

Abstract
Infection by the human T-cell leukemia virus type 1 (HTLV-1) is thought to cause dysregulated T-cell proliferation, which in turn leads to adult T-cell leukemia/lymphoma. Early cellular changes after HTLV-1 infection have been difficult to study due to the poorly infectious nature of HTLV-1 and the need for cell-to-cell contact for HTLV-1 transmission. Using a series of reporter systems, we show that HeLa cells cease proliferation within one or two division cycles after infection by HTLV-1 or transduction of the HTLV-1 tax gene. HTLV-1-infected HeLa cells, like their tax-transduced counterparts, expressed high levels of p21CIP1/WAF1 and p27KIP1, developed mitotic abnormalities, and became arrested in G1 in senescence. In contrast, cells of a human osteosarcoma lineage (HOS) continued to divide after HTLV-1 infection or Tax expression, albeit at a reduced growth rate and with mitotic aberrations. Unique to HOS cells is the dramatic reduction of p21CIP1/WAF1 and p27KIP1 expression, which is in part associated with the constitutive activation of the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) pathway. The loss of p21CIP1/WAF1 and p27KIP1 in HOS cells apparently allows HTLV-1- and Tax-induced G1 arrest to be bypassed. Finally, HTLV-1 infection and Tax expression also cause human SupT1 T cells to arrest in the G1 phase of the cell cycle. These results suggest that productive HTLV-1 infection ordinarily leads to Tax-mediated G1 arrest. However, T cells containing somatic mutations that inactivate p21CIP1/WAF1 and p27KIP1 may continue to proliferate after HTLV-1 infection and Tax expression. These infected cells can expand clonally, accumulate additional chromosomal abnormalities, and progress to cancer.
Adult T-cell leukemia/lymphoma (ATL) is a rare T-cell malignancy characterized by hypercalcemia, hepatosplenomegaly, lymphadenopathy, skin involvement, and presence of abnormal lymphocytes. ATL develops in 2 to 5% of human T-cell leukemia virus type 1 (HTLV-1)-infected individuals over a clinical latency of 20 to 40 years. The long incubation period and the low frequency of clinical progression to ATL suggest that complex viral and cellular events are involved in ATL development. It has been proposed that after HTLV-1 infection in vivo, at least five independent genetic changes are needed before the onset of ATL (33).
The viral determinant critical for the progression to T-cell malignancy in HTLV-1-infected persons is thought to be the HTLV-1 transactivator/oncoprotein, Tax. How Tax influences ATL development is incompletely understood. The effects that Tax exerts over cells are pleiotropic and include potent NF-κB activation, cell cycle perturbation, and cell transformation. ATL cells in general do not express HTLV-1 sequence, suggesting that Tax likely affects the early stage of the disease, and persistent Tax expression is not needed for maintenance of the neoplasm (9). Recent data have indicated that an antisense mRNA is transcribed from the 3′ end of the HTLV-1 provirus. This mRNA encodes a basic domain-leucine zipper protein known as HBZ, which negatively regulates Tax transactivation. The HBZ mRNA is widely expressed in ATL cells (29, 39). Intriguingly, the HBZ mRNA transcript has been reported to stimulate cell proliferation (39).
Tax impacts multiple steps in cell cycle progression. It can promote quiescent T cells to enter into G1/S (11, 14, 15, 32). The potent NF-κB activation induced by Tax is also mitogenic and antiapoptotic (43, 44). Tax has been shown to functionally inactivate p53 (36, 37) and inhibit DNA damage repair (12, 17, 27, 34). In spite of these activities of Tax, constitutive expression of tax in most cultured mammalian cell lines has been difficult to achieve. We have recently observed that Tax directly binds and activates the anaphase-promoting complex (APC) (20), an E3 ubiquitin ligase that becomes active during mitosis and controls the metaphase-to-anaphase transition and mitotic exit by targeting the destruction of cyclin A, securin, cyclin B1, and other cell cycle regulators. Premature activation of APC by Tax causes polyubiquitination and degradation of cyclin A, cyclin B1, and securin before the onset of M phase (20) and contributes to a delay in cell cycle progression, DNA aneuploidy, and formation of micro-, bi-, and multinucleated cells (19, 21, 24, 25). Most recently, we have found that the cell cycle demise that arises from unscheduled APC activation by Tax goes beyond mitotic dysfunction. In most Tax-expressing cells, the substrate-targeting subunit of the Skp-Cullin-F box (SCF) E3-ubiquitin ligase, Skp2, also becomes prematurely polyubiquitinated and degraded. The loss of Skp2 dramatically stabilizes a key substrate of SCFSkp2, the Cdk2 inhibitor, p27KIP1 (16). Through a mechanism not fully understood at present, p21CIP1/WAF1 mRNA and protein also became sharply increased concurrently with the unscheduled APC activation. The buildup of both p21CIP1/WAF1 and p27KIP1 commits cells into a state of permanent G1 arrest termed Tax-induced rapid senescence (16).
In this study, we have investigated whether HTLV-1 infection also leads to cell cycle arrest. To tackle the technical difficulty posed by the low infectivity of HTLV-1, reporter cell lines carrying the enhanced green fluorescent protein (EGFP) gene under the control of a highly Tax-responsive promoter were developed in the background of the HeLa cell line, a human osteosarcoma cell line (HOS), and the SupT1 human T-cell line to track HTLV-1 infection. Using these reporter cell lines, we demonstrate that HTLV-1-infected HeLa cells become G1 arrested immediately after viral infection. The infected HeLa cells expressed high levels of p21CIP1/WAF1 and p27KIP1 and displayed phenotypes indistinguishable from the phenotypes of cells in the state of Tax-induced rapid senescence (16). Likewise, SupT1 cells also underwent cell cycle arrest upon infection by HTLV-1 or after transduction of the tax gene via a lentivirus vector. In contrast, HOS cells continued to proliferate after HTLV-1 infection or tax gene transduction, albeit at a reduced rate and with development of mitotic abnormalities. Constitutive activation of the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) Akt pathway in HOS cells contributes to the downregulation and functional inactivation of p21CIP1/WAF1 and p27KIP1, thereby allowing Tax-induced rapid senescence to be averted. We suggest that HTLV-1 infection usually leads to arrest in the G1 phase of the cell cycle. A critical step of ATL development may involve HTLV-1 infection of or reactivation from T cells containing somatic mutations that constitutively inactivate p27KIP1 and p21CIP1/WAF1, which can be achieved in part through activation of the PI3K-Akt pathway. This type of T cells can expand clonally after HTLV-1 infection and be driven further into malignancy by Tax-induced NF-κB activation, p53 inactivation, defects in DNA repair, and chromosomal instability. Finally, a loss of HTLV-1 and tax expression eventually stabilizes the malignant T cells karyotypically and prevents them from cytotoxic T-lymphocyte killing, leading to ATL.
MATERIALS AND METHODS
Cell culture and media.
HeLa, 293T, and HOS cells were grown in Dulbecco's modified Eagle's medium (DMEM). MT2 and SupT1 T cells were grown in RPMI medium. All media were supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 100 U/ml each of penicillin and streptomycin.
HTLV-1 infection following coculture with MT2 cells.
HTLV-1-producing MT2 cells were collected by centrifugation and kept at a density of 2 × 105 cells/ml in RPMI medium supplemented with 10% fetal bovine serum. One day before coculture, HeLa and HOS cells in 2 ml DMEM plus 10% fetal bovine serum, were seeded in six-well plates at a density of 2 × 105 cells per well. On the following day, the culture medium was removed, and 1 ml of MT2 cells was added to each well and incubated for 16 h. MT2 cells were then removed by extensive shaking and washing. SupT1 cells were suspended at 2 × 106 cells/ml, and 0.1 ml of the cell suspension was mixed with 1 ml of MT2 cells (2 × 105 cells/ml) in each well of a six-well plate for 48 h. When appropriate, the reverse transcriptase inhibitor, zidovudine (AZT), was added to the medium at a final concentration of 1 μM concomitant with the application of MT2 cells and kept in the medium for the duration of the experiment.
Cell cycle analysis.
Two days after coculture with MT2 cells, HeLa/18x21-EGFP or HOS/18x21-EGFP cells were trypsinized and collected. Approximately 1 × 106 cells were plated and grown on each 10-cm plate. Twenty hours later, cells were harvested for flow cytometry as previously described (16, 21). Similarly, SupT1/18x21-EGFP cells transduced with lentivirus vector LV-Tax or LV-Puro were fixed, stained, and analyzed 48 h after lentiviral vector transduction. Simultaneous detection of EGFP and propidium iodide fluorescence was carried out using a fluorescence-activated cell sorter (Beckman-Coulter Epics XL-MCL flow cytometer). The fractions of cells in G0/G1, S, and G2/M in the gated EGFP-positive or EGFP-negative population were computed using the ModFit LT software package.
Production of lentivirus vectors and infection with lentivirus and adenovirus.
Lentiviral vectors were produced by transient transfection of HEK293T cells using a calcium phosphate transfection kit (Invitrogen Life Technologies, Carlsbad, CA) as reported previously (16). Infection or transduction with lentivirus and adenovirus vectors was also performed as described previously (16).
Immunofluorescence of p27KIP1 and Tax.
Two days after HTLV-1 infection, HeLa/18x21-EGFP cells were fixed with 1% formaldehyde, permeabilized with 0.5% Triton X-100, stained with an antibody against p27KIP1 (Santa Cruz Inc.) and a rhodamine-conjugated anti-mouse antibody (Jackson ImmunoResearch Inc.), and visualized and photographed using an Olympus IX81 fluorescence microscope equipped with a charge-coupled device camera. Immunofluorescence for Tax was done similarly except that LV-Tax-transduced HeLa/18x21-EGFP cells and a Tax monoclonal antibody, 4C5, were used.
Detection of the senescence-associated β-galactosidase in HTLV-1-infected HeLa/18x21-EGFP cells.
HeLa/18X21-EGFP cells (2.5 × 104 cells/well) and MT2 cells (2.5 × 104/well) were cocultured in a six-well plate as described above. MT2 cells were then removed by aspiration, and HeLa/18X21-EGFP cells were then washed three times with serum-free DMEM to remove residual MT2 cells. Attached HeLa/18X21-EGFP cells were then fixed and stained with a senescence cell histochemical staining kit (Sigma catalog no. CS0030) according to the vendor's instructions.
RESULTS
Derivation of cell lines containing stably integrated Tax-responsive 18x21-EGFP reporter cassette.
HTLV-1 can infect a wide variety of cells, including T lymphocytes, B lymphocytes, monocytes, endothelial cells, and fibroblasts. This is due in part to its use of a ubiquitous cell surface molecule, glucose transporter 1, as the receptor for virus entry (26). HTLV-1 infection is highly cell associated, however. Cell-to-cell transmission of virions through “virological synapses” formed, in part, through LFA1 and ICAM1 is required for infection of naïve cells (1, 13). For this reason, two approaches—coculture with HTLV-1-producing cells and cell-free infection with vesicular stomatitis virus G-pseudotyped viral particles—have been the mainstays for studying HTLV-1 infection in cell culture (7, 8). Both methods are plagued with a low efficiency of viral infection due to the poorly infectious nature of HTLV-1 (7, 8).
To track cellular changes that occur after HTLV-1 infection, we have generated several reporter cell lines using a recently described system derived from a self-inactivating lentivirus vector, SMPU (45). Briefly, a promoter element, termed 18x21, containing 18 copies of the Tax-inducible HTLV-1 21-bp repeat, the viral TATA element, the complete R region, and part of the U5 sequence was used to replace the cytomegalovirus (CMV) immediate-early promoter in the SMPU-CMV-EGFP lentiviral vector. Vesicular stomatitis virus G-pseudotyped particles produced from the resultant SMPU-18x21-EGFP readily integrate the 18x21-EGFP cassette into the chromosomes of most cultured mammalian cells. Because most of the U3 region in the 3′ long terminal repeat of the SMPU vector has been deleted, after a single round of lentiviral vector infection, the only functional enhancer/promoter element in the integrated DNA is the Tax-responsive 18x21 promoter, which alone drives the expression of the EGFP reporter. The ease of production and the high titer of the self-inactivating vector-based reporter greatly facilitated derivation of stable reporter cell lines (see below).
The SMPU-18x21-EGFP vector particles were produced as previously described and used to transduce human osteosarcoma and HeLa cells at a multiplicity of infection (MOI) of approximately 2. Two days postinfection, the infected cells were cloned by using limiting dilutions in 96-well plates. Individual cell clones were expanded and screened for the stable integration of the reporter cassette by infection with an adenovirus vector, Ad-Tax, carrying the tax gene (MOI of 3 to 5) or with a control vector, Ad-tTa, that contains the tet transactivator gene. Because the titer of SMPU-18x21-EGFP easily reached 5 × 106 infectious units/ml of culture supernatant, cell clones with the integrated 18x21-EGFP reporter appeared at a high frequency after lentiviral transduction and were readily obtained. As shown in Fig. 1A, HOS and HeLa cell lines harboring the stably integrated 18x21-EGFP cassette were found to express abundant EGFP after Ad-Tax infection, but not in its absence or after Ad-tTa infection. Furthermore, immunofluorescence of HeLa/18x21-EGFP cells transduced with LV-Tax, a lentivirus vector for Tax, indicated that Tax expression correlated directly with EGFP expression (Fig. 1B). Using the same strategy, we have also derived a reporter human T-cell line, SupT1/18x21-EGFP, by using limiting dilutions of SupT1 cells transduced with the SMPU-18x21-EGFP vector (see below).
FIG. 1.

Establishment of cell lines containing a stably integrated Tax-responsive 18x21-EGFP reporter cassette. (A) HOS and HeLa cell lines were transduced with the SMPU-18x21-EGFP lentivirus vector at an MOI of approximately 2. Infected cells were then cloned in 96-well plates by using limiting dilutions. Individual clones were expanded and screened for stable integration of the reporter cassette by infection with the Ad-Tax adenovirus vector. The reporter-containing clones usually ranged from 60 to 80%. Cloned HeLa/18x21-EGFP and HOS/18x21-EGFP cells infected with a control adenovirus vector, Ad-tTa, are shown for comparison. (B) HeLa/18x21-EGFP cells seeded on a coverslip were transduced with LV-Tax for 48 h. Immunofluorescence of Tax was carried out as described in Materials and Methods. DAPI, 4′,6′-diamidino-2-phenylindole.
HTLV-1 can infect HeLa and HOS cells.
To investigate the cellular changes that take place after HTLV-1 infection, HeLa and HOS reporter cell lines were cocultured with an HTLV-1-producer T-cell line, MT2, at a ratio of 1:1. Sixteen hours after coculture, MT2 cells were removed by extensive shaking and washing. For both HOS and HeLa cells, at 16 h after coculture, multinucleated giant cells (syncytia) that expressed EGFP could be easily detected. These syncytia arose as a result of cell-to-cell fusion, and as expected, their emergence was resistant to the reverse transcriptase inhibitor, AZT (Fig. 2). Apparently, the cytoplasmic mixing after cell fusion allowed the Tax protein expressed in MT2 cells to activate the integrated 18x21 EGFP cassette in the HeLa and HOS fusion partners. In contrast, at 2 days after coculture, 5 to 6% (HeLa and HOS) cells on the plate became EGFP positive, but with a “normal” appearance. Pretreatment with AZT drastically reduced the number of these EGFP-positive cells at this time point (Fig. 2A). This suggests that these EGFP-positive cells were newly infected by HTLV-1 after cell-cell contact. As expected, free HTLV-1 particles from the culture supernatant of MT2 cells when applied to the HeLa reporter cell line yielded few EGFP-positive cells (not shown). Similar results were also obtained using two other HTLV-1-producing T-cell lines, C91PL and MS9 in place of MT2 (unpublished data). It should be pointed out that while HeLa cells can be infected by HTLV-1, the infected HeLa cells are unable to transmit virus possibly because of a lack of LFA1 expression (unpublished results). For EGFP-positive HeLa and HOS cells, the AZT-sensitive population far outnumbered the AZT-resistant syncytia by 100- to 200-fold, strongly suggesting that HTLV-1 transmission via cell-to-cell contact was the major route of virus transmission during coculture. To confirm this conclusion, we measured the levels of HTLV-1 gp46 (SU) and p24 (CA) expression in the presence or absence of AZT. As shown in Fig. 2B, in HeLa and HOS cells cocultivated with MT2, gp46, p24, and Tax levels increased significantly in the absence of AZT. As expected, AZT treatment drastically inhibited viral antigen expression, albeit it did not affect syncytium formation. While extensive washing did remove most, if not all, of the MT2 cells from MT2-HeLa or MT2-HOS cocultures, some HTLV-1 particles apparently remained attached to the surfaces of HeLa and HOS cells or had entered the cells but did not reverse transcribe and integrate. This is reflected by the presence of gp46 and p24 in the AZT-treated samples (Fig. 2B); however, no Tax expression was detected in these samples, consistent with the inhibition of productive infection. These results indicate that cell-mediated infection of HeLa and HOS cells by HTLV-1 readily occurred and the amplification of HTLV-1 DNA template after reverse transcription and integration was principally responsible for the increase in the production of viral antigens.
FIG. 2.

Infection of HOS and HeLa cells following coculture with MT2 cells. (A) HOS and HeLa reporter cells were cocultured with MT2 cells (MT2+HOS and MT2+HeLa) as described in Materials and Methods at a cell-to-cell ratio of 1:1 for 48 h with or without AZT (1 μM). Strongly EGFP-positive clusters in the AZT-treated column are likely due to syncytium formation. (B) Immunoblots of HTLV-1 gp46 (SU), p24 (CA), Tax, and actin. Coculture with MT2 cells (+) was carried out as described above for panel A. After coculture overnight, MT2 cells were removed by extensive washing. Two days after coculture, lysates of infected cells were prepared for immunoblotting with gp46, p24, Tax, and actin antibodies, respectively. Cell lysates from cocultures treated with 1 μM AZT (+) or not treated with AZT (−) are compared side by side.
HeLa cells infected by HTLV-1 become arrested in G1 in a senescence-like state.
We have shown recently that Tax directly interacts with the APC and activates it in an unscheduled manner (20, 21). Most recently, we have found that APC activation by Tax leads to premature polyubiquitination/degradation of Skp2, inactivation of SCFSkp2, and dramatic stabilization of p27KIP1. Concurrent with these cell cycle changes, the levels of p21CIP1/WAF1 mRNA and protein also became greatly activated by Tax. The buildup of p21CIP1/WAF1 and especially p27KIP1 then commits the cells to senescence (16).
To determine whether HTLV-1 infection also leads to similar cell cycle abnormalities, HeLa/18x21-EGFP cells were again cocultured with MT2 cells at a cell ratio of 1 to 1 in the presence or absence of 1 μM AZT as described above. Sixteen hours after coculture, MT2 cells were removed, and the infected HeLa cells were washed extensively to remove any residual MT2 cells. Two days after infection, the cells were collected, fixed, stained with propidium iodide, and analyzed for their cell cycle profiles by flow cytometry. As shown in Fig. 3A, the majority of HTLV-1-infected/EGFP-positive cells in the AZT-free coculture were arrested at G1 (84% G1, 6.38% S, and 9.47% G2/M). This contrasted with the uninfected EGFP-negative HeLa cells in the same cell population (58.61% G1, 28.38% S, and 13.01% G2/M). AZT treatment effectively blocked HTLV-1 infection (as indicated by the absence of EGFP-positive cells from the coculture), and as expected, the cell cycle profile of HeLa cells from the AZT-treated coculture (Fig. 3A, 62.14% G1, 26.04% S, and 11.55% G2/M) was similar to the profile of the uninfected EGFP-negative cells in the AZT-free coculture (Fig. 3A, compare the bottom middle and bottom right panels).
FIG. 3.

HTLV-1-infected HeLa cells become arrested in G1. (A) HTLV-1-infected HeLa cells are arrested in G1. Two days after coculture, HeLa cells were trypsinized, plated at a density of 1 × 106 cells/10-cm plate, and grown for another 24 h. The cells were then harvested, fixed, and stained with propidium iodide for flow cytometry as detailed in Materials and Methods. Infected EGFP-positive (EGFP +) and uninfected EGFP-negative (EGFP −) cell populations in the AZT-free coculture (top left panel labeled w/o AZT [for without AZT]) were gated and further analyzed for cell cycle progression (two bottom left panels). The percentages of cells in the G0/G1, S, and G2/M phases of the cell cycle were computed using the ModFit LT software package. The uninfected EGFP-negative (EGFP −) cell population in the AZT-treated culture [AZT (1 μM), top right panel] was analyzed similarly (bottom right panel). (B) HTLV-1-infected HeLa cells cease proliferation. After removal of MT2 cells (see Materials and Methods), HeLa/18x21-EGFP cells that had been exposed to HTLV-1 were trysinized and plated as single cells on 10-cm plates on day 1 and grew for 6 days. Both infected (EGFP-positive) and uninfected (EGFP-negative) cells were then visualized using an Olympus IX81 fluorescence microscope. Individual EGFP-positive cells and cell clusters and the number of cells in each cluster were counted. The percentages of individual cells and cell clusters with cell number ranging from one to four and greater than four (>4) were plotted. (C) Increased expression of p21CIP1/WAF1 and p27KIP1 in HeLa cells cocultivated with MT2 cells. HeLa/18x21-EGFP-cells were cocultured with MT2 in the presence (+) or absence (−) of 1 μM AZT as described in the legend to panel A. Routinely, 5 to 6% of HeLa/18x21-EGFP cells were EGFP positive in the AZT-free culture. Cell lysates were prepared and immunoblotted with p21CIP1/WAF1, p27KIP1, Tax, and actin antibodies as described above. (D) Nuclear retention of p27KIP1 in HTLV-1-infected HeLa cells. HTLV-1 infection in HeLa cells was scored by EGFP expression (EGFP expression shown in green). The expression of p27KIP1 was detected by indirect immunofluorescence (in red) as described in Materials and Methods. The merged fluorescence image is displayed on the right. The correlation between EGFP and nuclear p27KIP1 signals is approximately 70%. (E) HTLV-1-infected HeLa cells expressed senescence-associated β-galactosidase (SA-β-Gal). HTLV-infected HeLa/18x21-EGFP cells were fixed and stained for SA-β-Gal activity using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) as a substrate (Materials and Methods). The hydrolysis of X-Gal by SA-β-Gal produced blue intracellular precipitates which appeared as dark spots in a bright-field black and white image (SA-β-galactosidase). HTLV-1 infection was indicated by EGFP expression. The two images were merged to show a direct correlation of infection and senescence.
In a separate approach, 16 h after coculture with MT2, HeLa cells were trypsinized and plated on 10-cm plates as single cells. They were then monitored by fluorescence microscopy over the course of 1 week. As shown in Fig. 3B, HTLV-1-infected HeLa cells (EGFP positive) quickly ceased cell division after infection; 72% remained as single cells, and 23% underwent only one or two cell division cycles (Fig. 3B, bar diagram) 7 days postinfection. The uninfected cells, in contrast, proliferated and formed large EGFP-negative colonies that were easily observed (Fig. 3B, compare the EGFP and Phase panels).
We next examined whether HTLV-1 infection also led to accumulation of p21CIP1/WAF1 and p27KIP1. To this end, HeLa/18x21-EGFP cells were plated at 30% confluence and cocultivated with MT2 cells for 16 h. Under this condition, the cell density reached 70% confluence 2 days postinfection, and approximately 5 to 6% of the cell population became infected with HTLV-1. Even though the efficiency of HTLV-1 infection is modest, the increase in p21CIP1/WAF1 and p27KIP1 in cells cocultivated with MT2 cells was readily apparent in the infected culture versus the AZT-treated control (Fig. 3C). Consistent with their being in a state of G1 arrest, the infected EGFP-positive cells expressed significant p27KIP1 in their nuclei compared to the neighboring uninfected EGFP-negative cells (Fig. 3D). Similar nuclear accumulation of p27KIP1 was also observed for HeLa cells transduced with LV-Tax (not shown). Finally, HTLV-1-infected HeLa/18x21-EGFP cells became enlarged, flattened, highly granulated, and expressed the senescence-associated β-galactosidase (Fig. 3E), much like what we have previously reported for LV-Tax-transduced HeLa cells (16). On the basis of these results, we conclude that HTLV-1-infected HeLa cells are in a senescence-like state.
HTLV-1-infected HOS cells escaped G1 arrest but developed mitotic abnormalities.
Efficient and persistent HTLV-1 infection of a human osteosarcoma cell line, HOS, following coculture with the C91PL cell line had been previously reported (6). This suggests that HOS cells may circumvent the senescence-like G1 arrest after HTLV-1 infection. To investigate the cell cycle effect of Tax expression on HOS cells, HOS/18x21-EGFP reporter cells were transduced with LV-Tax as previously described (16). As indicated in Fig. 4A, LV-Tax-transduced HOS/18x21-EGFP cells (scored as EGFP positive) progressed through cell division cycle without arrest (Fig. 4A), and a majority (around 60%) formed large colonies on plates (Fig. 4B). Consistent with the absence of G1 arrest, little increase in p21CIP1/WAF1 and p27KIP1 levels was observed in these cells over a course of 14 days compared to the untransduced control (Fig. 4C). While Tax-expressing HOS cells continued to divide, Tax-induced mitotic abnormalities persisted, as evidenced by the appearance of large multinucleated cells (Fig. 4B; also see Fig. 5B). Finally, the dampening effect of Tax on cell cycle progression was also readily apparent from the decrease in the EGFP-positive cell population during passage. Within 14 days after LV-Tax transduction, the population of EGFP-positive (tax-transduced) HOS cells declined from 80% to 60% (Fig. 4D), while the EGFP-negative (tax-negative) population increased in number.
FIG. 4.

LV-Tax-transduced HOS cells escape G1 arrest but develop mitotic abnormalities. (A) HOS cells escaped Tax-induced G1 arrest. HOS/18x21-EGFP cells were transduced with the LV-Tax vector as previously described (45) to achieve at least 80% gene transduction. The transduced cells were then subjected to flow cytometry as described in the legend to Fig. 3A. EGFP +, EGFP positive; EGFP −, EGFP negative. (B) LV-Tax-transduced HOS cells continued to proliferate and formed colonies. LV-Tax-transduced cells were seeded on 10-cm plates as described in the legend to Fig. 3B and allowed to grow for 6 days. EGFP-positive clusters/colonies were separated into three groups based on their sizes as indicated (growth arrested, small colony, and large colony) and counted. The percentage in each group was calculated. The numbers listed are derived from the results of two independent experiments. (C) Immunoblot of HOS cells transduced with LV-Tax at different times after gene transduction. Cell lysates were prepared from tax-transduced cells at the indicated times postinfection (Days pi) and immunoblotted (+) with Tax, cyclin B1, p27KIP1 (p27) p21CIP1/WAF1 (p21), and actin antibodies as shown. (D) LV-Tax-transduced HOS/18x21-EGFP cells were propagated in culture over a course of 2 weeks. The percentages of EGFP-positive (solid bars) and EGFP-negative (open bars) populations were determined by flow cytometry and plotted.
FIG. 5.

HTLV-1-infected-HOS cells escape G1 arrest. (A) HOS/18x21-EGFP cells were cocultivated with MT2 cells with or without (w/o) the addition of AZT as described in the legend to Fig. 3. Three days postinfection, infected and uninfected HOS cells were analyzed by flow cytometry as described in the legend to Fig. 3A for cell cycle profile. EGFP +, EGFP positive; EGFP −, EGFP negative. (B) HOS/18x21-EGFP cells were infected with HTLV-1 as described above for panel A. Cells were trypsinized and plated, grown for 6 days, photographed, and analyzed as described in the legend to Fig. 3B and 4B. The colonies/cell clusters of different sizes were counted as described in the legend to Fig. 4B. (C) HOS cells persistently infected by HTLV-1 were cloned by using limiting dilutions. A representative clone is shown. Cells with mitotic abnormalities are marked by arrows.
To assess the cell cycle effect of HTLV-1 infection, HOS/18x21-EGFP cells were cocultivated with MT2 cells in the presence or absence of 1 μM AZT as described above and analyzed by flow cytometry (Fig. 5A, top panels). Like their counterparts transduced by LV-Tax, HTLV-1-infected HOS cells (Fig. 5A, bottom left panel) continued to divide like uninfected cells (in both AZT-treated and untreated populations [Fig. 5A, top two panels]) with a comparable cell population in G1. HTLV-1-infected HOS cells, however, grew slower and showed a propensity to accumulate in G2/M with a corresponding decrease of cells in S phase. This apparent delay in G2/M is reminiscent of that of HTLV-1-transformed T cells previously characterized and attributed to the premature activation of APC by Tax (20). The persistently infected HOS cells were cloned by using limiting dilutions. Like their tax-transduced counterparts, cells with flattened, multinucleated, and dramatically altered morphology could be detected within a given clone (Fig. 5B and C).
Deficiency in p21CIP1/WAF1 and p27KIP1 allows HOS cells to evade HTLV-1-induced arrest.
We have recently shown that HTLV-1-transformed human T-cell lines invariably expressed a dramatically lower level of p27KIP1 compared to HTLV-1-unrelated T-cell lines. Furthermore, NIH 3T3 cells containing a homozygous deletion of p27KIP1 gene could stably express Tax without undergoing cell cycle arrest (16). We, therefore, surmise that functional inactivation and/or loss of expression of p21CIP1/WAF1 and p27KIP1 in particular is most likely responsible for the continued proliferation of Tax-expressing cells. In agreement with our previous prediction, the levels of p21CIP1/WAF1 and p27KIP1 in HOS cells were very low and not altered by Tax compared to those seen in HeLa cells (Fig. 6A, p21 and p27 blots).
FIG. 6.
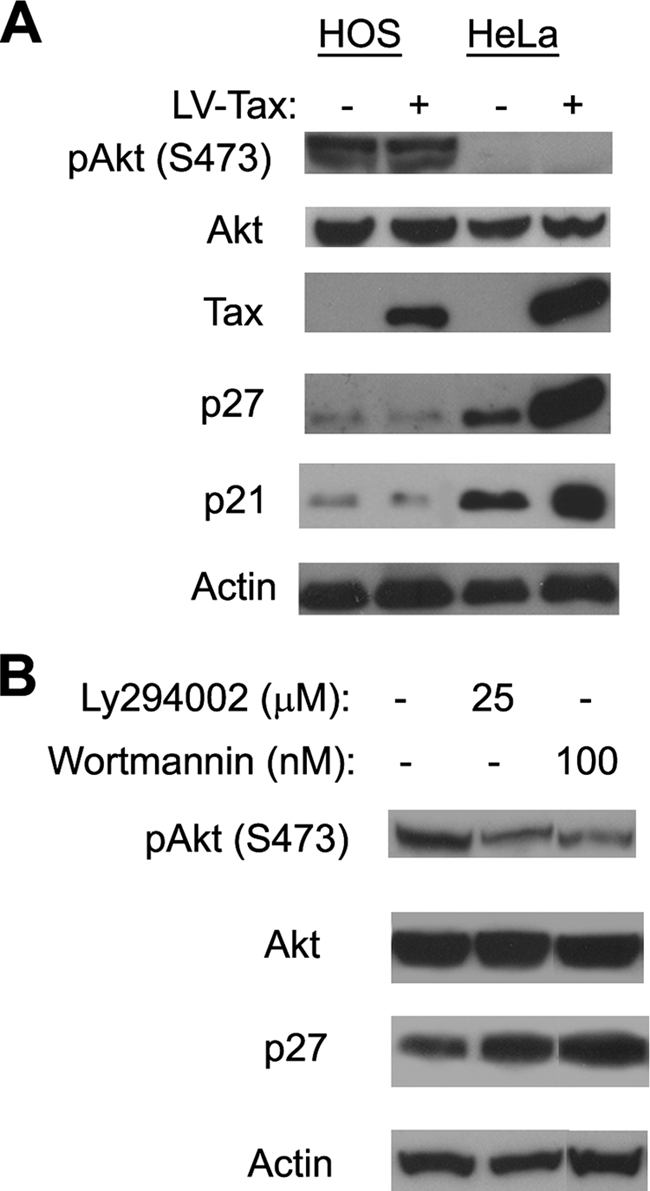
PI3K/Akt activation in HOS cells correlates with downregulation of p21CIP1/WAF1 and p27KIP1. (A) S473-phosphorylated Akt [pAkt (S473)], p21CIP1/WAF1, and p27KIP1 protein levels in HeLa and HOS cells. HOS and HeLa reporter cells were infected with LV-Tax or LV-SV40-puro. Two days postinfection, whole-cell lysates were prepared for immunoblotting with pAkt (S473) (S473-phosphorylated Akt), Akt (total Akt), Tax, p21CIP1/WAF1, p27KIP1, and actin antibodies. (B) PI3K inhibitors caused p27KIP1 level to increase in HOS cells. HOS cells were treated with the indicated concentration of PI3K inhibitor LY294002 or wortmannin for 36 h or not treated with LY294002 or wortmannin (−). Cell lysates were then prepared for immunoblotting using the serine-473-phosphorylated Akt, Akt, p27, and actin antibodies.
The control of p21CIP1/WAF1 and p27KIP1 is exerted at multiple levels. Their abundance is regulated by both transcription and translation, and the abundance of p27KIP1 is regulated by SCFSkp2-mediated degradation (30). Sequestration via complex formation with cyclin D/Cdk4 (4, 38) and cytoplasmic localization also functionally inactivate p21CIP1/WAF1 and p27KIP1 and facilitate S-phase entry (reviewed in reference 41). Activation of the PI3K-Akt (protein kinase B) pathway is known to phosphorylate and thereby inactivate the FOXO3 (Forkhead box, subgroup O) transcription factor that is required for p27KIP1expression (28). Direct phosphorylation of p27KIP1 by Akt also leads to its cytoplasmic sequestration and functional inactivation (18). Interestingly, Cereseto et al. had previously reported the activation of the PI3K-Akt pathway in HTLV-1-transformed, interleukin-2-independent T cells (3). For these reasons, we compared the status of Akt activation (phosphorylation) in HeLa and HOS cells using a phospho-Akt-specific antibody. As shown in Fig. 6A, the serine-473 phosphorylated Akt level is significantly higher in HOS cells. Furthermore, treatment of HOS cells with the PI3K inhibitors Ly294004 (25 μM) and wortmannin (100 μM) reduced Akt phosphorylation and elevated p27KIP1 expression (Fig. 6B). Together, these results suggest that the deficiency in p21CIP1/WAF1 and p27KIP1 in HOS cells is principally responsible for their escape from HTLV-1-induced G1 arrest/senescence and that this deficiency is due in part to the activation of the PI3K-Akt pathway.
SupT1 T cells infected by HTLV-1 or transduced with the tax gene undergo cell cycle arrest.
To determine the effect of HTLV-1 on the cell cycle progression of T lymphocytes, we tested five human T-cell lines, Jurkat, CEM, Molt4, H9, and SupT1 cells, for suitability for HTLV-1 infection. The cell line most suitable for such analysis is SupT1. The other cell lines either expressed high levels of phospho-Akt, which downregulated p27KIP1 or formed extensive syncytia with the HTLV-1-producing cell line MT2, making it difficult to know with certainty whether EGFP-positive cells arose from virus infection or syncytium formation (data not shown).
A SupT1 cell line, SupT1/18x21-EGFP, containing the stably integrated 18x21-EGFP reporter cassette was generated by using limiting dilutions after transduction with SMPU-18x21-EGFP. Half a million SupT1/18x21-EGFP cells were then cocultured with MT2 cells at a ratio of 1:1. Forty-eight hours after cocultivation, cells were dispersed into single cells and plated in a 12-well plate and monitored over a course of 5 to 10 days. As shown in Fig. 7A, approximately 0.5% of SupT1/18x21-EGFP cells became EGFP positive after coculture. As expected, the EGFP-positive SupT1 cells remained as single cells 5 days after coculture, while the surrounding EGFP-negative cells proliferated continuously. Because of the tendency of T cells to form clumps, after prolonged culture, the Tax-transduced single cells were often found attached to large clusters of EGFP-negative cells (Fig. 7A, day 5). Flow cytometry analysis further indicated that the EGFP-positive cells are mostly in the G1 phase of the cell cycle (not shown). Interestingly, most EGFP-positive cells were enlarged and expressed hair-like protrusions on their cell surface (Fig. 7B).
FIG. 7.

HTLV-1-infected Sup-T1 cells ceased proliferation. (A) Half a million 18X21-EGFP SupT1 reporter T cells were cocultured with the same number of MT2 cells in RPMI medium supplemented with 10% fetal bovine serum. After 48 h, cells were collected, counted, dispersed as single cells, and cultured in two 12-well plates at a density of approximately 2,000 cells/well in the same medium. Cells were visualized at 1, 3, or 5 days after limiting dilutions. (B) HTLV-1-infected Sup-T1 cells (EGFP positive) are enlarged and expressed hair-like surface protrusions. EGFP fluorescence and bright-field images and a merged image of the two (merge) of the same visual field are shown. Two randomly selected visual fields from two separate wells are shown.
To demonstrate that the cell cycle arrest of HTLV-1-infected EGFP-positive SupT1 is mediated by Tax, we transduced the SupT1/18x21-EGFP cells with LV-Tax or LV-Puro at an MOI of 1 (titered on HeLa/18x21-EGFP cells) and analyzed them by flow cytometry (Fig. 8A). As expected, the fraction of EGFP-positive SupT1 cells (transduced with tax) that were in S phase was significantly smaller (17%) than the fraction of untransduced EGFP-negative counterparts (33%). Indeed, most of the EGFP-positive SupT1 cells became accumulated at G1 in contrast to the untransduced cells (Fig. 8A, 74% versus 59%). As anticipated, the levels of p21CIP1/WAF1 and p27KIP in the Tax-transduced cells increased significantly, while that of cyclin B1 decreased (Fig. 8B). In a separate experiment, SupT1/18x21-EGFP cells were transduced with LV-Tax or LV-EGFP. The lentiviral vector-transduced cells were then dispersed and seeded at a density of 5 cells/well in a 96-well plate for 8 days. Here again, the LV-Tax-transduced (EGFP-positive) SupT1 cells failed to divide and remained as single cells over the entire time course of the experiment, while the untransduced (EGFP-negative) cells continued to divide. As in Fig. 7A, after prolonged culture, single EGFP-positive cells were often found attached to large clusters of EGFP-negative cells (Fig. 8C, LV-Tax day 4 and day 8). In contrast and as expected, the LV-EGFP-transduced SupT1 cells expressed EGFP and grew to form large clusters of EGFP-positive cells. In aggregate, these results strongly suggest that HTLV-1-infected T cells, like HeLa cells, are likely to be in a state of Tax-induced senescence/G1 arrest, contrary to the prevailing notion that HTLV-1 infection leads to T-cell proliferation.
FIG. 8.

LV-Tax-transduced-SupT1 cells ceased proliferation. (A) SupT1/18x21-EGFP reporter cells were transduced with the LV-Tax-SV-Puro or the LV-SV-Puro and cultured for 48 h in RPMI medium supplemented with 10% fetal bovine serum. Cells were collected, fixed, stained, and analyzed by flow cytometry as described in the legend to Fig. 3A. EGFP+, EGFP positive; EGFP−, EGFP negative. (B) SupT1/18x21-EGFP reporter cells were infected with the LV-Tax-SV-Puro or the LV-SV-Puro vectors such that more than 50% SupT1 cells became transduced (based on EGFP expression). Two days posttransduction, cells were harvested and analyzed by Western blotting for cyclin B1, p27, p21, and β-actin (protein control) as indicated. (C) SupT1/18x21-EGFP cells were infected with LV-Tax or LV-EGFP control and grown as described above for panel A. After 48 h, cells were collected, counted, and cultured in two 96-well plates at a density of 5 cells/per well by using limiting dilutions. Cell growth was monitored with an Olympus IX81 fluorescence microscope at 2, 3, 4, and 8 days after the limiting dilution.
DISCUSSION
It is generally thought that after HTLV-1 infection, viral proteins, such as Tax, promote cell proliferation. The ensuing oligoclonal expansion of infected T cells in turn leads to the onset of ATL. This line of thinking, however, cannot satisfactorily explain the low penetrance and long clinical latency of ATL following HTLV-1 infection. Furthermore, although HTLV-1 can immortalize primary T lymphocytes in culture, the fraction of T cells that become immortalized is low. Finally, one group of transgenic mice expressing Tax had been found to develop extensive thymic depletion and died rapidly after birth (31), and rabbits infected by HTLV-1 through injection of a virus-producing T-cell line, RH/K34, also showed thymic atrophy prior to developing ATL-like disease (42).
In this study, we have examined the biological effects of HTLV-1 infection on three different cell lines, HeLa, HOS, and SupT1. Contrary to the accepted paradigm, HTLV-1-infected HeLa cells ceased proliferation within one or two cell division cycles and became permanently arrested in G1. Increased expression of CDK2 inhibitors p21CIP1/WAF1 and p27KIP1 was readily detected even though only 5 to 6% of HeLa cells became infected, suggesting elevated production of both proteins in the infected cells, much like what had been previously observed in tax-transduced HeLa cells (16). Likewise, SupT1 cells infected by HTLV-1 or transduced with the tax gene via a lentivirus vector ceased proliferation and arrested in G1. In contrast, HOS cells became persistently infected by HTLV-1 and continued to grow and divide after infection, albeit at a slower rate and with mitotic abnormalities. This phenotype correlated with the loss of p21CIP1/WAF1 and p27KIP1 from HOS cells. We noted that the HTLV-1-infected HeLa and SupT1 cells persist in cell culture over a course of at least 2 weeks and likely could survive even longer. As HTLV-1 is transmitted by cell-to-cell contact, the survival of infected cells in a state similar to terminal differentiation may increase their longevity to facilitate viral spread.
The increase in p21CIP1/WAF1 and especially p27KIP1 seen in HTLV-1-infected HeLa cells is most likely due to the premature activation of the APC by Tax as described earlier (16). Induction of p21CIP1/WAF1 and p27KIP1 was also seen in Tax-expressing SupT1 cells (not shown). The same most likely occurred in HTLV-1-infected SupT1 T cells. We infer from these results and published literature that after HTLV-1 infection, quiescent T cells may be driven to enter G1/S by Tax (23, 32, 40). Once these cells enter into cell cycle, Tax-activated APC initiates the mitotic exit program ahead of schedule and commits HTLV-1-infected cells into a p21CIP1/WAF1/p27KIP1-mediated arrest/senescence (16). For the infected cells to escape from the arrest, a loss and/or inactivation of p21CIP1/WAF1 and p27KIP1 is necessary. On the basis of these results, we suggest that only when HTLV-1 infects or reactivates from T cells whose p21CIP1/WAF1 and p27KIP1 function and/or expression have/has been lost, could the carcinogenic potentials of Tax (NF-κB activation, inactivation of p53, perturbation of mitotic functions, and damage of DNA repair) be realized. This is likely followed by a repression of viral gene expression by epigenetic mechanisms, which allow transformed T cells not only to evade cytotoxic T lymphocyte killing but also to avert the Tax-induced mitotic aberrations that are detrimental to cell proliferation.
We have noticed that although HTLV-1-infected or tax-transduced HOS cells could continue to divide and proliferate, their number declined after passage compared to untransduced or uninfected counterparts in the same culture (Fig. 4D). Further, some cells (scored as EGFP positive and therefore Tax positive) eventually became flattened and enlarged, even though their levels of p21CIP1/WAF1 and p27KIP1 did not increase significantly (Fig. 5C). The cell cycle abnormalities in infected HOS cells likely originated from the various mitotic defects associated with Tax-induced APC activation, such as DNA aneuploidy and cytokinesis failure. The direct targeting of centrosomal components by Tax reported recently may also play an important role in this regard (5, 35). Finally, other viral proteins may contribute to this demise.
The gradual loss of proliferative potential from HTLV-1-infected or tax-transduced HOS cells contrasts with HTLV-1-producing and/or HTLV-1-transformed human T-cell lines, which express Tax abundantly, yet can proliferate indefinitely in culture. For this reason, we think the loss of p21CIP1/WAF1 and p27KIP1 expression and/or function is necessary but not sufficient for full cell transformation. Most likely, additional somatic changes had taken place in the transformed T cells to prevent the mitotic crisis and loss of ability to proliferate caused by Tax and HTLV-1. Whether Tax plays an active role in effecting these changes remains to be determined.
Signaling through the PI3K-Akt pathway is known to lower the levels of p21CIP1/WAF1 and p27KIP1 and functionally inactivates p21CIP1/WAF1 and p27KIP1 by localizing them to the cytoplasm (2). Our results indicate that a loss of p21CIP1/WAF1/p27KIP1expression and/or functions from HOS cells, in part through PI3K/Akt activation, may have allowed these cells to proliferate after HTLV-1 infection (Fig. 6). Constitutive activation of the PI3K pathway appears to be a common feature of HTLV-1- and Tax-transformed cells. Most, if not all, of these cells have barely detectable levels of p27KIP1 (3, 14). In addition, even though they all expressed high levels of p21CIP1/WAF1, the p21CIP1/WAF1 is primarily localized in the cytoplasm (unpublished data). Finally, treatment of interleukin-2-independent HTLV-1-transformed T cells with inhibitors of PI3 kinase leads to a p27KIP1-dependent cell cycle arrest (3, 14). Transformation of Rat-1 fibroblast cells by Tax is also associated with activation of PI3K and its downstream kinase, Akt (22). Most recently, Fukuda et al. have shown that signaling through the activation-inducible lymphocyte immunomediatory molecule (AILIM)/inducible costimulator (ICOS) leads to PI3K/Akt activation, microtubule rearrangement, and formation of multilobulated nuclei characteristic of the flower cells seen in acute ATL (10). These published reports and the results detailed herein are consistent with the notion that dysregulation of the PI3K pathway may constitute a critical step for escape from HTLV-1-induced cell cycle arrest and possibly collaborate with Tax to promote the progression to ATL. A direct examination of ATL cells in this regard may begin to shed light on the cellular changes that are needed to collude with HTLV-1 and Tax to cause T-cell transformation and ATL.
Acknowledgments
We thank K. Wolcott and K. Lund of the Uniformed Services University Biomedical Instrumentation Center for help with flow cytometry and K. S. Jones of SAIC-Frederick of National Cancer Institute for helpful discussions.
This work was supported by a grant (R01CA115884) from the National Institutes of Health.
Footnotes
Published ahead of print on 2 July 2008.
REFERENCES
- 1.Barnard, A. L., T. Igakura, Y. Tanaka, G. P. Taylor, and C. R. Bangham. 2005. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 106988-995. [DOI] [PubMed] [Google Scholar]
- 2.Brazil, D. P., Z. Z. Yang, and B. A. Hemmings. 2004. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 29233-242. [DOI] [PubMed] [Google Scholar]
- 3.Cereseto, A., P. R. Washington, E. Rivadeneira, and G. Franchini. 1999. Limiting amounts of p27Kip1 correlates with constitutive activation of cyclin E-CDK2 complex in HTLV-I-transformed T-cells. Oncogene 182441-2450. [DOI] [PubMed] [Google Scholar]
- 4.Cheng, M., P. Olivier, J. A. Diehl, M. Fero, M. F. Roussel, J. M. Roberts, and C. J. Sherr. 1999. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 181571-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ching, Y. P., S. F. Chan, K. T. Jeang, and D. Y. Jin. 2006. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat. Cell Biol. 8717-724. [DOI] [PubMed] [Google Scholar]
- 6.Clapham, P., K. Nagy, R. Cheingsong-Popov, M. Exley, and R. A. Weiss. 1983. Productive infection and cell-free transmission of human T-cell leukemia virus in a nonlymphoid cell line. Science 2221125-1127. [DOI] [PubMed] [Google Scholar]
- 7.Derse, D., G. Heidecker, M. Mitchell, S. Hill, P. Lloyd, and G. Princler. 2004. Infectious transmission and replication of human T-cell leukemia virus type 1. Front Biosci. 92495-2499. [DOI] [PubMed] [Google Scholar]
- 8.Derse, D., S. A. Hill, P. A. Lloyd, H. Chung, and B. A. Morse. 2001. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J. Virol. 758461-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franchini, G., F. Wong Staal, and R. C. Gallo. 1984. Human T-cell leukemia virus (HTLV-I) transcripts in fresh and cultured cells of patients with adult T-cell leukemia. Proc. Natl. Acad. Sci. USA 816207-6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukuda, R., A. Hayashi, A. Utsunomiya, Y. Nukada, R. Fukui, K. Itoh, K. Tezuka, K. Ohashi, K. Mizuno, M. Sakamoto, M. Hamanoue, and T. Tsuji. 2005. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL). Proc. Natl. Acad. Sci. USA 10215213-15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haller, K., T. Ruckes, I. Schmitt, D. Saul, E. Derow, and R. Grassmann. 2000. Tax-dependent stimulation of G1 phase-specific cyclin-dependent kinases and increased expression of signal transduction genes characterize HTLV type 1-transformed T cells. AIDS Res. Hum. Retrovir. 161683-1688. [DOI] [PubMed] [Google Scholar]
- 12.Haoudi, A., R. C. Daniels, E. Wong, G. Kupfer, and O. J. Semmes. 2003. Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem. 27837736-37744. [DOI] [PubMed] [Google Scholar]
- 13.Igakura, T., J. C. Stinchcombe, P. K. Goon, G. P. Taylor, J. N. Weber, G. M. Griffiths, Y. Tanaka, M. Osame, and C. R. Bangham. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2991713-1716. [DOI] [PubMed] [Google Scholar]
- 14.Iwanaga, R., K. Ohtani, T. Hayashi, and M. Nakamura. 2001. Molecular mechanism of cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene 202055-2067. [DOI] [PubMed] [Google Scholar]
- 15.Iwanaga, Y., T. Tsukahara, T. Ohashi, Y. Tanaka, M. Arai, M. Nakamura, K. Ohtani, Y. Koya, M. Kannagi, N. Yamamoto, and M. Fujii. 1999. Human T-cell leukemia virus type 1 Tax protein abrogates interleukin-2 dependence in a mouse T-cell line. J. Virol. 731271-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo, Y. L., and C. Z. Giam. 2006. Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. EMBO J. 251741-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemoine, F. J., and S. J. Marriott. 2002. Genomic instability driven by the human T-cell leukemia virus type I (HTLV-I) oncoprotein, Tax. Oncogene 217230-7234. [DOI] [PubMed] [Google Scholar]
- 18.Liang, J., J. Zubovitz, T. Petrocelli, R. Kotchetkov, M. K. Connor, K. Han, J. H. Lee, S. Ciarallo, C. Catzavelos, R. Beniston, E. Franssen, and J. M. Slingerland. 2002. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 81153-1160. [DOI] [PubMed] [Google Scholar]
- 19.Liang, M. H., T. Geisbert, Y. Yao, S. H. Hinrichs, and C. Z. Giam. 2002. Human T-lymphotropic virus type 1 oncoprotein Tax promotes S-phase entry but blocks mitosis. J. Virol. 764022-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, B., S. Hong, Z. Tang, H. Yu, and C. Z. Giam. 2005. HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc. Natl. Acad. Sci. USA 10263-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, B., M. H. Liang, Y. L. Kuo, W. Liao, I. Boros, T. Kleinberger, J. Blancato, and C. Z. Giam. 2003. Human T-lymphotropic virus type 1 oncoprotein Tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol. Cell. Biol. 235269-5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu, Y., Y. Wang, M. Yamakuchi, S. Masuda, T. Tokioka, S. Yamaoka, I. Maruyama, and I. Kitajima. 2001. Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene 202514-2526. [DOI] [PubMed] [Google Scholar]
- 23.Mahieux, R., C. A. Pise-Masison, P. F. Lambert, C. Nicot, L. De Marchis, A. Gessain, P. Green, W. Hall, and J. N. Brady. 2000. Differences in the ability of human T-cell lymphotropic virus type 1 (HTLV-1) and HTLV-2 Tax to inhibit p53 function. J. Virol. 746866-6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majone, F., and K. T. Jeang. 2000. Clastogenic effect of the human T-cell leukemia virus type I Tax oncoprotein correlates with unstabilized DNA breaks. J. Biol. Chem. 27532906-32910. [DOI] [PubMed] [Google Scholar]
- 25.Majone, F., O. J. Semmes, and K. T. Jeang. 1993. Induction of micronuclei by HTLV-I Tax: a cellular assay for function. Virology 193456-459. [DOI] [PubMed] [Google Scholar]
- 26.Manel, N., F. J. Kim, S. Kinet, N. Taylor, M. Sitbon, and J. L. Battini. 2003. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 115449-459. [DOI] [PubMed] [Google Scholar]
- 27.Marriott, S. J., and O. J. Semmes. 2005. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 245986-5995. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Gac, L., B. Alvarez, Z. Garcia, M. Marques, M. Arrizabalaga, and A. C. Carrera. 2004. Phosphoinositide 3-kinase and Forkhead, a switch for cell division. Biochem. Soc. Trans. 32360-361. [DOI] [PubMed] [Google Scholar]
- 29.Mesnard, J. M., B. Barbeau, and C. Devaux. 2006. HBZ, a new important player in the mystery of adult-T-cell leukemia. Blood 1083979-3982. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama, K., H. Nagahama, Y. A. Minamishima, M. Matsumoto, I. Nakamichi, K. Kitagawa, M. Shirane, R. Tsunematsu, T. Tsukiyama, N. Ishida, M. Kitagawa, K. Nakayama, and S. Hatakeyama. 2000. Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 192069-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nerenberg, M., S. H. Hinrichs, R. K. Reynolds, G. Khoury, and G. Jay. 1987. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science 2371324-1329. [DOI] [PubMed] [Google Scholar]
- 32.Ohtani, K., R. Iwanaga, M. Arai, Y. Huang, Y. Matsumura, and M. Nakamura. 2000. Cell type-specific E2F activation and cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. J. Biol. Chem. 27511154-11163. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto, T., Y. Ohno, S. Tsugane, S. Watanabe, M. Shimoyama, K. Tajima, M. Miwa, and K. Shimotohno. 1989. Multi-step carcinogenesis model for adult T-cell leukemia. Jpn. J. Cancer Res. 80191-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park, H. U., S. J. Jeong, J. H. Jeong, J. H. Chung, and J. N. Brady. 2006. Human T-cell leukemia virus type 1 Tax attenuates gamma-irradiation-induced apoptosis through physical interaction with Chk2. Oncogene 25438-447. [DOI] [PubMed] [Google Scholar]
- 35.Peloponese, J. M., Jr., K. Haller, A. Miyazato, and K. T. Jeang. 2005. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc. Natl. Acad. Sci. USA 10218974-18979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pise-Masison, C. A., R. Mahieux, H. Jiang, M. Ashcroft, M. Radonovich, J. Duvall, C. Guillerm, and J. N. Brady. 2000. Inactivation of p53 by human T-cell lymphotropic virus type 1 Tax requires activation of the NF-κB pathway and is dependent on p53 phosphorylation. Mol. Cell. Biol. 203377-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pise-Masison, C. A., R. Mahieux, M. Radonovich, H. Jiang, J. Duvall, C. Guillerm, and J. N. Brady. 2000. Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res. Hum. Retrovir. 161669-1675. [DOI] [PubMed] [Google Scholar]
- 38.Polyak, K., J. Y. Kato, M. J. Solomon, C. J. Sherr, J. Massague, J. M. Roberts, and A. Koff. 1994. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 89-22. [DOI] [PubMed] [Google Scholar]
- 39.Satou, Y., J. Yasunaga, M. Yoshida, and M. Matsuoka. 2006. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 103720-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt, I., O. Rosin, P. Rohwer, M. Gossen, and R. Grassmann. 1998. Stimulation of cyclin-dependent kinase activity and G1- to S-phase transition in human lymphocytes by the human T-cell leukemia/lymphotropic virus type 1 Tax protein. J. Virol. 72633-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 131501-1512. [DOI] [PubMed] [Google Scholar]
- 42.Simpson, R. M., T. M. Zhao, B. S. Hubbard, S. Sawasdikosol, and T. J. Kindt. 1996. Experimental acute adult T cell leukemia-lymphoma is associated with thymic atrophy in human T cell leukemia virus type I infection. Lab. Investig. 74696-710. [PubMed] [Google Scholar]
- 43.Tsukahara, T., M. Kannagi, T. Ohashi, H. Kato, M. Arai, G. Nunez, Y. Iwanaga, N. Yamamoto, K. Ohtani, M. Nakamura, and M. L. Fujii. 1999. Induction of Bcl-xL expression by human T-cell leukemia virus type 1 Tax through NF-κB in apoptosis-resistant T-cell transfectants with Tax. J. Virol. 737981-7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waldele, K., K. Silbermann, G. Schneider, T. Ruckes, B. R. Cullen, and R. Grassmann. 2006. Requirement of the human T-cell leukemia virus (HTLV-1) tax-stimulated HIAP-1 gene for the survival of transformed lymphocytes. Blood 1074491-4499. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, L., M. Liu, R. Merling, and C. Z. Giam. 2006. Versatile reporter systems show that transactivation by human T-cell leukemia virus type 1 Tax occurs independently of chromatin remodeling factor BRG1. J. Virol. 807459-7468. [DOI] [PMC free article] [PubMed] [Google Scholar]