, their prediction by means of a rapid in silico screen might be useful as a drug interaction screen or in the development of therapeutic compounds. While these ideas are more theoretical, more dramatic cases of heteroactivation of CYP2C9 have since been since reported. Lansoprazole has been shown to activate phenytoin hydroxylation by recombinant CYP2C9 ~10-fold11 and both of these compounds are marketed drugs. Even if a fraction of this activation was observed in vivo, it might dramatically lower the efficacy of a narrow therapeutic index drug such as phenytoin, or act to rescue overdose.
Herein, we describe a rapid docking methodology that can be used to screen virtual chemical libraries for novel P450 heteroactivators. In principle, the high-throughput filtering of drug-like molecules in silico could be used to quickly find ligands that bind to a biological target12. The method described here was developed using a representative substrate-effector system, namely CYP2C9-flurbiprofen-dapsone. In practice, the approach required a single molecular dynamics simulation with the substrate- and effector-bound P450 to generate a suitable docking receptor, followed by two rounds of automated docking. Despite the use of only a single enzyme structure, the docking methodology described here identified a new heteroactivator of CYP2C9 that had potencies similar to the prototypical CYP2C9 activator dapsone. Moreover, this new compound that tested positive for heteroactivation in vitro was structurally distinct from the control group of dapsone analogs. Hence, one major advantage to library screening is that chemical diversity is generated without the need for chemical synthesis, though confirmed hits resulting from a screen could be used to guide further synthesis and quantitative structure-activity relationship analysis. It is expected that most commercial or non-commercial docking program can be used for screening small molecule P450 effectors.
Results and Discussion
The heteroactivation of P450-mediated drug metabolism has been shown, in some cases, to result from the simultaneous binding of a drug substrate and an effector13–15. Though analogous to allosterism, the effect is thought to occur from substrate and effector binding to the same enzyme active site pocket. Furthermore, it is likely that simultaneous effector binding is not the only requirement for stimulating the P450 reaction rate. Dual ligand binding may change active site solvation, which is involved in protonating P450 iron-oxygen catalytic cycle intermediates. Herein, we describe in silico docking methods aimed at discovering novel heteroactivators of P450 drug metabolism to be used as probes in mechanistic studies of P450 activation, but also as a potential screens for drug-drug interactions.
Validation of the docking methodology
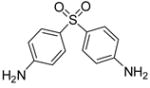
First, a data set of CYP2C9 heteroactivators with a range of biological activities was needed. Analogs of dapsone (4,4′-diaminophenylsulfone) were chosen because dapsone is the prototypical heteroactivator of non-steroidal antiinflammatory drugs by CYP2C9, a few analogs were already known to activate CYP2C9 to different degrees16, and variations of the dapsone scaffold were commercially available. In vitro screening was carried out with a single substrate concentration while the concentration of dapsone analog was varied. The degree of activation is modestly altered by substrate concentration while the concentration of heteroactivator will determine whether no effect or activation results17, or even if inhibition results as documented with some activators at high concentrations18. Analogs of dapsone were discovered that were both more and less effective activators of (S)-flurbiprofen hydroxylation, or inhibitors of (S)-flurbiprofen hydroxylation (Table 1). In total, nineteen compounds were screened for their ability to alter the rate of (S)-flurbiprofen metabolism in vitro.
Table 1.
Known activators and inhibitors of CYP2C9-mediated (S)-flurbiprofen hydroxylation and validation of docking to predict activators vs. inhibitors
| # | 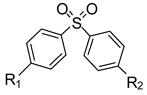 |
in vitro activation (%)a | GlideScore with no substrate present | Binding siteb | Prediction | False positive | False negative |
|---|---|---|---|---|---|---|---|
| 1 | R1=OH, R2=OH | 95.0 | −7.44 | Effector | Activator | ||
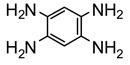
| 2 | R1=NH2, R2=NH2 | 64.1 | −7.69 | Effector | Activator | ||
| 3 | R1=CH3, R2=CH3 | 50.6 | −6.50 | Effector | Activator | ||
| 4 | R1=H, R2=H | 40.0 | −7.00 | Effector | Activator | ||
| 5 | R1=acetyl, R2=acetyl | 8.0 | −7.54 | Substrate | Inhibitor | X | |
| 6 |
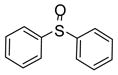
|
3.5 | −6.13 | Effector | Activator | ||
| 7 |
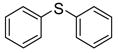
|
No effect | −4.76 | Inhibitor | |||
| 8 |
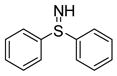
|
No effect | −6.95 | Substrate | Inhibitor | ||
| 9 | R1=COOH, R2=COOH | −5.0 | −7.74 | Substrate | Inhibitor | ||
| 10 |
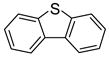
|
−10.6 | −5.86 | Inhibitor | |||
| 11 | R1=NO2, R2=NH2 | −10.6 | −7.67 | Effector | Activator | X | |
| 12 |
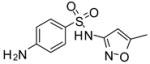
|
−17.1 | −6.47 | Substrate | Inhibitor | ||
| 13 |
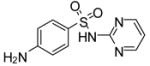
|
−17.1 | −7.34 | Substrate | Inhibitor | ||
| 14 |
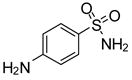
|
−17.8 | −5.79 | Inhibitor | |||
| 15 |
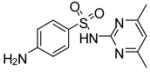
|
−18.8 | −8.18 | Effector | Activator | X | |
| 16 | R1=OCH3, R2=OCH3 | −25.0 | −6.65 | Substrate | Inhibitor | ||
| 17 |
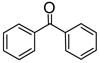
|
Inhibitorc | −5.79 | Inhibitor | |||
| 18 |
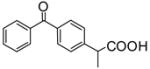
|
Inhibitorc | −6.85 | Substrate | Inhibitor | ||
| 19 |
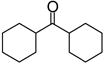
|
Inhibitorc | −7.00 | Substrate | Inhibitor |
Activation of 4′-OH flurbiprofen formation by reconstituted CYP2C9; negative numbers reflect percent inhibition
Activators were classified as compounds that docked at the effector site (described in the text) in at least one of the top three saved conformations; compounds with scores below the weakest activator (compound 6) were classified as inhibitors or as having no effect.
See reference39
Next, a docking protocol was developed to determine whether the known dapsone heteroactivators could be distinguished from dapsone analogs that inhibited CYP2C9 metabolism of (S)-flurbiprofen. However, a CYP2C9 enzyme receptor suitable for multiple ligand binding had to be generated. This is only now feasible given the availability of a CYP2C9-flurbiprofen complex crystal structure19, which differs from other mammalian structures significantly in the substrate recognition sequence regions20, including the other available CYP2C9 structures.
The complex crystal structure of CYP2C9-flurbiprofen indicated a binding mode for substrate (S)-flurbiprofen that places the hydroxylation site of (S)-flurbiprofen directly over the distal face of the heme. Though other binding conformations for (S)-flurbiprofen may exist, it seems likely metabolism would occur from the conformation suggested by the electron density. Therefore, the remaining active site volume located further away from the distal side of the heme prosthetic group is likely to be at least one of the effector binding sites in CYP2C9, but in the original crystal structure, may be too limited (223 Å3) to accommodate some potential heteroactivators based on the sizes of known activators amiodarone21 and delta-9-Tetrahydrocannabinol (Δ9-THC)22 (Fig. 1a). Hence, the CYP2C9 receptors used in docking were acquired from an equilibrated molecular dynamics trajectory that included both (S)-flurbiprofen and the prototypical (S)-flurbiprofen heteroactivator dapsone9. In this structure the volume of the unoccupied regions of the CYP2C9 active site was increased three-fold to 711 Å3. This degree of change is striking, but the movement of just a few residues creates a continuous pocket that extends from the heme group all the way to the substrate entrance channel near the F-G loop region. In addition, according to the documented variation in active site volumes of CYP3A4 whose volume approximately doubles upon binding of large substrates23, it is becoming more apparent that the large degree of P450 flexibility is integral in conferring the ability to bind a diverse array of drugs.
Figure 1.
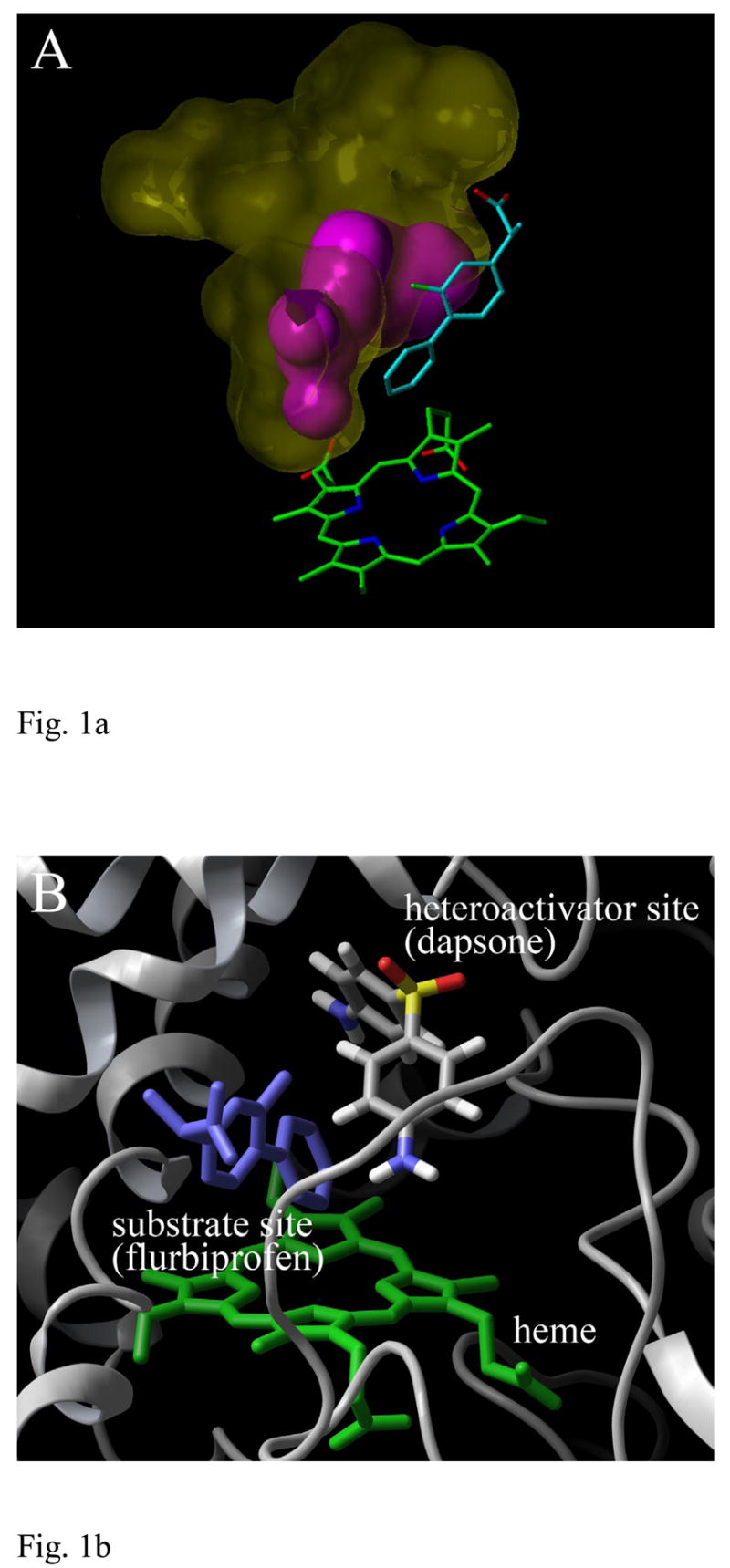
CYP2C9 structure used in the docking of heteroactivators of (S)-flurbiprofen metabolism. (A) The ligand binding pocket volume of CYP2C9 available for binding an effector with substrate (S)-flurbiprofen bound (cyan). A volume calculation was performed with the 1R9O.pdb coordinates40 (magenta) and subsequently, after molecular dynamics simulation with heteroactivator dapsone (yellow). The heme prosthetic group is shown in green. (B) A CYP2C9 binding site for acidic substrates identified by crystallography and the putative heteroactivator binding site identified by docking. The image is rotated approximately 90 degrees relative to panel A.
Docking of the dapsone analogs in the presence of (S)-flurbiprofen indicated no correlation between Glide docking scores and the degree of activation afforded by the compounds (data not shown). Alternatively, docking of the dapsone analogs in the CYP2C9 structure that contained no substrate resulted in the known heteroactivators binding preferentially to the effector site (Table 1). Five out of six heteroactivators were correctly predicted if one of the top three ligand conformations was bound to the effector site (only phenylsulfone (compound 4) was not bound to the effector site in its top scoring conformation) (Fig. 1b and Fig. 2). Only six out of thirteen inhibitors bound preferentially in the substrate site or in a manner that partially overlapped both sites (Fig. 2). However, the inhibitors tended to have GlideScores below that of the weakest heteroactivator, compound 6. Combining binding location preference and the GlideScore cutoff value of −6.13 resulted in the prediction of eleven out of thirteen inhibitors.
Figure 2.
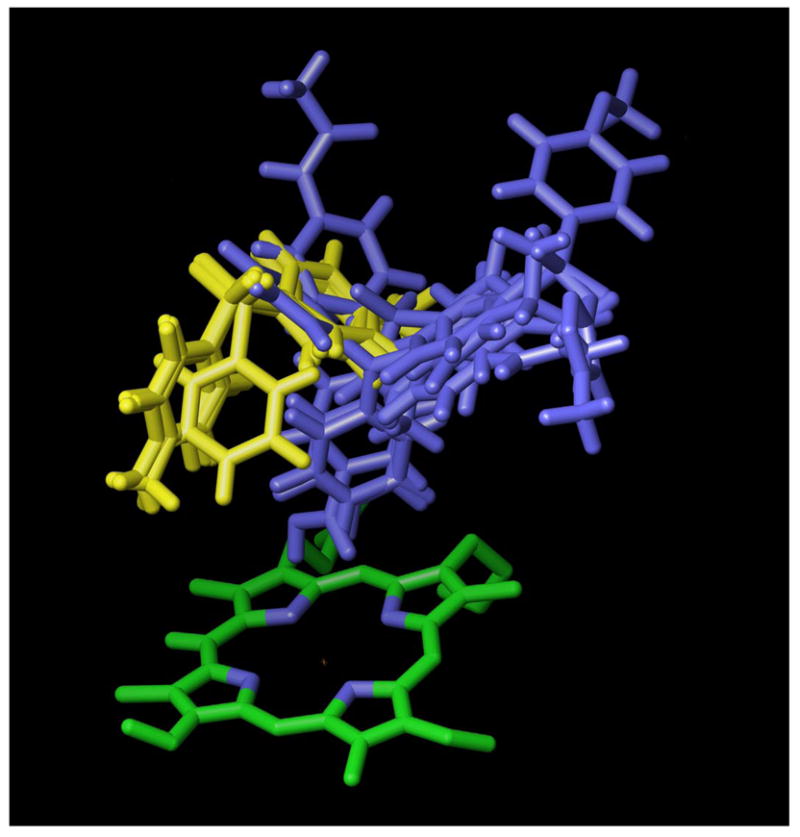
Top scoring docked poses of dapsone analogs correctly assigned as activators or inhibitors of CYP2C9-mediated (S)-flurbiprofen metabolism based on docking location in the free enzyme. Structures shown in yellow are known heteroactivators and their binding location corresponds to the free active site volume available when the substrate (S)-flurbiprofen is present (see Fig. 1A). Structures shown in blue correspond to known inhibitors of (S)-flurbiprofen metabolism.
Screening of a virtual library via docking
To further validate the docking method used to predict heteroactivators of a P450 enzyme, the same CYP2C9 structure was used in the docking of drug-like molecules compiled in the ZINC database24, for which biological activity was not known. As a control, a chemical catalog that contained the known CYP2C9 heteroactivator dapsone was chosen. Though several steps are needed to prepare the ligands for such screens (Table 2), the time required to screen tens of thousands of compounds is on the order of a few days using only two processors of a typical Unix or Linux workstation. The first docking step used rigid docking whereby rotamers of the minimized ligands are not scored. This was carried out in the enzyme receptor grid containing the substrate (S)-flurbiprofen, the activation of whose metabolism we were attempting to predict. Overall, this first docking step was deemed efficient since 76 % of the compounds screened were eliminated. Though rigid docking eliminated compounds that might otherwise bind favorably in an alternate conformation, the compounds that were scored still contained abundant chemical diversity. The presence of the substrate in the first docking step also aided in the elimination of compounds that would most likely have been too large to bind simultaneously with (S)-flurbiprofen to CYP2C9. Interestingly, the known CYP2C9 heteroactivator dapsone was, in fact, found to be included in the ZINC database of Sigma-Aldrich chemicals and it successfully made it past this initial screen, though 904 compounds were scored higher than dapsone.
Table 2.
Virtual screen protocol for identification of potential CYP2C9 heteroactivatorsa
| Number of compounds | Treatment or Results |
|---|---|
| 49020 | ZINC 2-D structures from the Sigma catalog |
| 81171 | Convert to 3-D structures, desalt, neutralize, generate up to four stereoisomers, minimization |
| 67894 | Molecular weight filter |
| 77420 | Generate alternate ionization states for groups with pKa 5–9 |
| 18558 | Rigid docking in presence of substrate (S)-flurbiprofen |
| 908 | Compounds with scores above the positive control dapsone (compound 2) (GlideScore = −7.47) |
| 5 | Flexible docking in absence of substrate and assignment of binding mode preference; five compounds predicted to be better than the positive control dapsone |
| 10 | Purchase and screen top ten scoring compounds in vitro |
For further details, consult Methods section.
The second docking step was carried out in the enzyme receptor grid that lacked substrate, incorporating the top scoring compounds that had been manually saved into a single file. Docking in Glide was then initiated in standard mode where bond rotation was used to optimize ligand binding energies. The top ten binding conformations, separated by an RMS deviation greater than 0.5, were saved for analysis. As observed with the dapsone control data set, some compounds clearly possessed preferences for binding to the substrate or effector site of CYP2C9. Nine compounds scoring higher than the control dapsone bound to the effector site in their highest scoring conformation. Since we wanted to evaluate potential inhibitors by screening compounds with scores above and below dapsone, the stringency of the screen was increased so that only those compounds that bound to the effector site in at least the top two scoring conformations were considered potential heteroactivators. This reduced the number of compounds scoring above dapsone to five out of the initial 77,420 compounds.
Ten of the top scoring compounds that bound to the effector site of the docking grid in their top two or three scoring conformations were next purchased to test their biological activity. One compound was no longer commercially available so that six of the compounds chosen had GlideScores below that of dapsone (in the absence of substrate (S)-flurbiprofen). Because the concentration of heteroactivator that gives optimal stimulation of P450 metabolism is slightly dependent on substrate concentration, but highly dependent on activator concentration9, a log scale of potential activator concentrations was tested in vitro against a single substrate concentration. Remarkably, compound 20 was nearly as effective as dapsone in stimulating the hydroxylation of (S)-flurbiprofen by CYP2C9 (Table 3) and statistically different from DMSO control (p<0.05). Four other compounds (21, 22, 23, and 24) appeared to be weak activators, but were not statistically different from the DMSO control (Fig. 2). Two of the five remaining compounds tested had no effect on (S)-flurbiprofen metabolism at concentrations as high as 10 mM. The remaining three compounds were inhibitors, though this might not rule out their simultaneous binding with (S)-flurbiprofen. Torimoto et al and others have suggested that multiple ligand binding may be responsible for partial inhibition of CYP3A425.
Table 3.
| # | in vitro activation (%)a | GlideScore with no substrate present | Consecutive conformations binding to effector siteb | |
|---|---|---|---|---|
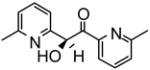
| 2 |
|
64.1 | −7.69 | 3 |
| 20 |
|
46.0 | −7.10 | 3 |
| 21 |
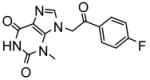
|
7.7 | −7.96 | 3 |
| 22 |
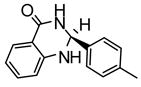
|
6.6 | −7.84 | 3 |
| 23 |
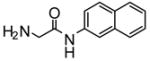
|
4.4 | −7.84 | 2 |
| 24 |
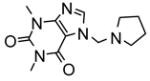
|
3.0 | −7.96 | 3 |
| 25 |
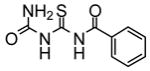
|
No effect | −7.65 | 2 |
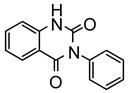
| 26 |
|
No effect | −7.36 | 3 |
| 27 |
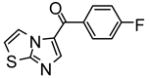
|
50% inhibition at 100μM | −7.24 | 3 |
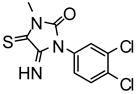
| 28 |
|
50% inhibition at 100μM | −6.97 | 2 |
| 29 |
|
50% inhibition at 1μM | −7.67 | 3 |
There are two caveats with the docking technique that should be mentioned. First, GlideScores of the new confirmed heteroactivators (Table 3) were generally above dapsone, but the order did not correlate with the percent activation. This was also true for docking scores obtained in the presence of substrate (data not shown), suggesting that binding alone is not sufficient for heteroactivation. In fact, there are quite possibly more reasons why multiple ligands binding to the same enzyme pocket would not result in enhanced turnover, including the hindering of binding, altered enzyme conformation, and disruption of water networks involved in catalysis. Therefore, it was not possible to fully evaluate heteroactivation based on the ability of the docking routine to rank known actives above non-actives by docking scores alone. Second, docking with a static receptor will most likely not provide the desired accuracy without further constraints or more intensive modeling methods, increasing the amount of required computation time. Preparation of the enzyme receptor via a molecular dynamics simulation increased the molecular weight range of compounds that could be screened, but use of a single receptor still limited the results to a particular compound geometry.
Identification of compound 20 exemplifies the potential in carrying out similar screens for P450 heteroactivators since a new series of leads with potency similar to previously characterized heteroactivators was discovered. None of the ketone-containing dapsone analogs were found to activate (S)-flurbiprofen hydroxylation. Yet, the new activator compound 20 formally contains a ketone, and is much more effective than the single oxygen-containing sulfoxide analog of dapsone. Upon closer examination, the crystal structure26 of this compound indicates it is predominantly in the enol form. In this tautomeric form it does resemble an alcohol except that the oxygen bearing carbon is still sp2 hybridized with a C-O bond length shorter (1.367 Å) than a normal alcohol C-O bond (1.43 Å). Furthermore, the hydrogen of the alcohol is hydrogen bonded to the pyridine nitrogen. This renders the enol oxygen somewhat negatively charged and thus similar to the sulfoxide oxygens of dapsone. Thus, the electronic environment between the two aromatic rings of 20 is similar to dapsone albeit with a distinctly different functionality. This result may help reveal the underlying interactions that are involved in rendering effector activity.
In addition, rather than favor H-bond donating groups on the aromatic rings (e.g. compounds 1 and 2), compound 20 clearly contains potential H-bond acceptors on its pyridine rings26. In fact, the chemical diversity in the new heteroactivators discovered here may help uncover the mechanisms(s) behind heteroactivation. Previously, heteroactivators were found to alter substrate binding conformation via steric effects9, but it also likely that H-bond donors and H-bond acceptors will have roles in P450 activation and catalysis in addition to binding to enzyme. Highly specific water networks that line the P450 active site and are dependent on highly conserved amino acids have been described27 and may be affected by heteroactivators. These networks are thought to act as proton-relay pathways that help protonate the iron-peroxo anion, iron-hydroperoxo, and ferryl-oxo P450 catalytic intermediates.
The proposed CYP2C9 effector binding region involved in the heteroactivation of non-steroidal antiinflammatory drug metabolism is made up of multiple P450 substrate recognition sites (SRS)28 including SRS-1 (B-C loop), 2 (F-helix), 4 (I-helix), and 6 (C-terminal β-strand region 4) (Fig. 1). These SRS regions define the segments of the P450 polypeptide, from the N- to C-terminus, that create the substrate binding pocket in P450s. Previously, a CYP2C9 heteroactivator pharmacophore29 was found to include an aromatic group separated from an H-bond acceptor by 2–3 bonds. All of the sulfones of the dapsone analogs are predicted to make polar interactions with the hydroxyl of S209 while an aromatic ring in all heteroactivators (except compound 24) stacks with F476. Hence, even though our study was carried out with a different substrate, the results may be describing the same binding region. As might be expected, SRS regions 2 and 6, the site of these residues, are located further away from the heme group where substrate oxidation occurs (Fig. 1a). However, only one of the new heteroactivators (21) made an H-bonding interaction with CYP2C9 in its lowest energy minimized conformation. Therefore, binding energy is expected to arise from additional hydrophobic interactions with the enzyme and even the substrate, whose shape will determine the complementarity of heteroactivator binding to the enzyme-substrate complex.
Conclusions
The current study demonstrates that simple high-throughput docking methods can be used to discover novel heteroactivators of cytochromes P450 from a large library of virtual compounds. In addition, our results suggest we have identified one of the heteroactivator binding sites for the metabolism of non-steroidal antiinflammatory drugs within the CYP2C9 enzyme. Docking scores, but also the binding location of ligands, were used to identify potential heteroactivators. The screening process was highly effective at eliminating compounds at each stage so that the total percentage of compounds scored as predicted heteroactivators was < 0.03 % of the total compounds screened. Most of the new heteroactivators were weak when tested in vitro so that the success rate was not ideal. However, we would stress two points. Only a single enzyme structure was used in the entire screening process, and docking is only a screen for binding. Binding of an effector to a P450 is not expected to be the only requirement for achieving heteroactivation since simultaneous ligand binding may also affect the P450 catalytic cycle in a negative manner. In summary, quickly identifying a diverse set of compounds in silico that are readily available for testing in vitro may benefit the study of P450 heteroactivation as it relates to P450 mechanism, drug-drug interactions, and drug design.
Experimental Methods
Materials
NADPH and other biochemicals, DMSO (99.8 %), dapsone (4,4′-diaminophenylsulfone) and its analogs (Table 1) were from Sigma (St. Louis, MO). HPLC grade acetonitrile was from Fisher Scientific (Fairlawn, NJ). (S)-flurbiprofen, the 4′-OH-flurbiprofen metabolite standard, and 2-fluoro-biphenylacetic acid (internal standard) were gifts from the former Pharmacia Corp. (Kalamazoo, MI). Compounds tested as potentially new heteroactivators of CYP2C9 were purchased from the Sigma-Aldrich Library of Rare Chemicals (St. Louis, MO).
Drug metabolism reactions
Purified, full-length human CYP2C9 and rat cytochrome P450 reductase were expressed in E. coli, then solubilized and purified as before9. CYP2C9 was added to two eq of rat cytochrome P450 reductase and the mixture reconstituted with extruded 1,2-dilauroyl-sn-glycero-3-phosphocholine lipid (1 μg/pmol P450) as detailed previously9. Five pmol of reconstituted CYP2C9 was added to 0.2 mL drug incubations containing 20 μM (S)-flurbiprofen and 50 mM potassium phosphate buffer (pH 7.4). Reactions were carried out in a 37 °C water bath for 20 min, quenched, and analyzed for flurbiprofen metabolite formation as before30. All potential heteroactivators were dissolved in DMSO to obtain 10 or 100 mM solutions and subsequently diluted further in DMSO to the desired concentrations. (S)-Flurbiprofen concentration was held constant while the potential heteroactivators were screened on a log scale of concentrations from 0.001–1000 μM, dependent upon solubility. The concentration of DMSO in all enzyme reactions, including controls, was 1 %, and all reactions were performed in triplicate. Potential heteroactivators were analyzed using the flurbiprofen HPLC assay to ensure no peaks overlapped with the flurbiprofen metabolite or internal standard.
Preparation of chemical library
The Sigma-Aldrich compounds (catalog version 2004-01-01) available in the ZINC database31 (http://blaster.docking.org/zinc/) were downloaded in .sdf format and prepared using the Schrödinger programs (2005 release) LigPrep, QikProp, MacroModel and the various utilities that accompany this software according to the Preparing a Library manual (Schrödinger, LLC, New York, NY). Further details can be found in Table 2. Conversion of 2-D structures to 3-D was carried out with retention of any specified stereochemistry and up to four different additional stereoisomers were generated. No tautomers were generated and only the lowest energy ring conformation was used for each compound. Next, a molecular weight filter was used to eliminate compounds with molecular weights below 120 or above 500. Lastly, alternate ionization states were generated for groups with pKa values between 5 and 9 and a final geometry optimization performed with the OPLS_2005 forcefield.
Molecular dynamics
The CYP2C9-flurbiprofen complex structure (1R9O.pdb) was obtained from the Protein Database (http://www.rcsb.org) and modified to fill in residues 38–42 and 214–220 using InsightII (Accelrys, San Diego, CA), followed by molecular dynamics equilibration with both (S)-flurbiprofen and dapsone present inside the enzyme as outlined previously9. The manually positioned starting conformation of dapsone was chosen based on the highest scoring docking pose of dapsone in the original crystal structure containing (S)-flurbiprofen, which was reported using the program AutoDock32, and reproduced here using Glide.
Molecular mechanics and dynamics were performed with Sander module of Amber 833 and the Cornell 94 force field34. The CYP2C9 structure was loaded into the Amber module xleap, hydrogen atoms added, 1 chloride atom added to neutralize the charge, and a water box (TIP3) added so that the dimensions of the box were at least 10 angstroms greater than the dimensions of the solute (CYP2C9). Initially, the water and solute were equilibrated by minimizing the water and counterion, with CYP2C9 fixed (500 kcal/mol·Å positional restraints on all atoms of the solute), to a gradient convergence of 10−3 kcal/mol·Å. This was followed by 25 ps of non-particle mesh Ewald (PME) dynamics with CYP2C9 fixed to allow for equilibration of water and counterion. All subsequent MD simulations were performed in the isothermal-isobaric ensemble (initial temp 100 K, 300 K after warm up, 1 atm) using periodic boundary conditions. The SHAKE algorithm was applied to hydrogen atoms with a tolerance limit of 0.0005 Å and a 2 femtosecond (fs) timestep. A 9 Å cutoff was applied to Lennard-Jones interactions. Simulations were performed using Berendsen temperature coupling algorithm and constant pressure with isotropic molecule based scaling (each with a 0.2 ps time constant). The non-bonded pair list was updated every 20 fs, and the particle mesh-Ewald method (PME), for inclusion of long-range electrostatic interactions without truncation, was used. The next step in the simulation protocol was 25 ps of PME dynamics for inclusion of long-range electrostatic interactions without truncation. The PME charge grid spacing was approximately 1.0 Å, and the charge grid was interpolated on a cubic grid with the direct sum tolerance set to 10−4.
Following the 25 ps PME dynamics, the entire structure was minimized for 1000 steps with 25 kcal/mole·Å of positional restrains on the solute followed by a 3 ps molecular dynamics run, which allowed water and the Cl− ion to relax around the solute. Subsequently, 600 steps of equilibration with gradual removal of positional restraints by 5 kcal/mole·Å on the solute were performed. During the final molecular dynamics run, the system was heated from 100 K to 300 K over 20 ps. After each molecular dynamics simulation, the PME box information was updated to match the final box coordinates from previous runs. Production runs of approximately 2 ns duration, at constant temperature and pressure (T=300 K/P=1 atm), were performed after the final equilibration step. All trajectories were stable over the time period of the molecular dynamics simulations.
Docking methodology
Glide (Schrödinger, LLC, New York, NY, 2005) was used in preparation of the CYP2C9 receptor and for all docking studies. A single representative structure was taken from the equilibrated portion (500–2000 ps) of the molecular dynamics trajectory and used to prepare two CYP2C9 receptors – one with (S)-flurbiprofen present and one with no ligands present. The most representative structure, which has the lowest average RMSD to all other members of its cluster, was determined using the cluster analysis routines in MOIL-view35. Receptors were prepared using the Protein Preparation and Grid Preparation tools in the Schrödinger Maestro interface. Values for cutoff, neutralization, scaling, dimensions of the binding pocket used for grid preparation, and treatment of the heme prosthetic group were previously described9.
Dapsone and its analogs (Table 1) were prepared and minimized using the same methodology as above with the Schrödinger software except that the steps were conducted using the Maestro graphic user interface. Four of the dapsone analogs possess pKa values close to physiological pH according to calculations with ACD/PhysChem Batch v8.14 (ACD/Labs, Toronto, ON, Canada). Hence, sulfamethoxazole (12), sulfadiazine (13), sulfamethazine (15), and ketoprofen (18), having pKas of 5.8, 6.5, 7.5, and 4.1 respectively, were also docked in their ionized states. Only the GlideScores representing the ionized states are provided in Table 1. Docking was carried out in standard docking mode with the CYP2C9 receptor that lacked the substrate (S)-flurbiprofen. Default values for van der Waals scaling, electrostatics, and ligand minimization were used. The top ten scoring conformations of each compound were saved. During analysis, the docking scores were noted and the frequency in the top three scoring conformations in which a compound was located in the effector (i.e. dapsone) or substrate (i.e. (S)-flurbiprofen) binding site were also recorded (Fig. 1 and 2). When the distinction between binding sites was not evident, the classification was made by preparing an additional receptor with the potential heteroactivator and ensuring (S)-flurbiprofen would dock in the CYP2C9 grid within the default energy cutoff (0 kcal/mol). Otherwise, if the energy was too high, (S)-flurbiprofen was not scored and the potential heteroactivator was therefore assigned as an inhibitor.
A slightly different protocol was used in screening of the virtual library (Table 2). The first stage of docking was conducted in rigid docking mode with the CYP2C9-flurbiprofen receptor and the minimized ligands, which were segmented into groups of 10,000. Compounds that made it through this initial screen based on the default energy cutoff (0 kcal/mol) were then evaluated. The ligands that had the highest GlideScores from each subset, minus those that differed only in ionization state, were recorded. Next, the remaining compounds were docked in the CYP2C9 receptor without substrate present using the standard docking mode of Glide, which allows bond rotation in the ligands. The first ten highest scoring ligands that were found to occupy the CYP2C9 dapsone/effector site in at least its first two top scoring (lowest energy) conformations and were currently commercially available, were chosen for in vitro testing.
Vermeulen and co-workers have demonstrated that the presence of explicit waters increases the accuracy of docking poses36. While Glide incorporates explicit waters to evaluate water binding interactions in the calculation of scores 37, it is possible the docking poses would be altered if a different treatment for water was used. However, we suspect a diverse set of ligands may have varying abilities to also displace predicted or crystallographic waters in addition to binding to waters. Therefore, for consistency, the effect of active site waters was not evaluated.
Estimation of molecular volumes
The AMBER interface xleap33 was used to solvate the 1R9O crystal structure38 containing (S)-flurbiprofen or the molecular dynamics equilibrated CYP2C9 structure generated with (S)-flurbiprofen and dapsone, but with the dapsone ligand removed. A glycerol molecule crystallized in the original structure was removed before the calculation. A 5 Å-thick shell of TIP3P water molecules that was allowed to come within 0.8 Å of the enzyme was added. This eliminated the enzyme active site volume calculation from extending out to the surface of the enzyme. The MOLCAD multi-channel surface feature in Sybyl v7.1 (Tripos Inc., St. Louis, MO) was then used to determine active site volume with a probe size of 1.5 Å.
Statistical analysis
Statistical comparisons were conducted using a repeated measures ANOVA with multiple comparisons (SigmaStat 3.1, Point Richmond, CA). Statistical significance was set at p = 0.05.
Figure 3.
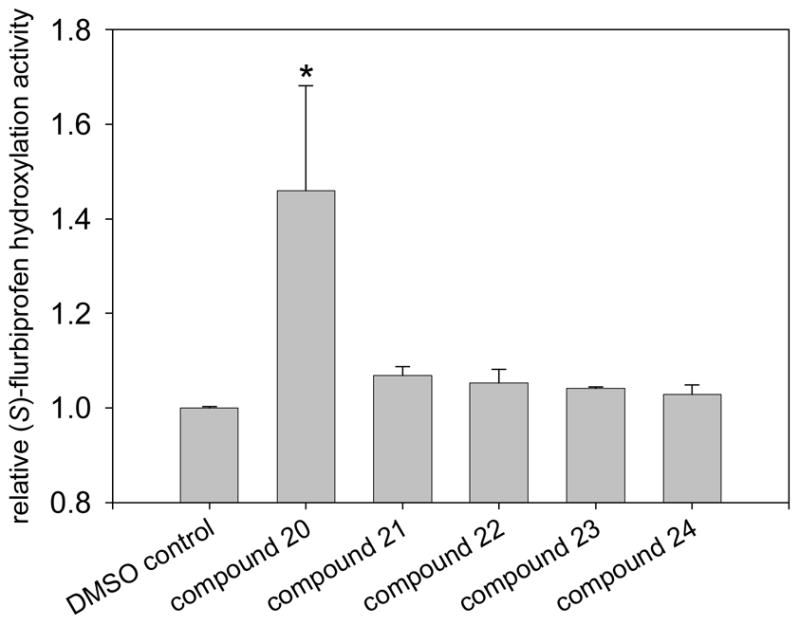
Heteroactivation of (S)-flurbiprofen hydroxylation via CYP2C9 by compounds identified through high-throughput docking of a virtual chemical library. Measurements were conducted in triplicate and the standard deviations shown as error bars. Optimal concentrations of heteroactivators were: compound 20 (100 μM), compound 21 (100 μM), compound 22 (0.1 μM), compound 23 (0.01 μM), compound 24 (0.01 μM). *Statistically different from controls p < 0.05.
Acknowledgments
We thank Drs. Yuk Y. Sham and Eric M. Bennet for technical help. In addition, we are grateful for resources from the University of Minnesota Supercomputing Institute. This work was supported in part by NIH grant GM 063215 (T.S.T.).
Abbreviations
- P450
cytochrome P450
Reference List
- 1.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 2.Bachmann KA. Inhibition constants, inhibitor concentrations and the prediction of inhibitory drug drug interactions: pitfalls, progress and promise. Curr Drug Metab. 2006;7:1–14. doi: 10.2174/138920006774832541. [DOI] [PubMed] [Google Scholar]
- 3.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 4.Atkins WM. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 5.Tracy TS, Hummel MA. Modeling kinetic data from in vitro drug metabolism enzyme experiments. Drug Metab Rev. 2004;36:231–242. doi: 10.1081/dmr-120033999. [DOI] [PubMed] [Google Scholar]
- 6.Dabrowski MJ, Schrag ML, Wienkers LC, Atkins WM. Pyrene. pyrene complexes at the active site of cytochrome P450 3A4: evidence for a multiple substrate binding site. J Am Chem Soc. 2002;124:11866–11867. doi: 10.1021/ja027552x. [DOI] [PubMed] [Google Scholar]
- 7.Hummel MA, Gannett PM, Aguilar JS, Tracy TS. Effector-mediated alteration of substrate orientation in cytochrome P450 2C9. Biochemistry. 2004;43:7207–7214. doi: 10.1021/bi036158o. [DOI] [PubMed] [Google Scholar]
- 8.Rock DA, Perkins BN, Wahlstrom J, Jones JP. A method for determining two substrates binding in the same active site of cytochrome P450BM3: an explanation of high energy omega product formation. Arch Biochem Biophys. 2003;416:9–16. doi: 10.1016/s0003-9861(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 9.Locuson CW, Gannett PM, Tracy TS. Heteroactivator effects on the coupling and spin state equilibrium of CYP2C9. Arch Biochem Biophys. 2006;449:115–129. doi: 10.1016/j.abb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Hutzler JM, Frye RF, Korzekwa KR, Branch RA, Huang SM, Tracy TS. Minimal in vivo activation of CYP2C9-mediated flurbiprofen metabolism by dapsone. Eur J Pharm Sci. 2001;14:47–52. doi: 10.1016/s0928-0987(01)00144-0. [DOI] [PubMed] [Google Scholar]
- 11.Liu KH, Kim MJ, Jung WM, Kang W, Cha IJ, Shin JG. Lansoprazole enantiomer activates human liver microsomal CYP2C9 catalytic activity in a stereospecific and substrate-specific manner. Drug Metab Dispos. 2005;33:209–213. doi: 10.1124/dmd.104.001438. [DOI] [PubMed] [Google Scholar]
- 12.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol. 2002;6:439–446. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 13.Dabrowski MJ, Schrag ML, Wienkers LC, Atkins WM. Pyrene. pyrene complexes at the active site of cytochrome P450 3A4: evidence for a multiple substrate binding site. J Am Chem Soc. 2002;124:11866–11867. doi: 10.1021/ja027552x. [DOI] [PubMed] [Google Scholar]
- 14.Hummel MA, Gannett PM, Aguilar JS, Tracy TS. Effector-mediated alteration of substrate orientation in cytochrome P450 2C9. Biochemistry. 2004;43:7207–7214. doi: 10.1021/bi036158o. [DOI] [PubMed] [Google Scholar]
- 15.Rock DA, Perkins BN, Wahlstrom J, Jones JP. A method for determining two substrates binding in the same active site of cytochrome P450BM3: an explanation of high energy omega product formation. Arch Biochem Biophys. 2003;416:9–16. doi: 10.1016/s0003-9861(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 16.Hutzler JM, Kolwankar D, Hummel MA, Tracy TS. Activation of CYP2C9-mediated metabolism by a series of dapsone analogs: kinetics and structural requirements. Drug Metab Dispos. 2002;30:1194–1200. doi: 10.1124/dmd.30.11.1194. [DOI] [PubMed] [Google Scholar]
- 17.Hutzler JM, Kolwankar D, Hummel MA, Tracy TS. Activation of CYP2C9-mediated metabolism by a series of dapsone analogs: kinetics and structural requirements. Drug Metab Dispos. 2002;30:1194–1200. doi: 10.1124/dmd.30.11.1194. [DOI] [PubMed] [Google Scholar]
- 18.Egnell AC, Eriksson C, Albertson N, Houston B, Boyer S. Generation and evaluation of a CYP2C9 heteroactivation pharmacophore. J Pharmacol Exp Ther. 2003;307:878–887. doi: 10.1124/jpet.103.054999. [DOI] [PubMed] [Google Scholar]
- 19.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 20.Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 21.Kumar V, Locuson CW, Sham YY, Tracy TS. Amiodarone analog-dependent effects on CYP2C9-mediated metabolism and kinetic profiles. Drug Metab Dispos. 2006;34:1688–1696. doi: 10.1124/dmd.106.010678. [DOI] [PubMed] [Google Scholar]
- 22.Bland TM, Haining RL, Tracy TS, Callery PS. CYP2C-catalyzed delta9-tetrahydrocannabinol metabolism: kinetics, pharmacogenetics and interaction with phenytoin. Biochem Pharmacol. 2005;70:1096–1103. doi: 10.1016/j.bcp.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torimoto N, Ishii I, Hata M, Nakamura H, Imada H, Ariyoshi N, Ohmori S, Igarashi T, Kitada M. Direct interaction between substrates and endogenous steroids in the active site may change the activity of cytochrome P450 3A4. Biochemistry. 2003;42:15068–15077. doi: 10.1021/bi034409n. [DOI] [PubMed] [Google Scholar]
- 26.Ashida T, Hirokawa S. The crystal structure of α-pyridoin,1,2-di-2-pyridylethenediol-1,2. Acta Cryst. 1965;18:122–127. [Google Scholar]
- 27.Nagano S, Poulos TL. Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 28.Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 29.Egnell AC, Eriksson C, Albertson N, Houston B, Boyer S. Generation and evaluation of a CYP2C9 heteroactivation pharmacophore. J Pharmacol Exp Ther. 2003;307:878–887. doi: 10.1124/jpet.103.054999. [DOI] [PubMed] [Google Scholar]
- 30.Tracy TS, Hutzler JM, Haining RL, Rettie AE, Hummel MA, Dickmann LJ. Polymorphic variants (CYP2C9*3 and CYP2C9*5) and the F114L active site mutation of CYP2C9: effect on atypical kinetic metabolism profiles. Drug Metab Dispos. 2002;30:385–390. doi: 10.1124/dmd.30.4.385. [DOI] [PubMed] [Google Scholar]
- 31.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 33.Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER. Vol. 8. University of California; San Francisco: 2004. [Google Scholar]
- 34.Cornell W, Cieplak P, Bayly C, Gould I, Merz K, Jr, Ferguson D, Spellmeyer D, Fox T, Caldwell J, Kollman P. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1999;117:5179–5197. [Google Scholar]
- 35.Simmerling C, Elber R, Zhang J. MOIL-View - A program for visualization of structure and dynamics of biomolecules and STO - A program for computing stochastic paths. In: Pullman A, editor. Modeling of biomolecular structure and mechanisms. Kluwer; Netherlands: 1995. pp. 241–265. [Google Scholar]
- 36.de Graaf C, Pospisil P, Pos W, Folkers G, Vermeulen NP. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J Med Chem. 2005;48:2308–2318. doi: 10.1021/jm049650u. [DOI] [PubMed] [Google Scholar]
- 37.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 38.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 39.Hutzler JM, Kolwankar D, Hummel MA, Tracy TS. Activation of CYP2C9-mediated metabolism by a series of dapsone analogs: kinetics and structural requirements. Drug Metab Dispos. 2002;30:1194–1200. doi: 10.1124/dmd.30.11.1194. [DOI] [PubMed] [Google Scholar]
- 40.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]