

Abstract
Human endogenous retroviruses (HERVs) comprise approximately 8% of the human genome, but all are remnants of ancient retroviral infections and harbor inactivating mutations that render them replication defective. Nevertheless, as viral “fossils,” HERVs may provide insights into ancient retrovirus-host interactions and their evolution. Indeed, one endogenous retrovirus [HERV-K(HML-2)], which has replicated in humans for the past few million years but is now thought to be extinct, was recently reconstituted in a functional form, and infection assays based on it have been established. Here, we show that several human APOBEC3 proteins are intrinsically capable of mutating and inhibiting infection by HERV-K(HML-2) in cell culture. We also present striking evidence that two HERV-K(HML-2) proviruses that are fixed in the modern human genome (HERV-K60 and HERV-KI) were subjected to hypermutation by a cytidine deaminase. Inspection of the spectrum of mutations that are found in HERV-K proviruses in the human genome and HERV-K DNA generated during in vitro replication in the presence of each of the human APOBEC3 proteins unequivocally identifies APOBEC3G as the cytidine deaminase responsible for hypermutation of HERV-K60 and HERV-KI. This is a rare example of the antiretroviral effects of APOBEC3G in the setting of natural human infection, whose consequences have been fossilized in human DNA, and a striking example of inactivation of ancient retroviruses in humans through enzymatic cytidine deamination.
Retroviruses can become endogenous when they infect germ line cells, or their progenitors, which subsequently constitute the gametes that give rise to viable progeny. Thereafter, endogenous retroviruses (ERVs) behave much like other host genomic DNA elements; they are inherited in a Mendelian manner and can become fixed or lost in the host population depending on their effect on the reproductive fitness of the host (40). As the presence of active, replication-competent proviruses in a host genome is most likely to be deleterious to host fitness through insertional mutagenesis, cytopathic virus production, ectopic recombination, and alteration of host gene transcription by viral promoters, endogenous retroviruses are often transcriptionally silenced (9, 39).
Human endogenous retroviruses (HERVs) comprise approximately 8% of the human genome, but all are remnants of ancient retroviral infections and harbor inactivating mutations that render them replication defective (reviewed in reference 2). Nevertheless, as viral “fossils,” HERVs may provide insights into the ancient retroviral-host interactions and their evolution. For example, several antiretroviral genes have been identified in recent years (24, 30, 34), including the APOBEC3 family of cytidine deaminase-encoding genes, which have proliferated during mammalian speciation and many members of which exhibit signs of positive (diversifying) selection in the primate lineage (28). This evolutionary legacy likely affects the course of viral infections in modern organisms, including humans. Indeed, human immunodeficiency virus type 1 (HIV-1) and hepatitis B virus (HBV) sequences in infected humans exhibit patterns of G-to-A hypermutations which suggest that APOBEC proteins have a current physiological role as an antiviral defense (6, 8, 22, 31, 35, 38). The source of the ancient evolutionary pressure on APOBEC proteins is unknown, but the ERVs that are fossilized in the genomes of modern humans and other primates are reasonable candidates, particularly given the recent observation that endogenous murine leukemia viruses bear signatures of mutation by the single murine APOBEC3 protein (18).
Among HERVs, the HERV-K(HML-2) family is thought to be the youngest, with multiple human-specific and polymorphic insertions that indicate replication in human ancestors within the past few million years (3, 4, 15, 37). None of the known proviruses in modern humans are capable of replication and, therefore, many aspects of HERV-K biology could not previously be studied. Recently, however, we and others have derived a consensus HERV-K(HML-2) sequence by aligning human specific proviruses and synthesized a pseudo-ancestral proviral construct (11, 21). We have shown that this virus, named HERV-KCON, encodes and expresses all viral proteins necessary for infection, and we have derived an experimental system in which HERV-KCON can be shown to infect numerous cell lines in a single-cycle assay. Using this approach, we also have shown that human APOBEC3F (hA3F) protein can inhibit HERV-KCON infection in single-cycle, transient-overexpression assays (21).
In this study, we expand our study of ancient retrovirus-host interactions to other APOBEC3 family members and find that several APOBEC3 proteins are intrinsically capable of mutating and inhibiting infection by HERV-K. We also present striking evidence that two HERV-K(HML-2) proviruses that are present in the human genome have been subjected to hypermutation by APOBEC3G (hA3G), in a manner that can be accurately recapitulated during HERV-K replication in vitro. Along with evidence of hA3G hypermutation in HIV-1- and HBV-infected patient samples (17, 35, 38), this is a rare example of the antiretroviral effects of hA3G in a natural infection setting and the first example where inactivation of ancient retroviruses in humans by hA3G can be demonstrated.
MATERIALS AND METHODS
Cell lines and transfection.
293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and gentamicin. CEM cells were maintained in RPMI supplemented with 10% fetal calf serum and gentamicin. For transfection, 293T cells were seeded at around 1.5 × 106 cells in six-well plates and transfected using polyethylenimine as previously described (21). Medium was changed 5 h after transfection with fresh medium containing 5 μM sodium butyrate, and virus-containing supernatants were collected after an additional 40 h.
Expression plasmids.
The HERV-KCON vector genome plasmid CCGBX is a derivative of the previously described CHKCG (21) and was constructed by inserting a CMVP-EGFP cassette into XbaI and SwaI sites of CHKCG, resulting in a vector genome that is slightly smaller than CHKCG and gives higher infectious titers. CCGBX-P was derived from CCGBX by inserting a 53-bp HIV-1-derived sequence (GATCTGAGCCTGGGAGCTCTCTGGCTTGTGACTCTGGTAACTAGAGATCCCTC) into the 5′ end of the 3′ long terminal repeat (LTR) to allow specific amplification of newly introduced HERV-KCON sequences upon infection of human cells. The insertion was made using overlap extension PCR and SwaI and NheI restriction sites in the HERV-KCON vector sequence. HERV-K protein expression vectors (pCRVI/Con Gag-PR-Pol and pCR3.1/K-Rev) have been previously described (21). CSGW, a packageable HIV-1 vector plasmid expressing enhanced green fluorescent protein (EGFP), and HIV-1NL4-3 GagPol expression plasmids have been described elsewhere (1, 13). Plasmids expressing various human APOBEC3 proteins, namely hA3A, hA3B, hA3C, hA3F, and hA3G, have been described previously (6). Plasmids expressing additional human APOBEC3 proteins (hA3DE and hA3H) were cloned using the same pCMV4-HA vector and HindIII and XbaI restriction sites.
Infection assays.
To generate infectious HERV-KCON-derived virions, 293T cells in six-well plates were transfected with 1.8 μg CCGBX, 0.5 μg pCRVI/Con Gag-PR-Pol, 0.5 μg pCR3.1/K-Rev, 0.2 μg G protein of vesicular stomatitis virus (VSV-G), and an APOBEC3 expression plasmid or empty vector control. To generate infectious HIV-1 virions, 293T cells were transfected in six-well plates with 0.75 μg of CSGW, 0.75 μg HIV-1 Gag-Pol, 0.2 μg VSV-G, and an APOBEC3 expression plasmid or empty vector control. No Vif protein was expressed during the generation of virions. Media were changed 5 h after transfection with fresh media containing 5 μM sodium butyrate (sodium butyrate for HERV-K only), and virus-containing supernatants were collected after an additional 40 h. For HERV-K infection, filtered (0.2-μm pore size) supernatant was layered onto 5 × 104 293T cells in 24-well plates with fresh medium supplemented with 5 μg of polybrene/ml and spinoculated at 2,000 rpm for 2 hours at room temperature. For HIV-1 infection, filtered supernatant was layered onto 2.5 × 104 CEM cells in 96-well plates with fresh medium and 5 μg of polybrene/ml. Two days after infection, GFP+ cells were quantified by fluorescence-activated cell sorter analysis.
HERV-K sequence analyses.
Full-length HERV-K(HML-2) sequences in the human genome were identified using a TBLASTX search (www.ensembl.org/Multi/blastview) of the human genome with the HERV-KCON Gag sequence as the query sequence. Sixteen unique (by chromosomal location) human-specific full-length HERV-K(HML-2) proviruses were identified by cross-referencing with insertions identified by Belshaw et al. and Romano et al. (4, 27). All identified insertions were included in subgroup N, as defined by Romano et al. (27). Specifically, the proviruses included K101, K102, K104, K106, K107, K108, K109, K113, K115, 11q22, 12q14, 19q12, 1p31 (K4), 3q27 (K50b), 3q21 (KI), and 21q21 (K60). All GenBank accession numbers for these sequences are found in the reports by Barbulescu et al. and Romano et al. In addition, the K60 sequence was deduced from GenBank entry AL109763 (3, 27). The proviruses were aligned to HERV-KCON using AlignX (Invitrogen) for comparison. Figures 2B and 4A, below, depict G-to-A differences between the provirus and HERV-KCON and were derived using the HYPERMUT program (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html).
FIG. 2.

Nucleotide changes in human-specific HERV-K proviruses relative to HERV-KCON. (A) The numbers of changes of each type from the HERV-KCON to sequences in endogenous proviruses (listed in Materials and Methods) are plotted. For comparison of sequences flanking HERV-K60 and HERV-KI, 4,000 nucleotides immediately proximal to the 5′ and 3′ ends of the proviruses were compared for changes from chimpanzee to the orthologous human sequence. For each sequence comparison, the numbers of changes were normalized to enable direct comparison with the numbers of changes in the individual HERV-K60 and HERV-KI proviruses. (B) Graphical representation of nucleotide changes relative to HERV-KCON in the 16 human specific proviruses. Red, GG to AG; cyan, GA to AA; green, GC to AC; magenta, GT to AT; black, non-G-to-A transitions; yellow, gaps in the sequence.
FIG. 4.

Hypermutation of HERV-K during in vitro infection. All changes in HERV-KCON reverse transcripts relative to preinfection sequence were analyzed for +1 site preference using HYPERMUT, and the sequences of 12 HERV-K clones generated during infection in the presence of each APOBEC protein are represented as horizontal lines. Mutations are indicated as vertical lines. Red, GG to AG; cyan, GA to AA; green, GC to AC; magenta, GT to AT; black, non-G-to-A transitions. (B) Flanking nucleotide sequence context surrounding mutations generated by APOBEC3 proteins during HERV-K infection in the presence of hA3A, hA3B, hA3F, and hA3G. The numbers of times that each nucleotide occurred at five positions 5′ and 3′ to each G-to-A change were plotted.
The P values that accompany the sequence analyses were calculated by a chi-square test of independence to determine whether the frequency at which each nucleotide occurred at each position flanking each mutation was significantly different relative to its expected frequency based on nucleotide composition.
HERV-K hypermutation assay.
Infectious HERV-KCON particles were generated as described above using CCGBX-P in place of CCGBX. Prior to infection, supernatant was supplemented with 10 mM MgCl2 and treated with DNase I (0.1 U per μl; Roche) for 1 h at 37°C to eliminate residual transfected DNA. Fresh 293T cells were infected as described above. Ten hours postinfection, total DNA was collected using the DNeasy blood and tissue kit (Qiagen). Partial EGFP (Clontech) and HERV-K sequence of 762 nucleotides were amplified using oligos designed to target EGFP (CGC ACC ATC TTC TTC AAG GAC GAC G) and the inserted HIV-1 sequence (GAG GGA TCT CTA GTT ACC AGA GTC ACA AGC C) using Phusion polymerase (Invitrogen) (98°C for 10 s, 55°C for 10 s, and 72°C for 15 s; 30 cycles). Amplified DNA was purified using a gel extraction kit (Qiagen) and cloned into pCR-Blunt II-TOPO according to the manufacturer's instructions (Invitrogen). To confirm the complete elimination of transfected DNA, we attempted to amplify plasmid sequence using primers targeting the plasmid backbone sequence and the untranslated region of HERV-K using similar PCR conditions. Also, to confirm that the amplified sequences were derived from de novo reverse-transcribed DNA, HERV-K particles containing a mutationally inactivated reverse transcriptase were subjected to the same infection procedure and PCR analysis. Twelve clones were sequenced for each APOBEC3 protein studied, as well as the empty vector control, and compared to the original CCGBX-P sequence for evidence of hypermutation.
Western blot analysis.
To generate an anti-CA polyclonal antiserum, the N-terminal cleavage site of CA was first determined by Edman sequencing of the putative CA protein isolated from purified HERV-KCON virion particles. As CA is known to be around 30 kDa, the position of the C-terminal CA cleavage site was estimated using this molecular mass and the determined position of the CA N terminus. The deduced CA-encoding sequence was cloned into pGEX-6P-1 (GE Healthcare Life Sciences) to express a glutathione S-transferase (GST)-tagged CA protein that was purified using glutathione-agarose beads. The GST tag was eliminated by PreScission protease cleavage as per the manufacturer's instructions (GE Healthcare Life Sciences). The purified recombinant CA protein was used to generate the antiserum (Covance).
Transfected 293T cells were lysed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer and separated on 4 to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gradient gels. Proteins were transferred onto nitrocellulose membranes and probed with anti-HERV-K Gag, anti-HIV-1 CA, or anti-hemagglutinin (anti-HA; Covance) primary antibody followed by anti-rabbit or anti-mouse antibody-peroxidase conjugate.
RESULTS
Inhibition of HERV-KCON infection by APOBEC3 proteins.
Given our previous observation that HERV-K infectivity can be inhibited by overexpressed hA3F (21), we determined whether other human APOBEC3 proteins that HERV-K might encounter during its replication could also inhibit HERV-K infection. Although it would be optimal to test infection under physiologically relevant protein levels, the expression level of most APOBEC3 proteins in various tissues is unknown. Moreover, the tissue tropism of HERV-K is unknown. Instead, by titrating the amount of expression vector in our assays, we expressed APOBEC3 proteins at various, and relatively low, levels that mimicked the range of protein levels at which hA3G, hA3F, and hA3B can inhibit HIV-1 infection in the absence of Vif (Fig. 1A and B).
FIG. 1.

Effects of APOBEC3 proteins on HERV-KCON infection. (A) Anti-HA (top panel) and anti-HERV-K CA (bottom panel) Western blot of 293T cell lysates transfected with HERV-KCON, VSV-G, and APOBEC-HA plasmids. (B) Infection of CEM cells with HIV-1 (left panel) or 293T cells with HERV-KCON virions, generated in the presence of the indicated APOBEC3-HA proteins. Infectious units were quantified as GFP+ cells using fluorescence-activated cell sorter analysis 2 days postinfection, and are expressed as a percentage of the number of infected cells (typically 15 to 30%) that were obtained in the absence of an APOBEC3 protein.
HERV-KCON virus-like particles were generated in 293T cells in the presence of each of the C-terminally HA-tagged human APOBEC3 proteins. Western blot analysis of cell lysates showed that APOBEC3 protein expression varied (Fig. 1A), but the range of expression levels for most of the proteins overlapped as a result of transfecting various levels of the corresponding expression plasmids. However, hA3DE and hA3H were comparatively poorly expressed (26). Importantly, HERV-K Gag was expressed equally in all samples (Fig. 1A). The only exception to this was at high levels of hA3B expression, which appeared to slightly reduce the levels of HERV-K Gag expression.
Fresh 293T cells were infected with HERV-K virions generated in the presence of each of the APOBEC3 proteins. As can be seen in Fig. 1B, hA3A, hA3B, and hA3F inhibited HERV-K-CON infection by approximately fivefold at the highest concentration tested. Only marginal inhibition of infection restriction was seen with hA3G, while hA3C, hA3DE, and hA3H did not inhibit infection. Clearly, the relative sensitivity of HERV-K to the various APOBEC3 proteins differed from that of HIV-1 (Fig. 1B).
Hypermutated HERV-K proviruses in modern human DNA.
While several APOBEC3 proteins appeared capable of inhibiting HERV-K infection in vitro, to determine whether restriction of HERV-K infection might have occurred in vivo, we investigated whether evidence of APOBEC3-induced mutation could be found in HERV-K proviruses that are present in modern human DNA. Specifically, we inspected 16 human-specific full-length HERV-K(HML-2) proviruses and looked for biases in the patterns of mutation therein, relative to the pseudo-ancestral HERV-KCON sequence. Overall, we found that G-to-A and C-to-T substitutions were the most abundant changes in the proviruses as a whole (Fig. 2A), as is typical of the general pattern of nucleotide substitutions that are found in the human genome.
Fourteen HERV-K proviruses showed a comparatively minor increase in the frequency of G-to-A changes and C-to-T changes relative to other changes (Fig. 2A). However, two proviruses, HERV-K60 and HERV-KI, were exceptional in the total quantity and type of changes. Overall, they exhibited similar frequencies of C-to-T mutation as did the other 14 proviruses, but both HERV-K60 and HERV-KI exhibited a very high frequency of G-to-A changes relative to the HERV-KCON (Fig. 2A and B). Indeed, each of these individual proviruses had more G-to-A mutations than the other 14 proviruses combined; two-thirds and one-half of all the changes in K60 and KI, respectively, were G-to-A mutations (Fig. 2A).
To ensure that the exceptional properties of the two apparently hypermutated proviruses were not due to their insertion into an unusually hypermutated region of the human genome, we examined 2 kb of flanking genomic sequence at each end of the two proviruses for evidence of hypermutation. Specifically, we examined changes between the equivalent loci in humans compared to chimpanzees. This is a conservative approach, since the HERV-K insertions are absent in chimpanzees and have, therefore, been resident in the human genome for less time than the flanking sequences have been diverging. We found, as expected, that G-to-A, C-to-T, and reciprocal A-to-G and T-to-C changes were the most abundant in comparisons of the human and chimpanzee flanking sequences (Fig. 2A). However, G-to-A changes did not greatly outnumber other changes, suggesting that that the apparent hypermutation in the inserted proviruses occurred independently of the genomic context sequence, likely prior to their integration.
In addition to the 16 aforementioned proviruses, additional human-specific partial HERV-K sequences that lacked LTRs, as well as HERV-K proviruses (defined as group N by Romano et al.) that were not human specific, and group N CERV-K proviruses (chimpanzee counterparts of HERV-K), including four chimpanzee-specific insertions, were also examined, as were related group O proviruses. No evidence of hypermutation was evident in these sequences (data not shown).
Flanking nucleotide characteristics of G-to-A changes in hypermutated HERV-K proviruses.
A common cause of G-to-A and C-to-T substitutions in genomic DNA is spontaneous cytosine deamination. This occurs most often after methylation of cytosines in CG dinucleotides, followed by spontaneous deamination of 5-methylcytosine to a thymine (CG to mCG to TG). This series of events would lead to an overabundance of plus-strand C-to-T changes with G in the +1 position relative to the C-to-T mutation (CG to TG). Conversely, the same deamination event on the minus strand would lead to plus-strand G-to-A changes with an overabundance of C at the −1 position relative to the mutated nucleotide (CG to CA). Indeed, we found that in most of the HERV-K proviruses examined (the 14 nonhypermutated proviruses), G-to-A changes were significantly enriched for C in the −1 position (Fig. 3A), suggesting most G-to-A changes were a result of spontaneous cytosine deamination, as would be expected of DNA elements that are long-term residents of the human genome.
FIG. 3.

Nucleotide sequence context bias accompanying G-to-A mutations in endogenous HERV-K proviruses. (A and B) Nucleotide occurrence at five positions 5′ and 3′ to G-to-A changes was catalogued, using the HERV-KCON sequence as a reference. The absolute number of times that each nucleotide occurred at each position relative to each G-to-A mutation in the nonhypermutated proviruses (A) as well as HERV-K60 and HERV-KI (B) is plotted. The P value in panel B for deviation from random nucleotides at +1 and +2 positions was <0.0001, calculated using a chi-square test of independence. (C and D) Frequencies of each di- and trinucleotide for all G-to-A and GG-to-AG changes, respectively, in HERV-K60 (C) and HERV-KI (D) are plotted as black bars. The expected numbers of G-to-A mutations in each sequence context, based on di- and trinucleotide composition, of HERV-KCON is represented as a horizontal gray line. The P value, for deviation from random di- and trinucleotide preference, was <0.0001, calculated by a chi-square test of independence.
To determine whether the excessive G-to-A changes present in HERV-K60 and HERV-KI were indicative of APOBEC3-induced hypermutation, and if so, to determine the identity of the responsible protein, we examined the nucleotides flanking the G-to-A changes (Fig. 3B). At least some of the APOBEC3 proteins have signature dinucleotide preferences; for example, hA3B and hA3F prefer to deaminate cytosines within TC dinucleotides (resulting in GA-to-AA mutations on the viral plus strand), while hA3G exhibits a bias for deaminating CC dinucleotide (leading to plus-strand GG-to-AG changes). In fact, of the human APOBEC3 proteins that have been examined, only hA3G exhibits the bias toward GG-to-AG changes. In both HERV-K60 and HERV-KI, a strong GG dinucleotide bias was detected at positions where G-to-A changes occurred (Fig. 3B to D). This preference indicates that hA3G was likely responsible for the excessive G-to-A mutations in HERV-K60 and HERV-KI. In other proviruses, and the genomic DNA flanking HERV-K60 and HERV-KI, no such dinucleotide preference was detected (Fig. 3A and data not shown). Furthermore, a strong, statistically significant bias for GGG trinucleotides at G-to-A mutated positions was also evident upon examination of the third nucleotide in all GG-to-AG substitutions (Fig. 3C and D). Notably, the GGG preference has been detected in previous studies of hA3G with a Vif-deficient HIV-1 (43), further supporting the notion that hA3G was responsible for the excessive G-to-A mutations in HERV-KI and HERV-K60.
Hypermutation of HERV-K by APOBEC3 proteins during in vitro replication.
To test whether the in vivo hypermutation changes could be recapitulated in vitro, we generated HERV-K virions in the presence of each human APOBEC3 protein using the HERV-K packaging construct CCGBX-P, which contains HIV-1 sequence in the 3′ LTR to allow selective amplification of newly synthesized HERV-K in a background of existing HERV-K proviruses present in human cells. A 762-bp sequence of nascent HERV-K DNA was thus amplified using primers targeting the EGFP insert in the vector genome and the inserted HIV-1 sequence. Two controls were done to establish the success of this strategy and to show that DNase treatment reduced contaminating plasmid DNA in the virion preparations to subdetectable levels. First, PCR amplifications were done using primers targeting the plasmid backbone. Second, infections were done using virions harboring an inactivating point mutation in the HERV-K reverse transcriptase. In both cases, PCR products were not detected, indicating that the sequences generated following HERV-K infection genuinely represented infection-dependent, de novo-synthesized HERV-K DNA.
We found that hA3A, hA3B, hA3F, and hA3G were capable of inducing hypermutation of HERV-K during in vitro infection. However the patterns and frequency of hypermutation were different (Fig. 4A). In particular, nearly all HERV-K clones generated in the presence of hA3G had G-to-A mutations, but each had a low to moderate number of changes (median of six G-to-A changes per clone). Conversely, hA3A, hA3B, and hA3F each induced hypermutation in a minority of clones, but hA3A and hA3B hypermutated clones had a very high burden of mutations (37 changes for the sole hA3A hypermutated clone, and a median of 43.5 G-to-A changes for hA3B hypermutated clones). Only a few changes were seen in HERV-K DNA generated in the presence of hA3F (median of four changes per clone).
Of the four human APOBEC3 proteins found to hypermutate HERV-K in vitro, only hA3G exhibited the GG dinucleotide and GGG trinucleotide bias for the generation of G-to-A mutations (Fig. 4A and B), as has previously been reported for hA3G mutation of HIV-1. Moreover, hA3F and hA3B exhibited the expected GA dinucleotide bias at positions where G-to-A mutations were generated (Fig. 4A and B). hA3A also showed the same GA bias, with the caveat that only a single HERV-KCON clone was found to be mutated by hA3A (Fig. 4A and B). Hence, among all seven of the human APOBEC3 proteins, four appear intrinsically capable of inducing hypermutation in HERV-K, but only hA3G was capable of hypermutating HERV-K during in vitro infection with a characteristic bias that very closely mimicked mutations found in the endogenous HERV-K60 and HERV-KI proviruses (compare Fig. 3B and 4B). Moreover, when we compared the 273 bp of HERVKCON sequence that was analyzed in the hA3G mutagenesis assay with the corresponding sequence in HERV-K60 and HERV-KI, 10 out of the possible 75 G-to-A mutations were found in either or both HERV-K60 and HERV-KI, while 15 out of the possible 75 G-to-A mutations were represented in corresponding sequences that were mutated by hA3G during in vitro infection (Fig. 5A and B). Notably, six of these G-to-A mutations were at identical positions, a highly significant correlation (P = 0.003, Fisher's exact test), lending further supporting to the notion that hA3G was responsible for hypermutation of HERV-K60 and HERV-KI.
FIG. 5.
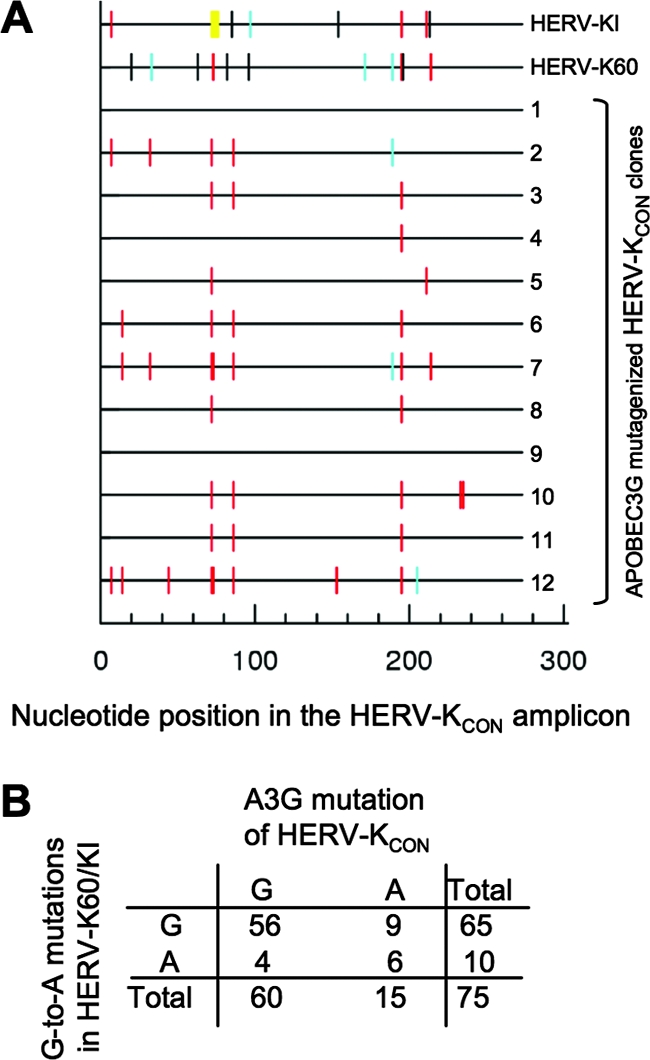
Comparison of mutations relative to HERV-KCON that appear in naturally hypermutated proviruses (HERV-K60 and HERV-KI) and in the 273 nucleotides of mutated HERV-K sequence generated in vitro in the presence of hA3G. Changes relative to HERV-KCON are represented graphically on horizontal lines and are color coded according to the nucleotide appearing at the +1 site for HERV-K60 and HERV-KI and 12 HERV-K clones generated during infection in the presence of hA3G. Mutations are indicated as vertical lines. Red, GG to AG; cyan, GA to AA; green, GC to AC; magenta, GT to AT; black, non-G-to-A transitions; yellow, gaps in sequence. (B) The 75 G nucleotides in the HERV-K-derived portion of the in vitro-hypermutated sequence and the number of positions at which the G residue was universally unaltered or sometimes mutated to A in the naturally and experimentally hypermutated HERV-K sequences.
DISCUSSION
In recent years, several gene products with antiretroviral activity have been identified, such as TRIM5, tetherin, ZAP, and the APOBEC3 family of proteins. In at least some cases, positive selection pressure has been placed on these genes during primate evolution (19, 28, 29, 32). As HERV-K has been repeatedly colonizing the genomes of Old World primates since the divergence of Old and New World monkeys approximately 35 million years ago, it is a potential source of recurrent selective pressure on primate hosts (2). By deducing a consensus sequence and reconstituting this ancient retrovirus, it has been possible to directly test the effects of antiretroviral proteins on HERV-K infection and uncover evidence of ancient host-virus interactions.
When comparing full-length human-specific HERV-K proviruses to HERV-KCON, which approximates an ancestral sequence for the most recent expansion of HERV-K proviruses in humans, we found an abundance of G-to-A and C-to-T substitutions. These substitutions, the most common change found in genomes, can occur when DNA methyltransferase methylates cytosines in CG dinucleotides to 5-methylcytosine, which spontaneously deaminates to thymine, resulting in a CG-to-TG change. These methylation events, important for development via genomic imprinting and X chromosome inactivation, can also silence retroelements. This effect has been demonstrated in mice, where knocking out DNA methyltransferase Dnmt1 or Dnmt3L leads to transcriptional activation of mouse retroelements intracisternal A particles and LINE-1 (9, 39). In addition, previous studies of a selection of HERV-K LTRs in the teratocarcinoma cell line Tera-1 showed that methylation and transcription are inversely related (20). Given these two facts, one would expect to find that HERV-K proviruses would be cytosine methylated by the host, and consequently G-to-A and C-to-T mutations might be abundant. This was indeed the case, and in 14 of 16 HERV-K proviruses examined, the CG dinucleotide methylation/spontaneous deamination pathway appeared to be the major source of G-to-A and C-to-T mutations.
Another common cause of G-to-A, and less frequently C-to-T (7), changes in viral DNA is APOBEC3-mediated cytidine deamination. Fortuitously, the two events are easily distinguished. DNA methyltransferases methylate cytosines in CG dinucleotides, while APOBEC proteins deaminate cytosines in XC dinucleotides, where X can differ depending on the APOBEC protein that is responsible for deamination. Hence, by examining the nucleotides immediately 5′ and 3′ to the mutated cytosine, we can largely assign G-to-A and C-to-T changes to either mechanism. One exception is when both DNA methyltransferase and APOBEC preferred nucleotides flank the altered cytosine, such as CCG trinucleotides, where CC represents the dinucleotide preference of hA3G and CG the preference of DNA methyltransferase. However, exclusion of these ambiguous samples in our analyses did not alter the conclusions.
The characteristics of HERV-K hypermutation found both in vivo and in vitro match several of the characteristics previously observed in the context of APOBEC-induced mutations in other retrovirus infections. First, G-to-A mutations constituted a large fraction of the total mutations in HERV-K60 and HERV-KI, as has previously been found for hypermutated viral sequences in HIV- and HBV-infected patients (17, 31, 36, 38). Furthermore, the GGG trinucleotide preference found during in vitro HERV-K infection in the presence of hA3G has been documented in HIV-1 infection assays by several groups (6, 41, 43). The combination of these two major characteristics found in HERV-K60 and HERV-KI, plus the failure of any other human APOBEC3 protein to induce a similar pattern of mutation during HERV-K replication in vitro, makes a strong argument for hA3G as the sole source of hypermutation in ancient HERV-K proviruses.
Another reported characteristic of viral hypermutation by hA3G is the gradient of changes along the viral genome. This characteristic is thought to derive from the position-dependent length of time that the nascent viral DNA is in the form of single-stranded DNA, the preferred nucleic substrate of hA3G. However, this was not observed in the HERV-K60 and HERV-KI sequences (data not shown). The reasons for this are unclear at present. Nonetheless, our ability to fairly precisely recreate the hypermutation patterns present in ancient proviruses specifically using hA3G during in vitro HERV-K replication assays suggests that the interaction between this protein and HERV-K occurred and was physiologically and evolutionarily relevant. HERV-K is therefore an eminently reasonable candidate for an infectious agent that has applied selective pressure on A3G during primate evolution. However, we do note that HERV-K is but one of a number of agents that could potentially have imposed selective pressure on antiretroviral defenses. Other abundant endogenous retroelements, such as Alu and LINE-1 elements in humans, have also been shown to be restricted by APOBEC3 proteins (10, 16, 25, 33), and these elements as well as other exogenous and endogenous retroviruses may also have contributed to the expansion and positive selection that is evident in APOBEC3 genes. Indeed, among the ancient retroviruses, only those that colonized the germ line are accessible to this type of analysis, and it is completely unknown what fraction of ancient retroviruses that replicated in ancestral primates are fossilized in modern DNA. Nonetheless, A3G has been under positive selection for many millions of years (28), and HERV-K could, potentially, have contributed to this pressure.
Given that HERV-K(HML-2) appears intrinsically mutable by hA3G and a hypermutated provirus is less likely to be harmful (and hence more likely to become fixed in a host genome) than an intact provirus, it is perhaps surprising that only 2 out of 16 HERV-K human-specific proviruses and 0 of 4 chimpanzee-specific proviruses were clearly hypermutated. Several factors may have contributed to this, and perhaps the most important influence would be viral tropism. The appearance of a hypermutated provirus in human DNA indicates that HERV-K likely replicated in an A3G-expressing maternal or paternal tissue immediately prior to deposition of the provirus into the germ line. Conversely, the deposition of a nonhypermutated virus suggests that the preceding generations involved replication in APOBEC3G-negative tissues. The simplest explanation for the appearance of hypermutated and nonhypermutated proviruses in the human genome is that HERV-K replicated in both A3G-expressing and nonexpressing somatic cells prior to germ line infection.
Moreover, the frequency of hypermutated proviruses in modern genomes may not reflect the frequency at which hypermutation occurred during ancient infections. Indeed, while hypermutation would generally inactivate a particular provirus, hypermutation itself is unlikely to be always necessary or sufficient to result in fixation of the element, where chance-influenced mechanisms, such as drift or bottlenecking, may play a dominant role in provirus fixation. Of note, older HERV-K sequences belonging to group O as defined by Romano et al. (27) did not exhibit signs of hypermutation compared to HERV-KCON. Moreover, a previous study of endogenous murine leukemia viruses also found that only a minority of proviruses were overtly hypermutated, perhaps for the same aforementioned reasons.
While there was a reasonable qualitative correlation between the appearance of APOBEC3-induced G-to-A mutations and infection inhibition during in vitro HERV-K replication, there was a notable lack of a quantitative correlation between the burden of mutations and the extent to which infection was inhibited. Specifically, hA3A, hA3B, and hA3F caused mutation in a minority of nascent HERV-K reverse transcripts yet inhibited infection to a greater degree than hA3G, which mutated the majority of nascent HERV-K DNA molecules. Since we found no evidence of hA3A, hA3B, or hA3F hypermutation in endogenous proviruses, inhibition of HERV-K infection by these cytidine deaminases may be physiologically irrelevant or might occur primarily via mechanisms (e.g., inhibition of DNA synthesis or integration) that would not leave remnants of the viral encounter with the APOBEC protein (5, 12, 14, 23, 42).
The lack of strong inhibition of HERV-K infection by hA3G in the single-cycle replication assay should not be overinterpreted as suggesting that hA3G lacks anti-HERV-K activity in vivo. As documented here and elsewhere, APOBEC3G appears to have evolved to target GG dinucleotides, especially GGG trinucleotides. This property makes it a particularly efficient mutator of tryptophan codons. G-to-A mutation of tryptophan codons invariably leads to the generation of new stop codons, which would almost always be lethal to a retrovirus, even if a provirus were successfully established with a relatively low burden of APOBEC3G-induced mutation. Importantly, the HERV-K infection assay requires a single cycle of infection by a reporter virus that encodes the commonly used EGFP as the reporter gene. EGFP contains only a single tryptophan, and thus a moderate level of hA3G-induced mutations might not score as strong inhibition during an in vitro single-cycle infection assay but would abolish further rounds of replication in an in vivo spreading infection. Indeed, HERV-K60 and HERV-KI represent clear examples of viral sequences that have been fossilized in the human genome in defective form as a consequence of hA3G-induced hypermutation.
Acknowledgments
We thank members of the Bieniasz, Hatziioannou, and Malim laboratories for reagents and advice. We thank Mark Schroeder for assistance with sequence analysis.
This work was supported by NIH grant R01AI64003 (P.D.B.) and the UK Medical Research Council (M.H.M.). P.D.B. and M.H.M. are Elisabeth Glaser Scientists.
Footnotes
Published ahead of print on 18 June 2008.
REFERENCES
- 1.Bainbridge, J. W., C. Stephens, K. Parsley, C. Demaison, A. Halfyard, A. J. Thrasher, and R. R. Ali. 2001. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 81665-1668. [DOI] [PubMed] [Google Scholar]
- 2.Bannert, N., and R. Kurth. 2004. Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. USA 10114572-14579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbulescu, M., G. Turner, M. I. Seaman, A. S. Deinard, K. K. Kidd, and J. Lenz. 1999. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr. Biol. 9861-868. [DOI] [PubMed] [Google Scholar]
- 4.Belshaw, R., A. L. A. Dawson, J. Woolven-Allen, J. Redding, A. Burt, and M. Tristem. 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J. Virol. 7912507-12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop, K. N., R. K. Holmes, and M. H. Malim. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 808450-8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S.-J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 141392-1396. [DOI] [PubMed] [Google Scholar]
- 7.Bishop, K. N., R. K. Holmes, A. M. Sheehy, and M. H. Malim. 2004. APOBEC-mediated editing of viral RNA. Science 305645. [DOI] [PubMed] [Google Scholar]
- 8.Borman, A. M., C. Quillent, P. Charneau, K. M. Kean, and F. Clavel. 1995. A highly defective HIV-1 group O provirus: evidence for the role of local sequence determinants in G→A hypermutation during negative-strand viral DNA synthesis. Virology 208601-609. [DOI] [PubMed] [Google Scholar]
- 9.Bourc'his, D., and T. H. Bestor. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 43196-99. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, Y. L., H. E. Witkowska, S. C. Hall, M. Santiago, V. B. Soros, C. Esnault, T. Heidmann, and W. C. Greene. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. USA 10315588-15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewannieux, M., F. Harper, A. Richaud, C. Letzelter, D. Ribet, G. Pierron, and T. Heidmann. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 161548-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo, F., S. Cen, M. Niu, J. Saadatmand, and L. Kleiman. 2006. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 8011710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatziioannou, T., S. Cowan, S. P. Goff, P. D. Bieniasz, and G. J. Towers. 2003. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 22385-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. J. Biol. Chem. 2822587-2595. [DOI] [PubMed] [Google Scholar]
- 15.Hughes, J. F., and J. M. Coffin. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 1011668-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulme, A. E., H. P. Bogerd, B. R. Cullen, and J. V. Moran. 2007. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene 390199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janini, M., M. Rogers, D. R. Birx, and F. E. McCutchan. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T Cells. J. Virol. 757973-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jern, P., J. P. Stoye, and J. M. Coffin. 2007. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 32014-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerns, J. A., M. Emerman, and H. S. Malik. 2008. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 4e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavie, L., M. Kitova, E. Maldener, E. Meese, and J. Mayer. 2005. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J. Virol. 79876-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, Y. N., and P. D. Bieniasz. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 3e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 141385-1391. [DOI] [PubMed] [Google Scholar]
- 23.Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S. Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A. Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 817099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451425-430. [DOI] [PubMed] [Google Scholar]
- 25.Niewiadomska, A. M., C. Tian, L. Tan, T. Wang, P. T. Sarkis, and X. F. Yu. 2007. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 819577-9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.OhAinle, M., J. A. Kerns, H. S. Malik, and M. Emerman. 2006. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 803853-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romano, C. M., R. F. Ramalho, and P. M. D. A. Zanotto. 2006. Tempo and mode of ERV-K evolution in human and chimpanzee genomes. Arch. Virol. 1512215-2228. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer, S. L., M. Emerman, and H. S. Malik. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawyer, S. L., L. I. Wu, M. Emerman, and H. S. Malik. 2005. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 1022832-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 31.Simon, V., V. Zennou, D. Murray, Y. Huang, D. D. Ho, and P. D. Bieniasz. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song, B., B. Gold, C. O'Huigin, H. Javanbakht, X. Li, M. Stremlau, C. Winkler, M. Dean, and J. Sodroski. 2005. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5α exhibits lineage-specific length and sequence variation in primates. J. Virol. 796111-6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenglein, M. D., and R. S. Harris. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 28116837-16841. [DOI] [PubMed] [Google Scholar]
- 34.Stremlau, M., C. M. Owens, M. J. Perron, M. Kiessling, P. Autissier, and J. Sodroski. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427848-853. [DOI] [PubMed] [Google Scholar]
- 35.Suspene, R., D. Guetard, M. Henry, P. Sommer, S. Wain-Hobson, and J.-P. Vartanian. 2005. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1028321-8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suspene, R., C. Rusniok, J.-P. Vartanian, and S. Wain-Hobson. 2006. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 344677-4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner, G., M. Barbulescu, M. Su, M. I. Jensen-Seaman, K. K. Kidd, and J. Lenz. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 111531-1535. [DOI] [PubMed] [Google Scholar]
- 38.Vartanian, J. P., A. Meyerhans, B. Asjo, and S. Wain-Hobson. 1991. Selection, recombination, and G→A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 651779-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh, C. P., J. R. Chaillet, and T. H. Bestor. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20116-117. [DOI] [PubMed] [Google Scholar]
- 40.Weiss, R. A. 2006. The discovery of endogenous retroviruses. Retrovirology 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 232451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, Y., F. Guo, S. Cen, and L. Kleiman. 2007. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology 36592-100. [DOI] [PubMed] [Google Scholar]
- 43.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11435-442. [DOI] [PubMed] [Google Scholar]