

Abstract
Y-27632, an inhibitor of the Rho-associated kinase ROCK, is a therapeutic lead for Huntington disease (HD). The downstream targets that mediate its inhibitory effects on huntingtin (Htt) aggregation and toxicity are unknown. We have identified profilin, a small actin-binding factor that also interacts with Htt, as being a direct target of the ROCK1 isoform. The overexpression of profilin reduces the aggregation of polyglutamine-expanded Htt and androgen receptor (AR) peptides. This requires profilin's G-actin binding activity and its direct interaction with Htt, which are both inhibited by the ROCK1-mediated phosphorylation of profilin at Ser-137. Y-27632 blocks the phosphorylation of profilin in HEK293 cells and primary neurons, which maintains profilin in an active state. The knockdown of profilin blocks the inhibitory effect of Y-27632 on both AR and Htt aggregation. A signaling pathway from ROCK1 to profilin thus controls polyglutamine protein aggregation and is targeted by a promising therapeutic lead for HD.
Huntington disease (HD) and spinobulbar muscular atrophy (SBMA) cause devastating neurodegeneration. HD derives from an expanded CAG codon repeat that produces an elongated polyglutamine tract in the huntingtin (Htt) protein, and SBMA derives from an elongated tract in the androgen receptor (AR) (19, 23). A toxic, aggregation-prone conformation is favored by the expanded polyglutamine tract but does not always occur in cells, implying a role for additional protein interactions (11, 50). The basic mechanisms that influence intracellular polyglutamine protein aggregation and toxicity are not well understood. There is no effective therapy for any polyglutamine disease, and thus a better understanding of fundamental mechanisms that might be targeted by new drugs is crucial.
Protein aggregation plays an important role in the cytotoxicity of polyglutamine proteins such as AR and Htt and is associated closely, but not invariably, with inclusion formation. Inclusions are macromolecular structures that represent an adaptive cellular response to large quantities of misfolded proteins (25, 38, 46). We previously developed a quantitative assay to detect intracellular aggregation and inclusion formation that is based on fluorescence resonance energy transfer (FRET) (36). We used this system to identify multiple biologically active small molecules that reduce intracellular AR and Htt aggregation and toxicity (10, 36). One lead compound, Y-27632, an inhibitor of the Rho-associated kinase ROCK (47), reduced AR and Htt aggregation in cultured cells and Htt-mediated neurodegeneration in Drosophila melanogaster (36). We recently validated ROCK and another Rho-associated kinase, PRK2, as being intracellular targets of Y-27632 that regulate polyglutamine aggregation (43). Here, we have investigated the molecular mechanism of ROCK inhibition and, in doing so, have elucidated a novel signaling pathway, from ROCK1 to the actin-binding factor profilin, which regulates polyglutamine aggregation.
MATERIALS AND METHODS
Reagents.
Protease inhibitor cocktail tablets (Complete Mini, catalog number 11-836-153-001, and Complete Mini EDTA free, catalog number 11-836-170-001) were purchased from Roche Diagnostics. An ECL Plus Western blotting detection kit (catalog number RPN2132) was purchased from GE Healthcare. Phosphatase inhibitor cocktail 1 (catalog number P2850) was purchased from Sigma. Control small interfering RNA (siRNA) (catalog number sc-37007), profilin-1-specific siRNAs (catalog number sc-36316), and profilin-1/profilin-2a-specific siRNAs (catalog number sc-44045) were purchased from Santa Cruz Biotechnology. Profilin-1-specific siRNAs contain three different sequences: GUGUCCUGGUUGGCAAAGA, CACGGUGGUUUGAUCAACA, and CCCCAUACCCCUUAUUGCU. Profilin-1/profilin 2a siRNAs contain four different sequences: two targeting profilin-1 (GCAAAGACCGGUCAAGUUU and CACGGUGGUUUGAUCAACA) and the other two targeting profilin-2a (GUAGAGCAUUGGUUAUAGU and CCAGGGACAUUCCAUCAUU). Lysophosphatidic acid (LPA) was purchased from Sigma.
Constructs.
cDNAs encoding ARN127(Q65) or Htt exon 1 fused to cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP) (36) were subcloned from p6R (36) into the backbone of pEYFP.N1 (Clontech) to drive expression under the cytomegalovirus promoter. For bacterial expression, glutathione S-transferase (GST)-ARN127(Q25) YFP was cloned into the pGEX-4T-1 vector (Amersham Biosciences). pGEX-Htt exon 1 (Q20 or Q53) constructs were provided by Paul J. Muchowski (31). Human His6-profilin-1 (wild type [wt], S137A, and S137D) was PCR amplified and cloned into the bacterial expression vector pRK172. For expression in HEK293 cells, human profilin-1 was PCR amplified and cloned into pcDNA3.1. Mutations at Ser-137 were introduced into the PCR primers, and others were introduced by QuikChange mutagenesis (Stratagene). Human profilin-2a was PCR amplified and cloned into the gWIZ blank vector. For expression in primary neurons, profilin-1 (wt versus mutants) and Htt exon 1(Q72) YFP were cloned into vector pCAGGS (provided by Robert Edwards) to drive expression under the chicken β-actin promoter. pCAG-ROCK1(wt) or pCAG-ROCK1(KDIA) was provided by Shuh Narumiya (20). pXJ40-ROCK2 was provided by Thomas Leung (24).
Cell culture and transfection.
HEK293 cells were cultured in Dulbecco's modified Eagle's medium-5% fetal bovine serum (FBS) and transfected as described previously (10, 36). For LPA treatment and phospho-profilin-1 detection, primary basal ganglion neurons were dissected from embryonic day 14 rat embryos and maintained in Dulbecco's modified Eagle's medium-10% FBS for 4 days prior to use. For effects of profilin on aggregation, primary cortical neurons were dissected from embryonic day 19 rat embryos and electroporated with Htt exon 1(Q72) YFP and profilin-1 (wt versus mutants) using a rat neuron Nucleofector kit (catalog number DPG-1003; Amaxa). They were cultured in Neurobasal medium (Gibco) supplemented with 1% FBS, 2% B27, and 2 mM Glutamax for 4 days prior to use.
FRET measurements and calculations.
FRET measurements were made as described previously (10). The relative FRET/donor ratio was calculated as follows: relative FRET/donor ratio = [(FRET/donor)a − (FRET/donor)b]/(FRET/donor)b, where a represents cells cotransfected or treated with aggregation modulators and b represents control cells untreated or cotransfected with empty vector or control siRNAs.
Protein expression and affinity purification.
pGEX constructs were transformed into Rosetta(DE3) (Novagen) or Sure competent cells (Stratagene). Cells were grown at 37°C until an optical density at 600 nm of 0.6 to 0.9 was reached and induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 30°C for 5 h. Cells were lysed by sonication in phosphate saline buffer, and lysate was mixed with glutathione-Sepharose beads (Amersham Biosciences) at 4°C for 2 h. Beads were washed and eluted with 50 mM Tris-HCl (pH 8.0) containing 10 mM reduced glutathione. pRK172/His-profilin (wt, S137A, and S137D) was transformed into Rosetta(DE3) cells and induced with 0.5 mM IPTG at 30°C for 5 h. The lysate was sonicated and bound to Ni-nitrilotriacetic acid agarose (Qiagen) at 4°C for 2 h. Beads were washed and eluted with 250 mM imidazole. Proteins were dialyzed against a solution containing 50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 0.2 mM ATP, 5% sucrose, and 0.5 mM dithiothreitol (DTT) for in vitro kinase assays or against a solution containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 1 mM DTT for pull-down assays.
Immunoprecipitation and pull-down assays.
For coimmunoprecipitation of endogenous actin with Myc-profilin, HEK293 cells were lysed in a solution containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail (Roche). Cleared lysates were mixed with anti-mouse immunoglobulin G (IgG) beads (Sigma) bound with mouse anti-Myc antibody. After 2 h of mixing at 4°C, beads were washed with lysis buffer and analyzed by Western blotting using anti-Myc and antiactin antibodies. For interactions between GST-Htt exon 1 and profilin, Htt exon 1 was immobilized on glutathione-Sepharose and incubated with bacterial lysate or HEK293 cell lysate. Beads were washed with phosphate-buffered saline-1 mM EDTA-0.1% Tween 20 and analyzed by Coomassie blue staining and Western blotting against profilin. For interactions between His-profilin and Htt exon 1, His6-profilin was immobilized on Ni-nitrilotriacetic acid agarose and mixed with bacterial lysate containing GST-HA-Htt exon 1 (Q25). Beads were washed with phosphate-buffered saline and analyzed by Coomassie blue staining and Western blotting against Htt. For immunoprecipitation of phospho-profilin, HEK293 cells were lysed with a solution containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 mM NaF, 1% Triton X-100, and protease inhibitor cocktail. The cleared lysate was incubated with anti-rabbit IgG beads (Sigma) bound with rabbit antiphosphoserine antibody. After 2 h of mixing at 4°C, beads were washed with a solution containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 mM NaF, and 1% Triton X-100 and analyzed by Western blotting against the Myc tag.
In vitro kinase assay.
Immunoprecipitated Myc-ROCK1(wt) or Myc-ROCK1(KDIA) was mixed with 50 ng/μl His6-profilin (wt or S137A) in 25 μl kinase reaction buffer (25 mM Tris-HCl [pH 7.4], 4 mM MgCl2, 3.6 mM EDTA, 1 mM DTT, 0.1 μM calyculin A [Calbiochem], phosphatase inhibitor cocktail 1 [Sigma], 50 μM arachidonic acid [Sigma], 80 μM cold ATP, 20 μM [γ-32P]ATP [5 μCi; Perkin-Elmer]) for 60 min at 30°C. Reactions were stopped with sodium dodecyl sulfate (SDS) sample buffer and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and autoradiography.
Antibodies.
Commercial primary antibodies purchased were as follows: mouse anti-Myc tag (catalog number sc-40; Santa Cruz), mouse anti-Htt (MAB5374, clone mEM48; Chemicon), rabbit anti-actin (catalog number sc-1616-R; Santa Cruz), rabbit anti-phosphoserine (AB1603; Chemicon), and mouse anti-tetra-His (catalog number 34670; Qiagen). Secondary antibodies used for Western blotting include alkaline phosphatase-conjugated secondary antibodies (catalog number A3562; Sigma) and horseradish peroxidase-conjugated secondary antibodies (NA9340V for anti-rabbit and NA931V for anti-mouse antibodies; Amersham Biosciences). Mouse true blot secondary antibody was purchased from eBioscience. Secondary antibody-conjugated agarose beads used for immunoprecipitation are anti-mouse IgG-agarose (catalog number A6531; Sigma) and anti-rabbit IgG immunoprecipitation beads (catalog number 00-8800-25; eBioscience). A rabbit polyclonal antibody (P3490) selective for phospho-Ser-137 of profilin was raised using a synthetic phosphopeptide [Ac-CMASHLRR(pS)QY-OH] derived from the C terminus of human profilin-1 and affinity purified by New England Peptide, Inc. Polyclonal antibodies against profilin-1 and -2a were generated by Open Biosystems (Huntsville, AL) by injecting the linear peptides KCYEMASHLRRSQY and KAYSMAKYLRDSGF, respectively, conjugated to keyhole limpet hemocyanin into rabbits. Serum was purified with an affinity column containing the respective profilin peptide. A polyclonal antibody against the N-terminal portion of profilin-1 was purchased from Cell Signaling.
RESULTS
Profilin inhibits polyglutamine aggregation.
ROCK is a key player in actin cytoskeleton remodeling. It does so via phosphorylating several well-known targets, including LIM kinase (reviewed in references 1, 32, and 37). LIM kinase is activated by ROCK (26, 34, 45) and in turn phosphorylates cofilin at Ser-3 to inactivate cofilin's filamentous (F) actin-severing and -depolymerizing activity (reviewed in reference 3). We coexpressed constitutively active (S3A) or inactive (S3E) forms of cofilin with polyglutamine-expanded peptides derived from the first 127 amino acids of AR [ARN127(Q65) CFP/YFP] or the first exon of Htt [Htt exon 1(Q72) CFP/YFP] in HEK293 cells. Western blot analysis suggested an approximately twofold increase in levels of cofilin upon overexpression (data not shown). We quantified aggregation via FRET. Cofilin(S3A) increased the aggregation of both AR and Htt, whereas cofilin(S3E) suppressed AR aggregation and had no effect on Htt aggregation (Fig. 1A). These effects indirectly implicated the actin cytoskeleton in polyglutamine aggregation but were contrary to those expected if cofilin were the key secondary downstream target of ROCK mediating its effect.
FIG. 1.

Profilin inhibits AR and Htt aggregation in cultured HEK293 cells. (A) HEK293 cells were cotransfected with constitutively active cofilin(S3A) or inactive cofilin(S3E) with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP. Aggregation was measured by FRET, and the effects were expressed as relative values as described in Materials and Methods. Cofilin(S3A) increased the aggregation of both Htt exon 1(Q72) and ARN127(Q65), whereas cofilin(S3E) had little effect on Htt exon 1(Q72) aggregation (**, P < 0.005 by paired t test) and decreased ARN127(Q65) aggregation. Error bars represent the standard errors of the means (SEM). (B and C) HEK293 cells were cotransfected with Htt exon 1(Q72) CFP/YFP or ARN127(65) CFP/YFP and increasing amounts of profilin-1 (which will subsequently be referred to as profilin, unless otherwise stated) (B) or profilin-2a (C). Relative aggregation was measured by FRET. Both profilin isoforms dose-dependently reduced aggregation. Error bars represent the SEM. (D) HEK293 cells were cotransfected with Htt exon 1(Q72) YFP or ARN127(Q65) YFP and profilin-1. Cells were cultured for 2 days, fixed, and analyzed using fluorescence microscopy. Profilin-1 reduced the number of inclusions formed by both Htt exon 1(Q72) YFP and ARN127(Q65) YFP. Profilin-2a had the same effect (data not shown). Representative images (magnification, ×10) are shown.
We next focused on profilin, an actin binding protein previously identified as being an Htt-interacting protein in yeast two-hybrid and in vitro binding assays (17) and which was recently reported to ameliorate polyglutamine toxicity in cells and Drosophila (5). It is generally believed that profilin promotes F-actin polymerization by exchanging the adenosine nucleotide on G actin and facilitating its addition to growing filament ends (51), although contradictory data regarding its effect on intracellular F-actin content have been reported (40, 54). Two major profilin isoforms with similar biochemical properties exist: profilin-1 and profilin-2a (51). Profilin-1 is expressed in most tissues, whereas profilin-2a is largely restricted to the brain (52). We cotransfected HEK293 cells with increasing amounts of profilin-1 or profilin-2a along with ARN127(Q65) CFP/YFP or Htt exon 1(Q72) CFP/YFP. Transfection achieved a twofold increase in profilin-1 levels; endogenous profilin-2a is expressed at very low levels in these cells (data not shown). Both isoforms dose-dependently inhibited AR and Htt aggregation (Fig. 1B and C) and reduced the number of microscopically visible inclusions (Fig. 1D). All subsequent experiments were performed using profilin-1 unless otherwise stated.
Inhibition of Htt aggregation by profilin requires direct interaction.
Profilin has been reported to directly interact with Htt (17). We confirmed this via GST pull-down assays. Purified GST-Htt exon 1(Q25) bound recombinant profilin from bacterial lysate, whereas GST or GST-ARN127(Q25) did not (Fig. 2A). We observed a similar result using HEK293 cell lysates containing transfected profilin (Fig. 2B). Previously reported data suggest that profilin directly binds polyproline motifs similar to those found in Htt (51). Cocrystals of profilin and polyproline peptides (27, 28), along with mutagenesis studies, indicate that the polyproline binding site of profilin is in a hydrophobic patch formed by both the amino- and carboxy-terminal helices (4, 30). We tested whether profilin might utilize this region to interact with Htt by introducing two point mutations, W31F and H133S, that each block the interaction of profilin with its polyproline ligand (4, 35). GST pull-downs confirmed that these mutations eliminated profilin's interaction with Htt exon 1 (Fig. 2C) but had no effect on its interaction with G actin (Fig. 2D). When overexpressed in HEK293 cells, each mutation reduced profilin's ability to inhibit Htt exon 1(Q72) CFP/YFP aggregation by approximately 50% (Fig. 2E). However, neither mutation affected profilin's ability to reduce ARN127(Q65) CFP/YFP aggregation (Fig. 2E). Thus, the inhibition of Htt aggregation by profilin is based in part on a direct interaction.
FIG. 2.

Inhibition of Htt aggregation by profilin requires direct interaction. (A) Sepharose-bound GST, GST-ARN127(Q25) YFP, or GST-Htt exon 1(Q25) was mixed with bacterial lysate containing His-profilin. GST-Htt exon 1(Q25) interacted with His-profilin, whereas GST and GST-ARN127(Q25) YFP did not. (B) Sepharose-bound GST, GST-Htt exon 1(Q20), or GST-Htt exon 1(Q53) was mixed with HEK293 cell lysate containing profilin. GST-Htt exon 1(Q53) bound 85% less profilin than GST-Htt exon 1(Q20). Asterisks indicate two bands nonspecifically reacting with the anti-profilin antibody. (C) Sepharose-bound GST or GST-Htt exon 1(Q25) was mixed with HEK293 cell lysate containing profilin(wt) or the polyproline binding mutants (H133S and W31F). Profilin(wt) bound GST-Htt exon 1(Q25), and the two mutants did not. (D) HEK293 cells were transfected with untagged (first lane) or Myc-tagged profilin (wt, H133S, or W31F). Cell lysate was immunoprecipitated using an anti-Myc antibody, followed by Western blotting against actin. Polyproline binding-deficient profilin mutants bound amounts of actin similar to those of profilin(wt). (E) Profilin (wt, H133S, or W31F) was cotransfected into HEK293 cells with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP. Relative aggregation was measured by FRET. Both mutants inhibited ARN127(Q65) aggregation comparably to profilin(wt) but were significantly less effective in inhibiting Htt exon 1(Q72) aggregation (***, P < 0.0005; **, P < 0.005 [unpaired t test]). Error bars represent the SEM.
Actin binding by profilin is required to inhibit polyglutamine aggregation.
Profilin has well-defined actin binding activities, and analyses of actin/profilin cocrystals (42) and mutagenesis (41, 53) have defined a crucial residue (Tyr-59) within its actin binding domain. Profilin(Y59A) cannot bind actin in vitro (41, 53). We confirmed this in cultured HEK293 cells by coimmunoprecipitation (Fig. 3A). Profilin(Y59A) interacted with Htt exon 1 comparably to the wild-type form (Fig. 3B), but its ability to inhibit Htt exon 1(Q72) CFP/YFP aggregation was reduced by 50%, and it actually increased the aggregation of ARN127(Q65) CFP/YFP (Fig. 3C). These experiments demonstrate that the binding of G actin by profilin is essential for its inhibition of AR aggregation but is only partially required for profilin's inhibition of Htt aggregation, which also requires a direct interaction.
FIG. 3.

Actin binding by profilin is required to inhibit polyglutamine aggregation. (A) Myc-tagged profilin(wt) or profilin(Y59A) was immunoprecipitated from HEK293 cells, and coprecipitated actin was detected by Western blotting. Untagged profilin was used as the negative control (first lane). Profilin(Y59A) failed to bind actin. IP, immunoprecipitation. (B) Sepharose-bound GST or GST-Htt exon 1(Q25) was incubated with HEK293 cell lysate containing profilin(wt) or profilin(Y59A). Both profilin proteins bound Htt exon 1 comparably. (C) HEK293 cells were cotransfected with profilin(wt) or profilin(Y59A) with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP. Relative aggregation was measured by FRET. Profilin(wt) reduced the aggregation of Htt exon 1(Q72) and ARN127(Q65), whereas profilin(Y59A) partially reduced Htt exon 1(Q72) aggregation (***, P < 0.0005 by unpaired t test) and increased ARN127(Q65) aggregation. Error bars represent the SEM.
ROCK1 phosphorylates profilin at Ser-137.
In a previously reported study, Y-27632 treatment increased the isoelectric focusing point of profilin-2a, suggesting a decrease in its overall phosphorylation level and a possible link to ROCK (7). However, this study left uncertain whether profilin is a direct target of ROCK, and if so, what the specific phosphorylation site is. Sequence analysis of profilin revealed a potential ROCK phosphorylation consensus site near the C terminus: 135RRSQY139 (boldface indicates the phosphoserine targeted by ROCK1) (22, 37, 45). Ser-137 of profilin can be phosphorylated by protein kinase C in vitro (44, 48). However, no physiological protein kinase has yet been assigned for profilin, nor has its regulation by phosphorylation at any residue been demonstrated. Thus, we first tested whether ROCK phosphorylates profilin at Ser-137 in vitro. ROCK1(wt) (the ubiquitously expressed ROCK isoform) or its kinase-dead mutant, ROCK1(KDIA) (20), was immunoprecipitated from HEK293 cells and incubated with recombinant profilin. Radiolabeling with [γ-32P]ATP revealed the phosphorylation of profilin by ROCK1(wt) but not by ROCK1(KDIA) (Fig. 4A). Profilin(S137A) incorporated approximately 40% less 32P than did profilin(wt) (Fig. 4A), indicating that ROCK1 phosphorylates Ser-137, in addition to other sites, in vitro.
FIG. 4.

ROCK1 phosphorylates profilin at Ser-137 in vitro and in vivo. (A) Immunoprecipitated Myc-tagged ROCK1(wt) or ROCK1(KDIA) was mixed with profilin(wt) or profilin(S137A) in an in vitro kinase assay and analyzed by autoradiography and Western blotting. While the kinase-dead ROCK1(KDIA) caused little 32P incorporation into profilin, ROCK1(wt) phosphorylated profilin(wt) and, to a 40% lesser extent, profilin(S137A) (average of three experiments) (*, P < 0.05 by unpaired t test). Error bars represent the SEM. (B) Myc-tagged profilin(wt) or profilin(S137A) was cotransfected into HEK293 cells with ROCK1, ROCK2, or pcDNA3 and treated with or without 50 μM Y-27632 for 24 h. Immunoprecipitation was performed with a phosphoserine-specific antibody (P-Ser) or normal IgG (control [Ctrl]), followed by Western blotting for the Myc tag. No profilin(wt) bound to the P-Ser antibody without ROCK1, whereas ROCK1 overexpression increased binding, which was inhibited by Y-27632. Profilin(S137A) failed to bind the P-Ser antibody regardless of ROCK1 overexpression. ROCK2 overexpression did not increase the binding of profilin(wt) to the P-Ser antibody but increased the binding of profilin(S137A).
Next, we tested for intracellular ROCK-induced phosphorylation of profilin at Ser-137. We expressed Myc-tagged profilin(wt) or profilin(S137A) in HEK293 cells with or without ROCK1 or ROCK2 (an isoform highly expressed in brain), followed by immunoprecipitation with an anti-phosphoserine antibody and Western blotting with an anti-Myc antibody. We observed very little immunoprecipitation of profilin in the absence of ROCK1 or ROCK2 (Fig. 4B). The coexpression of ROCK1 increased the amount of profilin bound by the anti-phosphoserine antibody, and this effect was blocked by the ROCK inhibitor Y-27632 (Fig. 4B). No immunoprecipitation of profilin was induced by ROCK2 overexpression, suggesting that an isoform-specific relationship might exist between ROCK1 and profilin-1, which are expressed together in most tissues. No immunoprecipitation of profilin(S137A) was observed, regardless of ROCK1 overexpression (Fig. 4B). Unexpectedly, more profilin(S137A) was immunoprecipitated upon ROCK2 overexpression, suggesting that other sites might be more accessible to ROCK2 (but not ROCK1) phosphorylation when Ser-137 is mutated. Regardless, these data strongly suggested that ROCK1 regulates profilin via phosphorylation at Ser-137.
ROCK is a well-known effector of the small GTPase RhoA (1, 32, 37). To evaluate how Rho/ROCK signaling impacts the phosphorylation of endogenous profilin, we created a polyclonal phospho-specific antibody against p-Ser-137 of profilin (P3490). P3490 recognized both endogenous and exogenously expressed profilin in HEK293 cells by Western blotting (Fig. 5A). Its reactivity was sensitive to treatment of samples with alkaline phosphatase, indicating its preference for phosphoprofilin (Fig. 5B). Indeed, P3490 preferentially recognized the phosphomimetic profilin(S137D) versus the phospho-resistant profilin(S137A) (Fig. 5C). It also recognized bacterially expressed wild-type profilin, indicating a low level of reactivity with unphosphorylated profilin (Fig. 5C). P3490 reactivity with HEK293 cell lysate overexpressing ROCK1 increased by twofold, and this was blocked by Y-27632 treatment (Fig. 5D). LPA, a known activator of RhoA, increased P3490 reactivity by ∼58% without changing the total levels of profilin, and this effect was also blocked by Y-27632 treatment (Fig. 5E). Next, we tested for the presence of the Rho/ROCK/profilin signaling pathway in primary cultured neurons. The treatment of these cells with LPA increased profilin phosphorylation at Ser-137 by ∼60%, and Y-27632 treatment blocked this effect (Fig. 5F). Taken together, these results indicate a signaling pathway in which Rho/ROCK signaling increases profilin phosphorylation at Ser-137 in both neural and nonneural cells.
FIG. 5.

Rho/ROCK signaling regulates endogenous profilin phosphorylation at Ser-137. (A to C) Characterization of phosphospecific profilin antibody selective for P-Ser-137 (P3490). (A) HEK293 cells were transfected with Myc-tagged profilin(wt) or profilin(S137A). The cell lysate was analyzed by SDS-PAGE and Western blotting using P3490 or the anti-Myc antibody. While equal amounts of Myc-profilin (wt versus S137A) were present, P3490 preferentially recognized Myc-profilin(wt) versus Myc-profilin(S137A). (B) HEK293 cell lysate was incubated with a fixed amount of calf intestinal phosphatase and increasing concentrations of urea at 37°C for 6 h. Urea was used to partially denature profilin to make Ser-137 accessible to calf intestinal phosphatase. Equal amounts of lysate were analyzed by SDS-PAGE and Western blotting using P3490 or the anti-profilin antibody. Alkaline phosphatase treatment decreased the reactivity of P3490 with endogenous profilin in HEK293 cell lysate. (C) Equal amounts of bacterially expressed His-tagged profilin (wt, S137A, or S137D) were analyzed by SDS-PAGE and Western blotting using P3490 or the anti-His tag antibody. P3490 preferentially recognizes profilin(S137D). (D) HEK293 cells were transfected with pcDNA3 or ROCK1, followed by Y-27632 treatment for 24 h. Lysates were probed for phospho-profilin at Ser-137 (P3490) or total profilin. ROCK1 increased P3490 reactivity by over twofold without changing the total profilin level and was inhibited by Y-27632. (E) HEK293 cells or rat primary striatal neurons were treated with or without 50 μM Y-27632 for 24 h, followed by 10 μM LPA treatment for 45 min. Lysates were analyzed for phospho-profilin or total profilin as above (D). LPA increased P3490 reactivity in both cell types (58% for HEK293 and 60% for neurons) without changing the total profilin level. This was inhibited by Y-27632.
Phosphorylation at Ser-137 negatively regulates profilin.
To evaluate the effect of phosphorylation at Ser-137 on profilin functions, we expressed Myc-tagged wild-type, phospho-resistant (S137A), or phosphomimetic (S137D) profilin mutants in HEK293 cells and compared their interactions with endogenous G actin by coimmunoprecipitation. Under low-salt conditions (50 mM NaCl), all three proteins bound a similar amount of G actin (Fig. 6A). However, under high-salt conditions (500 mM NaCl), profilin(S137A) bound ∼50% more G actin than profilin(wt), whereas profilin(S137D) bound ∼30% less (Fig. 6A). We next tested whether phosphorylation at Ser-137, which is near the polyproline binding region, would inhibit profilin's interaction with Htt. Recombinant His-tagged profilin(wt) and profilin(S137A) bound equal amounts of GST-Htt exon 1, whereas profilin(S137D) failed to bind (Fig. 6B). These data suggested that phosphorylation at Ser-137 reduces profilin's affinity for G actin and blocks its interaction with Htt.
FIG. 6.

Phosphorylation at Ser-137 inhibits profilin activities. (A) Myc-tagged profilin(wt) or mutants (S137A or S137D) were immunoprecipitated (IP) from HEK293 cells. In 50 mM NaCl, all three profilin mutants bound a similar amount of actin; in 500 mM NaCl, profilin(S137A) bound more actin than profilin(wt), and profilin(S137D) bound less (significant difference based on three experiments) (P < 0.01 by unpaired t test). (B) Nickel resin-bound His6-profilin (wt, S137A, or S137D) was mixed with bacterial lysate containing GST-Htt exon 1(Q25). GST-Htt exon 1(Q25) bound profilin(wt) and profilin(S137A) but not profilin(S137D) or nickel resin alone (first lane). (C) Profilin (wt, S137A, or S137D) was cotransfected into HEK293 cells with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP. Relative aggregation was measured by FRET. Profilin(S137A) inhibited aggregation as effectively as did profilin(wt). Profilin(S137D) partially failed to inhibit Htt exon 1(Q72) aggregation (***, P < 0.0005 by unpaired t test) and completely failed to inhibit ARN127(Q65) aggregation. Error bars represent the SEM.
Next, we coexpressed profilin(wt), profilin(S137A), or profilin(S137D) with ARN127(Q65) CFP/YFP or Htt exon 1(Q72) CFP/YFP in HEK293 cells. Profilin(S137D) failed to inhibit AR aggregation and was only partially effective in reducing Htt aggregation (Fig. 6C). In contrast, profilin(S137A) was as effective as the wt protein (Fig. 6C). Next, we coexpressed Htt exon 1(Q72) YFP with either an empty vector or profilin constructs (wt, S137A, and S137D) in primary cultured rat embryonic cortical neurons. After 4 days, we counted the number of inclusions formed by Htt exon 1(Q72) YFP. As we observed in HEK293 cells, profilin(wt) or profilin(S137A) each reduced the number of inclusions, whereas the S137D mutant did not (Fig. 7). Thus, phosphorylation at Ser-137 reduces profilin's antiaggregation activity in primary neurons as it does in nonneural cells. Taken together, these data imply that while unphosphorylated profilin suppresses polyglutamine aggregation, ROCK1 promotes aggregation by phosphorylating and inactivating profilin. Moreover, profilin functions in both neural and nonneural cells.
FIG. 7.

Phosphorylation at Ser-137 inhibits profilin as an aggregation suppressor in primary neurons. Rat embryonic cortical neurons were cotransfected with Htt exon 1(Q72) YFP with either an empty vector or different profilin constructs (wt, S137A, or S137D). Cells were cultured for 4 days and fixed prior to counting inclusions in 10 to 20 fields per coverslip. (A) Representative images of transfected cells showing inclusions formed by Htt exon 1(Q72) YFP (magnification, ×10). Profilin(wt) and profilin(S137A) each reduced the number of inclusions, while profilin(S137D) did not. (B) Averages of three independent experiments showing the effect of profilin expression on inclusion formation. The y axis represents relative inclusion numbers normalized against those in the cells cotransfected with the empty vector. Profilin(wt) and profilin(S137A) each reduced inclusions, whereas profilin(S137D) did not (*, P < 0.05 by paired t test).
Profilin is required for Y-27632 activity.
It remained possible that the effects of Y-27632 on polyglutamine aggregation might result from blocking the phosphorylation of other ROCK targets. Thus, we tested whether profilin is required to mediate the inhibitory effect of Y-27632 on polyglutamine aggregation. We transfected HEK293 cells with two different pools of siRNAs: one targeting human profilin-1 alone and the other targeting both profilin-1 and profilin-2a. A scrambled siRNA sequence was used as a negative control. Western blotting showed different extents of profilin-1 knockdown by the two siRNA pools: ∼50% by profilin-1-specific siRNAs and ∼95% by profilin-1/profilin-2a siRNAs (Fig. 8A). Profilin-1/profilin-2a siRNAs also knocked down endogenous profilin-2a expression by 90%, whereas profilin-1 siRNAs did not (Fig. 8A). Following the first round of transfection with siRNAs alone, cells were transfected again with the same siRNAs along with ARN127(Q65) CFP/YFP or Htt exon 1(Q72) CFP/YFP. Profilin-1-specific siRNAs did not significantly affect the basal aggregation of either ARN127 or Htt exon 1, while profilin-1/profilin-2a-specific siRNAs increased the aggregation of both proteins (Fig. 8B).
FIG. 8.

Profilin is required for the aggregation-inhibitory activity of Y-27632. (A) Control (ctrl), profilin-1 (Prof-1), or profilin-1/profilin-2a-targeting siRNAs were transfected into HEK293 cells, and lysate was evaluated for profilin-1 or profilin-2a by Western blotting. Profilin-1 siRNAs knocked down profilin-1 by 56% and had no effect on profilin-2a; profilin-1/profilin-2a siRNAs knocked down both isoforms by more than 90%. (B) HEK293 cells were transfected with control, profilin-1, or profilin-1/profilin-2a siRNAs along with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP. Relative effects on the aggregation of profilin-1 or profilin-1/profilin-2a siRNAs versus control siRNAs were measured by FRET. Profilin-1 siRNAs had little effect on aggregation; profilin-1/profilin-2a siRNAs increased the aggregation of both Htt exon 1(Q72) (**, P < 0.005) and ARN127(Q65) (***, P < 0.0001 by unpaired t test). Error bars represent the SEM. (C) HEK293 cells were transfected with control, profilin-1, or profilin-1/profilin-2a siRNAs along with Htt exon 1(Q72) CFP/YFP or ARN127(Q65) CFP/YFP with or without Y-27632 (50 μM) treatment. Note that the relative effects of Y-27632 on aggregation in the presence of different siRNAs, not the direct effects of siRNAs on aggregation, as in B, are shown here. Profilin knockdown by both pools of siRNA partially blocked the effect of Y-27632 on ARN127(Q65) aggregation (***, P < 0.0005 by unpaired t test) and nearly eliminated (profilin-1 siRNAs) or reversed (profilin-1/profilin-2a siRNAs) the effect on Htt exon 1(Q72) aggregation (**, P < 0.005; ***, P < 0.0005 [unpaired t test]). Error bars represent the SEM.
We next assessed the effect of profilin knockdown on the activity of Y-27632 as an aggregation inhibitor. Despite a minimal effect on basal aggregation, profilin-1 siRNAs inhibited the Y-27632 effect on ARN127 aggregation by 46% and Htt exon 1 aggregation by 80% compared to cells transfected with the control siRNA (Fig. 8C). Thus, profilin is required for the inhibitory effect of Y-27632 on aggregation. The knockdown of profilin-1 and -2a together inhibited the Y-27632 effect on ARN127 aggregation by 63% and slightly increased Htt exon 1 aggregation in response to the compound (Fig. 8C). Taken together with the preceding experiments, these data place profilin directly downstream from ROCK signaling in its regulation of polyglutamine aggregation. They suggest a model in which ROCK1-induced phosphorylation at Ser-137 inhibits profilin's binding to G actin and Htt and thus simultaneously reduces profilin's ability to block AR and Htt aggregation (Fig. 9).
FIG. 9.
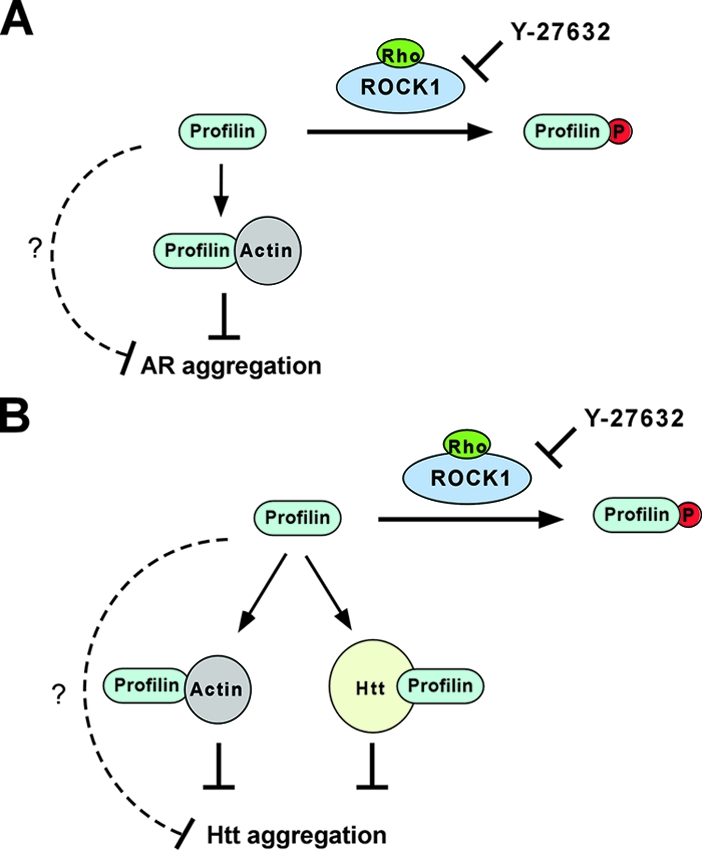
Rho/ROCK1/profilin signaling regulates AR and Htt aggregation via overlapping but distinct mechanisms. (A) Rho activation of ROCK1 leads to its phosphorylation of profilin at Ser-137. This inhibits profilin's interaction with G actin and inactivates profilin as an inhibitor of AR aggregation. (B) In addition to G actin binding, phosphorylation at Ser-137 also inhibits the polyproline binding of profilin and disrupts its direct interaction with Htt. In combination, this inactivates profilin as an inhibitor of Htt aggregation. In both cases, Y-27632 blocks the phosphorylation of profilin by ROCK1, which restores profilin's ability to bind G actin and Htt and its antiaggregation activity. For both AR and Htt, phosphorylation at Ser-137 could also affect other unknown functions of profilin (broken lines) that are important for its antiaggregation activity.
DISCUSSION
Y-27632 is a therapeutic lead for HD and SBMA. It diminishes AR and Htt aggregation in cultured cells and reduces Htt exon 1 toxicity in vivo (36). In this work, we have elucidated a signaling pathway that mediates these effects in neural and nonneural cells. We find that ROCK1, under the control of Rho-GTPase, phosphorylates the actin regulatory factor profilin at Ser-137, and this inactivates profilin as an aggregation inhibitor. Phosphorylation at Ser-137 modestly interferes with profilin's binding to actin, which is required for the maximal inhibition of both AR and Htt aggregation. Phosphorylation at Ser-137 also blocks profilin's interaction with Htt, which is necessary for profilin's maximal inhibition of Htt aggregation. The knockdown of profilin strongly reduces the inhibitory effect of Y-27632 on AR and Htt aggregation. Taken together, these data suggest that profilin is directly downstream of ROCK1, and the Rho/ROCK/profilin signaling pathway constitutes an important therapeutic target. More effective inhibitors of ROCK and other enzymes that modulate profilin phosphorylation at Ser-137 may prove to be effective HD therapies.
In contrast to profilin, cofilin, an actin-severing and -depolymerizing factor, increases AR and Htt aggregation. These data, combined with the known activities of ROCK as an actin-remodeling factor, suggest that the actin cytoskeleton might somehow play a role in regulating the aggregation process. This is supported by recent observations that the inhibition of actin polymerization pharmacologically or by the depletion of Arp2 promotes intracellular Htt aggregation (29). The precise mechanism by which the actin cytoskeleton influences polyglutamine aggregation awaits further study.
Profilin, a novel target of ROCK and an aggregation inhibitor with dual mechanisms.
ROCK regulates myriad cellular processes including smooth-muscle contraction, cell adhesion and migration, cytokinesis and mitosis, and axon growth. These functions are largely attributed to its reorganization of the actin cytoskeleton, and many of the responsible downstream targets have been identified (reviewed in references 1, 15, 32, 33, and 37). In this work, we have identified the actin binding protein profilin as being a novel ROCK1 target. We propose a mechanism whereby the phosphorylation of profilin at Ser-137 reduces its affinity for G actin, and thus actin remodeling by profilin might be downregulated by ROCK1 phosphorylation. Our data also imply that ROCK1 phosphorylation may affect profilin's interaction with other polyproline-rich proteins (in addition to Htt), many of which are known actin binding factors (51).
Our data suggest that profilin uses at least two separable mechanisms to inhibit AR and Htt aggregation: G-actin binding and polyproline binding. Based on mutagenesis studies, profilin's ability to reduce AR aggregation appears to depend entirely on its ability to bind G actin, whereas this only partially accounts for its effects on Htt. Profilin also uniquely interacts with Htt (via its polyproline binding region) to reduce Htt aggregation. This interaction may sequester Htt or stabilize it in a less aggregation-prone conformation. The expanded polyglutamine tract interferes with this interaction. This may, at least in part, explain the importance of the polyproline tract in Htt aggregation and toxicity. Indeed, a recent study in yeast suggested a protective role for the Htt polyproline tract, possibly mediated by its interaction with certain cellular factors (9). Our studies, however, do not exclude the possibility that other functional aspects of profilin might also be important for its antiaggregation activity: although the phosphomimetic mutant of profilin is only modestly affected in G-actin binding, it is completely inactive in suppressing AR aggregation (Fig. 5). Thus, phosphorylation at Ser-137 may alter profilin's interaction with other ligands (e.g., phosphatidylinositol-4,5-bisphosphate binding) in addition to G actin and polyprolines (Fig. 9).
Mechanism of a therapeutic lead.
The development of effective treatments for HD and other polyglutamine diseases will likely depend on a basic understanding of their therapeutic mechanisms of action. Multiple compounds have now demonstrated efficacy in mouse models of HD, such as minocycline (6, 49), creatine (2, 13), coenzyme Q (12, 39), cystamine (8, 21), and histone deacetylase inhibitors (14, 16, 18), but optimization of these compounds may be difficult because the specific mechanisms by which they inhibit Htt toxicity are still unknown. The work described here suggests a molecular mechanism for Y-27632 as a therapeutic lead: the inhibition of ROCK1-mediated phosphorylation of profilin at Ser-137. We have recently observed that the antiaggregation effect of Y-27632 is mediated in part by PRK2 inhibition (43). It is of future interest to investigate whether PRK2 also phosphorylates profilin at Ser-137 to regulate its activity. It is unknown whether a ROCK inhibitor might some day show efficacy in HD or SBMA patients; however, the elucidation of a signaling pathway inhibited by the compound, and a molecular model that makes predictions for its activity, may greatly improve the prospects for devising a mechanism-based therapy.
Acknowledgments
We thank Paul Muchowski for generously providing the pGEX-Htt exon 1 (Q25 or Q53) constructs, Shuh Narumiya for pCAG-ROCK1 (wt or KDIA) plasmids, Thomas Leung for pXJ40-ROCK2, Robert Edwards for the pCAGGS vector, and Donald C. Lo for the gWIZ blank vector. We also thank Venu Nemani and Ken Nakamura (in the laboratory of Robert Edwards) for kindly providing the rat brain tissues and Mei Li for helping with the dissections.
This study was supported by the Sandler Family Supporting Foundation (M.I.D.), the Taube Family Foundation Program in Huntington's Disease Research (M.I.D.), the Muscular Dystrophy Association (J.S. and M.I.D.), and the National Institutes of Health (N.A.D., W.J.W., and M.I.D.).
Footnotes
Published ahead of print on 23 June 2008.
REFERENCES
- 1.Amano, M., Y. Fukata, and K. Kaibuchi. 2000. Regulation and functions of Rho-associated kinase. Exp. Cell Res. 26144-51. [DOI] [PubMed] [Google Scholar]
- 2.Andreassen, O. A., A. Dedeoglu, R. J. Ferrante, B. G. Jenkins, K. L. Ferrante, M. Thomas, A. Friedlich, S. E. Browne, G. Schilling, D. R. Borchelt, S. M. Hersch, C. A. Ross, and M. F. Beal. 2001. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol. Dis. 8479-491. [DOI] [PubMed] [Google Scholar]
- 3.Bamburg, J. R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15185-230. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkegren, C., M. Rozycki, C. E. Schutt, U. Lindberg, and R. Karlsson. 1993. Mutagenesis of human profilin locates its poly(L-proline)-binding site to a hydrophobic patch of aromatic amino acids. FEBS Lett. 333123-126. [DOI] [PubMed] [Google Scholar]
- 5.Burnett, B. G., J. Andrews, S. Ranganathan, K. H. Fischbeck, and N. A. Di Prospero. 2008. Expression of expanded polyglutamine targets profilin for degradation and alters actin dynamics. Neurobiol. Dis. 30365-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, M., V. O. Ona, M. Li, R. J. Ferrante, K. B. Fink, S. Zhu, J. Bian, L. Guo, L. A. Farrell, S. M. Hersch, W. Hobbs, J. P. Vonsattel, J. H. Cha, and R. M. Friedlander. 2000. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 6797-801. [DOI] [PubMed] [Google Scholar]
- 7.Da Silva, J. S., M. Medina, C. Zuliani, A. Di Nardo, W. Witke, and C. G. Dotti. 2003. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J. Cell Biol. 1621267-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dedeoglu, A., J. K. Kubilus, T. M. Jeitner, S. A. Matson, M. Bogdanov, N. W. Kowall, W. R. Matson, A. J. Cooper, R. R. Ratan, M. F. Beal, S. M. Hersch, and R. J. Ferrante. 2002. Therapeutic effects of cystamine in a murine model of Huntington's disease. J. Neurosci. 228942-8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dehay, B., and A. Bertolotti. 2006. Critical role of the proline-rich region in huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 28135608-35615. [DOI] [PubMed] [Google Scholar]
- 10.Desai, U. A., J. Pallos, A. A. Ma, B. R. Stockwell, L. M. Thompson, J. L. Marsh, and M. I. Diamond. 2006. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 152114-2124. [DOI] [PubMed] [Google Scholar]
- 11.Diamond, M. I., M. R. Robinson, and K. R. Yamamoto. 2000. Regulation of expanded polyglutamine protein aggregation and nuclear localization by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 97657-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrante, R. J., O. A. Andreassen, A. Dedeoglu, K. L. Ferrante, B. G. Jenkins, S. M. Hersch, and M. F. Beal. 2002. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J. Neurosci. 221592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrante, R. J., O. A. Andreassen, B. G. Jenkins, A. Dedeoglu, S. Kuemmerle, J. K. Kubilus, R. Kaddurah-Daouk, S. M. Hersch, and M. F. Beal. 2000. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J. Neurosci. 204389-4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrante, R. J., J. K. Kubilus, J. Lee, H. Ryu, A. Beesen, B. Zucker, K. Smith, N. W. Kowall, R. R. Ratan, R. Luthi-Carter, and S. M. Hersch. 2003. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 239418-9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukata, Y., M. Amano, and K. Kaibuchi. 2001. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 2232-39. [DOI] [PubMed] [Google Scholar]
- 16.Gardian, G., S. E. Browne, D. K. Choi, P. Klivenyi, J. Gregorio, J. K. Kubilus, H. Ryu, B. Langley, R. R. Ratan, R. J. Ferrante, and M. F. Beal. 2005. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J. Biol. Chem. 280556-563. [DOI] [PubMed] [Google Scholar]
- 17.Goehler, H., M. Lalowski, U. Stelzl, S. Waelter, M. Stroedicke, U. Worm, A. Droege, K. S. Lindenberg, M. Knoblich, C. Haenig, M. Herbst, J. Suopanki, E. Scherzinger, C. Abraham, B. Bauer, R. Hasenbank, A. Fritzsche, A. H. Ludewig, K. Bussow, S. H. Coleman, C. A. Gutekunst, B. G. Landwehrmeyer, H. Lehrach, and E. E. Wanker. 2004. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol. Cell 15853-865. [DOI] [PubMed] [Google Scholar]
- 18.Hockly, E., V. M. Richon, B. Woodman, D. L. Smith, X. Zhou, E. Rosa, K. Sathasivam, S. Ghazi-Noori, A. Mahal, P. A. Lowden, J. S. Steffan, J. L. Marsh, L. M. Thompson, C. M. Lewis, P. A. Marks, and G. P. Bates. 2003. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA 1002041-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huntington's Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72971-983. [DOI] [PubMed] [Google Scholar]
- 20.Ishizaki, T., M. Naito, K. Fujisawa, M. Maekawa, N. Watanabe, Y. Saito, and S. Narumiya. 1997. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404118-124. [DOI] [PubMed] [Google Scholar]
- 21.Karpuj, M. V., M. W. Becher, J. E. Springer, D. Chabas, S. Youssef, R. Pedotti, D. Mitchell, and L. Steinman. 2002. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 8143-149. [DOI] [PubMed] [Google Scholar]
- 22.Kawano, Y., Y. Fukata, N. Oshiro, M. Amano, T. Nakamura, M. Ito, F. Matsumura, M. Inagaki, and K. Kaibuchi. 1999. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 1471023-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.La Spada, A. R., E. M. Wilson, D. B. Lubahn, A. E. Harding, and K. H. Fischbeck. 1991. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 35277-79. [DOI] [PubMed] [Google Scholar]
- 24.Leung, T., X. Q. Chen, E. Manser, and L. Lim. 1996. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 165313-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, M., E. S. Chevalier-Larsen, D. E. Merry, and M. I. Diamond. 2007. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J. Biol. Chem. 2823157-3164. [DOI] [PubMed] [Google Scholar]
- 26.Maekawa, M., T. Ishizaki, S. Boku, N. Watanabe, A. Fujita, A. Iwamatsu, T. Obinata, K. Ohashi, K. Mizuno, and S. Narumiya. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285895-898. [DOI] [PubMed] [Google Scholar]
- 27.Mahoney, N. M., P. A. Janmey, and S. C. Almo. 1997. Structure of the profilin-poly-L-proline complex involved in morphogenesis and cytoskeletal regulation. Nat. Struct. Biol. 4953-960. [DOI] [PubMed] [Google Scholar]
- 28.Mahoney, N. M., D. A. Rozwarski, E. Fedorov, A. A. Fedorov, and S. C. Almo. 1999. Profilin binds proline-rich ligands in two distinct amide backbone orientations. Nat. Struct. Biol. 6666-671. [DOI] [PubMed] [Google Scholar]
- 29.Meriin, A. B., X. Zhang, I. M. Alexandrov, A. B. Salnikova, M. D. Ter-Avanesian, Y. O. Chernoff, and M. Y. Sherman. 2007. Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains. FASEB J. 211915-1925. [DOI] [PubMed] [Google Scholar]
- 30.Metzler, W. J., A. J. Bell, E. Ernst, T. B. Lavoie, and L. Mueller. 1994. Identification of the poly-L-proline-binding site on human profilin. J. Biol. Chem. 2694620-4625. [PubMed] [Google Scholar]
- 31.Muchowski, P. J., G. Schaffar, A. Sittler, E. E. Wanker, M. K. Hayer-Hartl, and F. U. Hartl. 2000. hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 977841-7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller, B. K., H. Mack, and N. Teusch. 2005. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 4387-398. [DOI] [PubMed] [Google Scholar]
- 33.Noma, K., N. Oyama, and J. K. Liao. 2006. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 290C661-C668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohashi, K., K. Nagata, M. Maekawa, T. Ishizaki, S. Narumiya, and K. Mizuno. 2000. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 2753577-3582. [DOI] [PubMed] [Google Scholar]
- 35.Ostrander, D. B., E. G. Ernst, T. B. Lavoie, and J. A. Gorman. 1999. Polyproline binding is an essential function of human profilin in yeast. Eur. J. Biochem. 26226-35. [DOI] [PubMed] [Google Scholar]
- 36.Pollitt, S. K., J. Pallos, J. Shao, U. A. Desai, A. A. Ma, L. M. Thompson, J. L. Marsh, and M. I. Diamond. 2003. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 40685-694. [DOI] [PubMed] [Google Scholar]
- 37.Riento, K., and A. J. Ridley. 2003. Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4446-456. [DOI] [PubMed] [Google Scholar]
- 38.Ross, C. A., and M. A. Poirier. 2004. Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl)S10-S17. [DOI] [PubMed] [Google Scholar]
- 39.Schilling, G., M. L. Coonfield, C. A. Ross, and D. R. Borchelt. 2001. Coenzyme Q10 and remacemide hydrochloride ameliorate motor deficits in a Huntington's disease transgenic mouse model. Neurosci. Lett. 315149-153. [DOI] [PubMed] [Google Scholar]
- 40.Schluter, K., B. M. Jockusch, and M. Rothkegel. 1997. Profilins as regulators of actin dynamics. Biochim. Biophys. Acta 135997-109. [DOI] [PubMed] [Google Scholar]
- 41.Schluter, K., M. Schleicher, and B. M. Jockusch. 1998. Effects of single amino acid substitutions in the actin-binding site on the biological activity of bovine profilin I. J. Cell Sci. 1113261-3273. [DOI] [PubMed] [Google Scholar]
- 42.Schutt, C. E., J. C. Myslik, M. D. Rozycki, N. C. Goonesekere, and U. Lindberg. 1993. The structure of crystalline profilin-beta-actin. Nature 365810-816. [DOI] [PubMed] [Google Scholar]
- 43.Shao, J., W. J. Welch, and M. I. Diamond. 2008. ROCK and PRK-2 mediate the inhibitory effect of Y-27632 on polyglutamine aggregation. FEBS Lett. 5821637-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh, S. S., A. Chauhan, N. Murakami, J. Styles, M. Elzinga, and V. P. Chauhan. 1996. Phosphoinositide-dependent in vitro phosphorylation of profilin by protein kinase C. Phospholipid specificity and localization of the phosphorylation site. Recept. Signal Transduct. 677-86. [PubMed] [Google Scholar]
- 45.Sumi, T., K. Matsumoto, and T. Nakamura. 2001. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276670-676. [DOI] [PubMed] [Google Scholar]
- 46.Taylor, J. P., F. Tanaka, J. Robitschek, C. M. Sandoval, A. Taye, S. Markovic-Plese, and K. H. Fischbeck. 2003. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12749-757. [DOI] [PubMed] [Google Scholar]
- 47.Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389990-994. [DOI] [PubMed] [Google Scholar]
- 48.Vemuri, B., and S. S. Singh. 2001. Protein kinase C isozyme-specific phosphorylation of profilin. Cell. Signal. 13433-439. [DOI] [PubMed] [Google Scholar]
- 49.Wang, X., S. Zhu, M. Drozda, W. Zhang, I. G. Stavrovskaya, E. Cattaneo, R. J. Ferrante, B. S. Kristal, and R. M. Friedlander. 2003. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc. Natl. Acad. Sci. USA 10010483-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welch, W. J., and M. I. Diamond. 2001. Glucocorticoid modulation of androgen receptor nuclear aggregation and cellular toxicity is associated with distinct forms of soluble expanded polyglutamine protein. Hum. Mol. Genet. 103063-3074. [DOI] [PubMed] [Google Scholar]
- 51.Witke, W. 2004. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 14461-469. [DOI] [PubMed] [Google Scholar]
- 52.Witke, W., A. V. Podtelejnikov, A. Di Nardo, J. D. Sutherland, C. B. Gurniak, C. Dotti, and M. Mann. 1998. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 17967-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wittenmayer, N., B. Jandrig, M. Rothkegel, K. Schluter, W. Arnold, W. Haensch, S. Scherneck, and B. M. Jockusch. 2004. Tumor suppressor activity of profilin requires a functional actin binding site. Mol. Biol. Cell 151600-1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yarmola, E. G., and M. R. Bubb. 2006. Profilin: emerging concepts and lingering misconceptions. Trends Biochem. Sci. 31197-205. [DOI] [PubMed] [Google Scholar]