

Abstract
An increase in angiotensin II (ANG II) under conditions of high salt intake can result in renal damage. The extent to which ANG II does this directly or by way of stimulating aldosterone (Aldo) secretion is a subject of some debate. In the present study, we sought to determine the separate effects of Aldo and ANG II on the expression of plasminogen activator inhibitor-1 (PAI-1) and other factors related to renal fibrosis in the stroke-prone spontaneously hypertensive rat (SHRSP). Saline-drinking male SHRSPs underwent adrenalectomy (ADX) or sham operation (Sham). Treatment groups consisted of ADX + ANG II (25 ng/min sc) and ADX + Aldo (40 μg·kg−1·day−1 sc). After 2 wk of treatment, circulating Aldo levels were reduced to the limit of detection, renal PAI-1, transforming growth factor-β1 (TGF-β1), and osteopontin expression, and phospho-Smad2 (p-Smad2) level were decreased severalfold, and Smad7 (an inhibitory regulator of TGF-β1 action) expression was increased in ADX compared with Sham rats. Infusion of Aldo into ADX SHRSPs restored the renal mRNA expression of PAI-1, TGF-β1 (along with restored p-Smad2 level), and osteopontin and reduced that of Smad7, whereas ANG II had no or a lesser effect. The findings were confirmed by histological examination of renal tissue. In summary, in the saline-drinking SHRSP, Aldo increased renal profibrotic factors and produced renal injury whereas ANG II in the absence of the adrenals had no effect.
Keywords: plasminogen activator inhibitor-1, osteopontin, transforming growth factor-β1
an increase in angiotensin ii (ANG II) under conditions of high salt intake produces renal damage (5, 19). At least part of the effect of ANG II appears to be mediated by aldosterone (Aldo) (6), whose secretion is increased by ANG II. A better understanding of how Aldo affects the potential mediators of renal injury would lend additional insight into its supremacy as the final mediator. In the present study, three different promoters of tissue fibrosis and potential mediators of the renal injury conveyed by ANG II and/or Aldo were studied. Plasminogen activator inhibitor-1 (PAI-1) was selected since it is known to be stimulated by ANG II (7). There is growing evidence that an increase in Aldo may also be required, but this remains controversial (7, 8, 36). Transforming growth factor-β1 (TGF-β1) was studied since on being activated (converted from the latent form) it promotes fibroblast proliferation and differentiation, increases matrix synthesis, and decreases matrix metalloproteases and other proteases that attenuate the progression of the fibrosis (26). In previous studies of uninephrectomized rats, Aldo infusion for 6 wk increased TGF-β1 expression at sites of fibrosis in damaged kidneys (39). Chronic injection of anti-TGF-β1 antibody into salt-fed Dahl salt-sensitive rats attenuated renal injury (12). In addition, TGF-β1 has been shown to induce PAI-1 synthesis (23), and the human PAI-1 promoter region contains TGF-β1 response elements (17). Finally, antibody neutralization of TGF-β1 was shown to decrease the Aldo-induced increase in PAI-1 expression in cardiac cells (11).
A third factor studied was osteopontin (OPN), a chemoattractant that becomes elevated with vascular injury and inflammation (14, 16, 21). It is synthesized by numerous cell types including macrophages, activated T cells, and renal tubular epithelial cells (27). Renal vascular injury and fibrosis have been associated with increased expression of OPN (2), and studies in OPN-knockout mice suggest that upregulation of OPN plays a role in Aldo-induced cardiac remodeling (35, 40).
To further evaluate the singular actions of ANG II and Aldo on the renal expression of PAI-1, TGF-β1, and OPN, we studied the stroke-prone spontaneously hypertensive rat (SHRSP), an animal model highly susceptible to renal injury (9). After bilateral adrenalectomy (ADX) was performed to remove the influences of endogenous Aldo, the animals were treated with either ANG II or Aldo for 2 wk.
MATERIALS AND METHODS
Chemicals.
ANG II was purchased from American Peptide (Sunnyvale, CA), and Aldo and dexamethasone were obtained from Sigma (St. Louis, MO). SYBR Green Supermix with ROX reference was purchased from Bio-Rad (Hercules, CA). Real-time PCR primers and random hexamers were from Invitrogen (Carlsbad, CA). Anti-PAI-1 antibody was purchased from BD Transduction Laboratory (San Jose, CA). Anti-phospho-Smad2 antibody was from Cell Signaling Technology (Danvers, MA), and anti-Smad2/3 and actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Animals.
Male SHRSP/A3N (generations F76–F78 and F84; n = 21) obtained from the colony at New York Medical College (NYMC) were used in these experiments. This study was carried out in accordance with National Institutes of Health (NIH) guidelines and was approved by the NYMC Institutional Animal Care and Use Committee. All animals were housed in a room lighted 12 h per day at an ambient temperature of 22 ± 1°C in the Animal Care Facility at NYMC. Animals were weaned at 4 wk of age and allowed free access to Purina Lab Chow 5001 (Ralston Purina, St. Louis, MO) and tap water until the experiments were initiated.
Animal study protocol.
At 60 days of age, animals were placed on the Stroke-Prone Rodent Diet (no. 39-288; Zeigler Bros., Gardners, PA) and 1% NaCl drinking solution ad libitum. Two to seven days later, animals were anesthetized with pentobarbital (60 mg/kg ip) and ADX was performed in 18 SHRSPs through a flank incision. During this procedure, rats were assigned to one of three different groups, ADX, ADX+ANG II, or ADX+Aldo, and were infused with 0.5% ethanol vehicle (n = 6), ANG II at 25 ng/min (n = 6), or Aldo at 40 μg·kg−1·day−1 (n = 6), respectively, via Alzet osmotic minipumps implanted subcutaneously at the nape of the neck for 2 wk. Glucocorticoid replacement with dexamethasone (12 μg·g−1·day−1 in sesame oil) was instituted after surgery. Three rats received 0.5% ethanol as the vehicle control with adrenal glands left intact (Sham). The doses of Aldo and ANG II were selected based on the amount required to undo the prevention of renal damage in captopril-treated, saline-drinking SHRSPs (30). Two to three days before the termination of the experiment, animals were placed in individual metabolic cages and quantitative 24-h urine collections were obtained for the assessment of proteinuria. Urinary protein concentration was determined by the sulfosalicylic acid turbidity method, and urinary protein excretion was calculated as the product of urinary protein concentration times the urine flow rate. Systolic blood pressure (SBP) was measured by tail-cuff plethysmography with a Natsume KN-210 manometer and tachometer (Peninsula Laboratories, Belmont, CA); 24 h later the animals were killed by decapitation, trunk blood was collected, and the kidneys were removed. Plasma Aldo concentration was measured by standard RIA (Diagnostic Products, Los Angeles, CA), and plasma OPN concentration was measured with an Enzyme Immunometric Assay (Assay Designs, Ann Arbor, MI).
RNA isolation and quantitative real-time polymerase chain reaction assay.
RNA was isolated from kidney with TRIzol reagent (Invitrogen). cDNA was synthesized by the reverse transcriptase reaction, which contained 5.5 mM MgCl2, 2 mM dNTP, 0.4 mM random hexamer, 1 μg of sample RNA, and 2.5 U of reverse transcriptase at 42°C for 2 h. Quantitative real-time polymerase chain reaction (QRT-PCR) analysis was conducted in optical 96-well strips with optical caps, using the MX-4000 multiplex quantitative PCR system (Stratagene, La Jolla, CA) and SYBR Green Supermix (Bio-Rad). Amplification plots were analyzed with MX4000 software version 3 (Stratagene). Briefly, 2× 12.5 μl of iTaq SYBR Green Supermix, 0.2 μM sense and antisense primers, 2.5 μl of cDNA, and 9 μl of nuclease-free water were mixed for QRT-PCR analysis. PCR conditions consisted of an initial denaturation at 95°C for 3 min, followed by amplification for 40 cycles of 15 s at 95°C, 1 min at 60°C. Amplification was immediately followed by a denaturation (melt) program consisting of 1 min at 95°C, 30 s at 55°C with fluorescence acquisition at each temperature transition. The primers for target genes were PAI-1 forward (F): GAT GGG CAC GAG TAC GAC ATC, PAI-1 reverse (R): TGC AAT GAA CAT GCT GAG GG; Smad7 F: TTC GGA CAA CAA GAG TCA GCT GGT, Smad7 R: AGC CTT GAT GGA GAA ACC AGG GAA; and OPN F: TGA ACC AAG CGT GGA AAC ACA CAG, OPN R: TTT GGA ACT CGC CTG ACT GTC GAT (Invitrogen). Rodent TGF-β1 and GAPDH primers were purchased from Applied Biosystems (Foster City, CA). The threshold cycle (Ct) number was determined with the MX4000 software version 3 software program, and relative expression level of each gene was calculated compared with GAPDH expression (2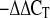 ).
).
Western blotting analysis.
PAI-1 protein expression was measured in fractions of kidney samples that were homogenized in the presence of T-PER tissue protein extraction reagent (Pierce, Rockford, IL) according to the manufacturer's suggested procedure. Samples of whole cell protein (20 μg) were separated in 10% acrylamide gel by SDS-PAGE. After transfer to a polyvinylidene difluoride membrane (Millipore, Bedford, MA), 10% milk in Tris-buffered saline-Tween (TBST) buffer (10 mM Tris·HCl, pH 8.0, 150 mM NaCl, and 0.1% Tween 20) was added to block nonspecific binding sites. A primary antibody at a 1:2,000 dilution in 5% skim milk in TBST was added for 2 h at room temperature. After extensive washing with TBST, horseradish peroxidase-linked secondary antibodies at 1:3,000 dilution were added for 2 h. After washing, target protein bands were detected by the ECL detection system (Amersham, Piscataway, NJ). The PAI-1 band density was measured with a FluorChem 8900 densitometer (Alpha Innotech, San Leandro, CA) and calculated with AlphaEaseFC software. Analysis of variance was used to test for differences in band intensity among the groups, with prespecified follow-up comparisons. A random block effect was included in the models.
Immunohistochemistry.
Immunohistochemical staining for OPN and the monocyte/macrophage marker ED-1 were performed on formalin-fixed, paraffin-embedded kidney sections cut 2–3 μm thick. Sections were dewaxed in xylene, rehydrated, and incubated in 3% H2O2 to inactivate endogenous peroxidase activity. Nonspecific antibody binding was blocked by incubating the slides with 10% or 20% normal goat serum in phosphate-buffered saline (PBS) for 25 min. Rabbit polyclonal anti-OPN (LF-123, Dr. Larry Fisher, NIH, Bethesda, MD) and ED-1 (Chemicon International, Temecula, CA) antibodies were diluted in PBS (1:100 and 1:150, respectively) and applied to each section for 1 h at room temperature. Negative controls were carried out by omitting the primary antibodies and incubating the sections with PBS alone. Sections were then washed thoroughly with PBS and incubated with either biotinylated anti-rabbit (1:500) or anti-mouse (1:500) secondary antibodies for 30 min at room temperature, followed by incubation with horseradish peroxidase-conjugated streptavidin (Dako Cytomation, Carpenteria, CA) for 1 h at room temperature and developing with 3,3′-diaminobenzidine (DAB, Dako Cytomation) to produce a brown color. The sections were then counterstained with hematoxylin.
Histological evaluation.
Coronal sections through the midportion of the kidneys were prepared for examination by light microscopy. Sections were cut at 3 μm and stained with hematoxylin and eosin and periodic acid Schiff reagent. Renal damage was scored on a scale of 0 to 4 based on the severity and prevalence of damage as previously described (37). Glomerular injury was characterized by ischemic or thrombonecrotic change, while microvessels showed proliferative vasculopathy and/or fibrinoid necrosis with extravasation of fragmented erythrocytes with or without thrombosis. A score of 4 indicated extensive lesions affecting >20% of glomeruli and vessels; 3 indicated 15–20% involvement of moderate degree; 2 was assigned to 10–15% involvement with mild to moderate lesions; and 1 was assigned to <10% involvement of mild degree. A score of 0 meant no overt morphological damage. The percentage of ischemic tubules was also evaluated.
Statistical analysis.
Comparisons among the four groups were performed with one-way ANOVA. Results of the pairwise comparisons between groups are shown with adjusting for multiple comparisons (multiple comparisons were performed with Tukey's method to control the overall significance level). Scatter plot and correlation coefficients for relationship between Aldo and profibrotic factors were analyzed with GraphPad software (San Diego, CA).
RESULTS
Blood pressure and plasma Aldo.
Table 1 summarizes the results for SBP, proteinuria, and plasma Aldo in the four groups of SHRSPs. ADX did not significantly affect SBP compared with Sham. Neither ANG II nor Aldo infusion further increased SBP in ADX rats. This result differs from our previous observation, where ADX significantly reduced SBP, which could be restored by either ANG II or Aldo infusion (9), and may reflect the higher baseline in the present series. Urinary protein excretion was reduced in the ADX and ADX+ANG II groups compared with the Sham and ADX+Aldo groups. Plasma Aldo levels were at or below the detection limit in ADX and ADX+ANG II groups compared with the Sham group, but Aldo infusion restored plasma Aldo to levels greater than those of the Sham group (P < 0.05). Histopathological analyses of the kidneys further revealed that Aldo, but not ANG II, was responsible for restoration of renal pathogenesis in ADX SHRSPs. The pattern of renal histology change was similar to that described in our previous publication (9). Staining for ED-1, a monocyte/macrophage marker, is displayed in Fig. 1. Sham-operated animals revealed the presence of occasional inflammatory cells surrounding various glomeruli. Few ED-1-positive cells were found in either the ADX or ADX+ANG II groups. Consistent with the previous patterns, Aldo infusion resulted in a moderate increase in inflammatory cell infiltration, mainly in the cortical regions.
Table 1.
Characteristics of animals
| Sham (n = 3) | ADX (n = 6) | ADX+ANG II (n = 6) | ADX+Aldo (n = 6) | |
|---|---|---|---|---|
| Systolic blood pressure, mmHg | 280.7±4.2 | 273.9±8.7 | 272.5±11.9 | 271.7±8.4 |
| Urinary protein excretion, mg/24 h | 78±36 | 12±1b | 38±15b | 64±14c |
| Plasma Aldo, pg/ml | 612±153 | 13±6b | 16±5b | 945±144a,d,e |
| Histopathological renal score | 2.08±0.72 | 0.11±0.11b | 0.29±0.08b | 2.08±0.41d,e |
Values are means ± SE for n rats. Sham, sham operation; ADX, adrenalectomy; ANG II, angiotensin II; Aldo, aldosterone.
P < 0.05,
P < 0.01 vs. Sham.
P < 0.05,
P < 0.01 vs. ADX.
P < 0.01 vs. ADX+ANG II.
Fig. 1.

Representative photomicrographs of immunocytochemical staining for monocyte/macrophage infiltration (ED-1-positive cells) in the kidney cortex of stroke-prone spontaneously hypertensive rats (SHRSPs). A: mild infiltration in sham-operated (Sham) SHRSP (arrows). B and C: accumulation of inflammatory cells was prevented in the adrenalectomy (ADX) and ADX + angiotensin II (ANG II) groups when aldosterone (Aldo) was absent. D: when Aldo was given back (ADX+Aldo), a substantial increase in ED-1-positive cells was observed (arrows).
PAI-1 expression.
The results for QRT-PCR analysis of PAI-1 expression in kidney samples of the four SHRSP groups are shown in Fig. 2A. ADX substantially decreased PAI-1 expression compared with the Sham group (P < 0.0001). Aldo infusion fully restored PAI-1 expression to the level of the Sham group; however, ANG II infusion in ADX SHRSPs did not alter PAI-1 expression levels compared with ADX SHRSPs. To determine whether these differences in PAI-1 mRNA expression were also present at the protein level, we performed Western blot analyses (Fig. 2B). As in the QRT-PCR analysis, the PAI-1 protein levels were lower in ADX and ADX+ANG II groups compared with the Sham group. Aldo infusion increased PAI-1 protein to levels that exceeded those of the Sham group (Fig. 2, B and C).
Fig. 2.

Relative plasminogen activator inhibitor-1 (PAI-1) gene expression by quantitative RT-PCR (QRT-PCR) analysis (A) and PAI-1 Western blot analysis (B). A: QRT-PCR result is expressed as means ± SE. Sham and ADX+Aldo groups showed higher expression of PAI-1 mRNA than ADX and ADX+ANG II groups. B: there was a higher PAI-1 protein level in Sham and ADX+Aldo groups than in ADX and ADX+ANG II groups (1 representative blot of 3 independent experiments). C: densitometric quantification of 3 separate blots from 3 different experiments (means ± SE). *P < 0.0001 vs. ADX+ANG II; †P < 0.0001 vs. ADX; ‡P < 0.05 vs. Sham.
TGF-β1, phospho-Smad2, and Smad7 expression.
To determine whether the increased PAI-1 expression was associated with increased TGF-β1 activity in SHRSPs, we also measured transcripts of TGF-β1 (Fig. 3A). TGF-β1 expression was 1.5- to 2-fold higher in the Sham and ADX+Aldo groups compared with the ADX and ADX+ANG II groups (P < 0.01 for all comparisons). However, the difference between ADX and ADX+ANG II groups was not statistically significant. Since in many cases TGF-β1 activity cannot be solely determined by its RNA expression or total protein level (18), we measured Smad7, a negative regulator of renal fibrosis, which inhibits TGF-β1 activity by inactivating Smad2 (20) (Fig. 3B). Smad7 mRNA expression was significantly higher in ADX and ADX+ANG II groups compared than in the Sham group (P < 0.001 and P < 0.05, respectively). Aldo infusion (ADX+Aldo) decreased Smad7 mRNA expression compared with the ADX control group (P < 0.001), suggesting that Aldo increased TGF-β1 activity. Smad7 expression was slightly lower in the ADX+ANG II group compared with the ADX group (P < 0.05), suggesting that TGF-β1 activity may have increased in ADX+ANG II rats although TGF-β1 mRNA expression was unaffected. To further confirm the level of biologically active TGF-β1, we measured phospho-Smad2 (p-Smad2) level (Fig. 3C). The p-Smad2 level was significantly lower in ADX and ADX+ANG II groups than in the Sham group (P < 0.01). Aldo infusion (ADX+Aldo) greatly increased the level of p-Smad2 compared with the ADX group (P < 0.01), suggesting that Aldo increased TGF-β1 activity. There was a small but significant increase of p-Smad2 level with ANG II infusion (ADX+ANG II) compared with the ADX group (P < 0.05), suggesting increased TGF-β1 activity as shown in Smad7 expression analysis.
Fig. 3.

Relative gene expression of transforming growth factor-β1 (TGF-β1; A) and Smad7 (B) by QRT-PCR analysis and phospho-Smad2 (p-Smad2) by Western blot analysis (expressed as means ± SE) (C). A: Sham and ADX+Aldo groups showed higher expression of TGF-β1 mRNA than ADX and ADX+ANG II groups. B: Sham and ADX+Aldo groups showed lower expression of Smad7 mRNA than ADX and ADX+ANG II groups. C: there was a higher p-Smad2 level in Sham and ADX+Aldo groups than in ADX and ADX+ANG II groups and a slightly increased level in the ADX+ANG II group compared with the ADX group. Densitometric quantification is also shown (1 representative blot of 3) *P < 0.001 vs. ADX+ANG II; †P < 0.01 vs. ADX; ‡P < 0.05 vs. ADX; §P < 0.001 vs. ADX; ‖P < 0.05 vs. ADX+ANG II.
Renal OPN expression.
OPN mRNA expression was markedly reduced in ADX compared with Sham SHRSPs (Fig. 4A). Infusion of Aldo, but not ANG II, into ADX SHRSPs increased OPN expression fivefold (P < 0.05), such that the levels of OPN mRNA expression in the Sham and ADX+Aldo groups were not different. The levels of OPN were measured by ELISA in plasma obtained from SHRSPs on termination of the study and were found to exhibit the same pattern as was obtained for the levels of OPN mRNA expression (Fig. 4B). Sham SHRSPs displayed OPN levels of 178 ± 31 ng/ml, whereas both the ADX and ADX+ANG II groups exhibited significantly lower plasma OPN levels (95 ± 12 and 93 ± 39 ng/ml, respectively; P < 0.05 vs. Sham). On the other hand, OPN levels were significantly higher in ADX+Aldo compared with ADX or ADX+ANG II groups (205 ± 25 ng/ml; P < 0.05). Representative immunohistochemical staining for OPN is displayed in Fig. 5. Sham-operated SHRSPs displayed moderate to intense expression of OPN in renal thick ascending limb cells and distal cortical tubules and de novo staining in ischemic proximal tubules surrounding areas exhibiting lesions of malignant nephrosclerosis (Fig. 5A). Elevated expression of OPN was absent in both ADX and ADX+ANG II groups, with immunostaining noted in only a few outer medullary tubules (Fig. 5, B and C). When aldosterone was infused into ADX SHRSPs, the same intensity and pattern of OPN expression was observed as seen in Sham SHRSPs (Fig. 5D). The intensity of immunostaining showed a pattern similar to that observed with OPN plasma levels in SHRSPs as seen in Fig. 5. The restoration of OPN expression with Aldo, and not ANG II, infusion suggests that Aldo directly modulates OPN expression in the kidney of SHRSPs. De novo OPN expression in ischemic cortical tubules in Sham and ADX+Aldo, but not in ADX and ADX+ANG II without malignant nephrosclerosis, suggests that OPN in these injured tubules is also modulated by Aldo.
Fig. 4.

Relative osteopontin (OPN) gene expression by QRT-PCR analysis (A) and plasma OPN quantification (B). A: QRT-PCR results are expressed as means ± SE. Sham and ADX+Aldo groups showed higher expression of OPN mRNA than ADX or ADX+ANG II groups. B: plasma OPN was measured by ELISA as described in materials and methods. *P < 0.05 vs. ADX+ANG II; †P < 0.05 vs. ADX.
Fig. 5.

OPN immunohistochemistry in SHRSP. Representative photomicrographs demonstrate moderately intense staining for OPN in cortical distal and ischemic proximal tubules in sham-operated (Sham) animals. A: OPN is localized to the basolateral aspects of tubular cells. Both ADX (B) and ADX+ANG II (C) infusion prevented the upregulation of OPN expression as seen in the Sham group. The intensity of OPN expression in the Sham group was similar to the intensity detected in the ADX+Aldo infusion group (D).
Correlation between Aldo level and expression of profibrotic factors.
To determine whether the plasma Aldo level correlates with the expression of profibrotic factors, plasma Aldo level (each value representing a single animal) was plotted against the level of expression of each of the profibrotic factors (Fig. 6). There was a strong linear relationship between Aldo and OPN expression (r2 = 0.3550, P < 0.005; Fig. 6A), Aldo and PAI-1 expression (r2 = 0.8142, P < 0.0001; Fig. 6B), and Aldo and TGF-1β expression (r2 = 0.5385, P < 0.0001; Fig. 6C). There were also correlations between the expression levels of TGF-β1 and PAI-1 (r2 = 0.4311, P = 0.005), between PAI-1 and OPN (r2 = 0.3057, P < 0.01), and between TGF-β1 and OPN (r2 = 0.2244, P < 0.05) (data not shown). The data further indicate that Aldo regulates profibrotic factors and that each appears to interact with each other to produce the tissue damage.
Fig. 6.

Correlation between Aldo level and mRNA expression of profibrotic factors. The scatter plot and correlation coefficient were analyzed and calculated with GraphPad software. There were significant relationships between Aldo and OPN expression (r2 = 0.3550, P < 0.005; A), Aldo and PAI-1 expression (r2 = 0.8142, P < 0.0001; B), and Aldo and TGF-1β expression (r2 = 0.5385, P < 0.0001; C).
DISCUSSION
In the present study, Aldo increased PAI-1, TGF-β1, and OPN gene expression in kidney. In the absence of adrenals, ANG II had no measurable effect on expression of any of these promoters of fibrosis. Aldo increased the active form of TGF-β1, as evidenced by decreased Smad7 expression and increased p-Smad2 levels in Sham and ADX+Aldo rats (ANG II alone increased TGF-β1 activity slightly). The increased TGF-β1 activity in Sham and ADX+Aldo rats was further confirmed by decreased Smad7 expression. The findings further indicate that the adverse renal effects of ANG II may in large part depend on increases in levels of Aldo.
The role of Aldo as a principal component of the renin-angiotensin-aldosterone system (RAAS) in provoking renal pathology in saline-drinking SHRSPs was previously suggested by the reduction in urinary protein excretion, nephrosclerosis, and cerebrovascular lesions when the animals were treated with spironolactone (29). Furthermore, the effect of the angiotensin-converting enzyme (ACE) inhibitor captopril to prevent renal damage (31) and stroke (25) was reversed by chronic Aldo infusion. The present findings confirm and extend the earlier observations by demonstrating that only Aldo increased the expression of key prothrombotic and profibrotic factors and led to the development of the pathology in SHRSPs.
Both TGF-β1 and ANG II are known to activate renal fibrosis by increasing the synthesis and decreasing the degradation of matrix proteins (1, 3, 4). An increase in PAI-1 leads to a decrease in generation of plasmin, thereby reducing plasmin-mediated activation of matrix metalloproteinases that normally break down matrix protein to reduce fibrosis (22). The induction of PAI-1 by ANG II has been demonstrated both in vivo and in vitro (28, 41). ANG II infusion increased PAI-1 in the endothelium of the vasculature in kidney and other organs. Studies by Brown and coworkers (7) demonstrated that direct application of ANG II to cultured rat aortic smooth muscle cells and human umbilical vein endothelial cells stimulated PAI-1 gene expression and PAI-1 release within 24 h. This effect was not thought to be mediated by Aldo since its direct application was without an effect despite the fact that these cells express the mineralocorticoid receptor (MR) (7). Studies in vivo suggest an action of Aldo that is independent of ANG II and suggest that it may mediate the actions of ANG II on PAI-1. Specifically, in experiments performed in animals with intact adrenal glands, either spironolactone or an AT1 receptor blocker was capable of reducing PAI-1 expression (8). Likewise in human subjects treated with an MR antagonist, an intervention known to increase levels of plasma renin activity and ANG II, PAI-1 was decreased (36), although this might also result from conversion to a low-salt state. In the present study, neither Aldo nor ANG II infusion had an effect on SBP in ADX SHRSPs, and only Aldo increased PAI-1 expression, suggesting that of the two major hormone products of the RAAS, Aldo mediated the renal injury.
We previously showed that TGF-β1 mediated Aldo's effects on inducible nitric oxide synthase (10) and PAI-1 (11) in cardiac myocytes. In those studies, Aldo appeared to transform a latent TGF-β1 to the biologically active form without at the same time affecting transcription or translation. Overexpression of Smad7 has been demonstrated to block TGF-β1-induced Smad2 activation and prevent collagen synthesis and myofibroblast transformation in rat kidney tubular epithelial cells (20). The lower Smad7 expression observed in Sham and ADX+Aldo SHRSPs is consistent with higher TGF-β1 activity in these groups compared with the ADX and ADX+ANG II SHRSPs. The slightly lower Smad7 expression in ADX+ANG II SHRSPs may be due to an increased TGF-β1 activity at the protein level, as suggested by the small increase in levels of p-Smad2, although it is also possible that ANG II might regulate Smad7 independently of TGF-β1 as has been shown in vascular smooth muscle cells (33). Whether Aldo-induced PAI-1 expression in SHRSP kidney is mediated by TGF-β1 remains to be established. Interestingly, recent studies using PAI-1-knockout mice showed higher renal TGF-β1 expression and activity than in wild-type mice (24), which may be due to increased plasmin-induced TGF-β1 activation in the absence of PAI-1. It is noteworthy that TGF-β1 and PAI-1 affect each other and that the loss of their tight interregulation might also contribute to renal fibrosis (15). Additional studies suggest that there are species-specific interactions between PAI-1 and TGF-β1 on tissue fibrosis that are complex (13, 24).
Aldo administration has been shown to increase OPN expression in rat heart (32) and kidney (2) as in the present study. Myocardial fibrosis and remodeling in response to Aldo infusion was markedly diminished in OPN-knockout mice compared with wild-type control mice, consistent with OPN being one of the mediators of the proinflammatory and profibrotic actions of Aldo (35). Aldo-induced OPN was inhibited by MR antagonists but not by the glucocorticoid receptor antagonist RU486 in rat aortic endothelial cells (2, 38). In PAI-1-knockout mice, Aldo-activated renal OPN expression was decreased compared with that in wild-type mice (24), suggesting an interaction between PAI-1 and OPN in Aldo-dependent renal fibrosis.
We obviously cannot from these experiments discount an important role for ANG II in certain human conditions where it has been amply demonstrated that ACE inhibitors and AT1 receptor blockers provide benefit. The findings do, however, reemphasize the potential renal damaging effects of Aldo. This was made clearer in the recent PAPY Study, a large-scale clinical trial of individuals with primary aldosteronism (high aldosterone levels and suppressed ANG II levels), where significant renal damage as assessed from urinary albumin excretion was more common than in individuals with primary hypertension (34).
In summary, administration of Aldo, but not ANG II, increased multiple profibrotic factors in ADX SHRSPs. The findings lend additional support for an important role of Aldo in promoting renal injury.
GRANTS
Support was provided by National Heart, Lung, and Blood Institute Grants R01-HL-35795 and R01-HL-67360 and by the Department of Veterans Affairs.
Acknowledgments
We thank George Eckert for help with performing the statistical analyses.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Anderson PW, Zhang XY, Tian J, Correale JD, Xi XP, Yang D, Graf K, Law RE, Hsueh WA. Insulin and angiotensin II are additive in stimulating TGF-beta 1 and matrix mRNAs in mesangial cells. Kidney Int 50: 745–753, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int 63: 1791–1800, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Border WA, Okuda S, Languino LR, Ruoslahti E. Transforming growth factor-beta regulates production of proteoglycans by mesangial cells. Kidney Int 37: 689–695, 1990. [DOI] [PubMed] [Google Scholar]
- 4.Border WA, Okuda S, Nakamura T, Languino LR, Ruoslahti E. Role of TGF-beta 1 in experimental glomerulonephritis. Ciba Found Symp 157: 178–189, 1991. [PubMed] [Google Scholar]
- 5.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Brown NJ Aldosterone and end-organ damage. Curr Opin Nephrol Hypertens 14: 235–241, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Brown NJ, Kim KS, Chen YQ, Blevins LS, Nadeau JH, Meranze SG, Vaughan DE. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab 85: 336–344, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, Vaughan DE, Fogo AB. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 58: 1219–1227, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Chander PN, Rocha R, Ranaudo J, Singh G, Zuckerman A, Stier CT Jr. Aldosterone plays a pivotal role in the pathogenesis of thrombotic microangiopathy in SHRSP. J Am Soc Nephrol 14: 1990–1997, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Chun TY, Bloem LJ, Pratt JH. Aldosterone inhibits inducible nitric oxide synthase in neonatal rat cardiomyocytes. Endocrinology 144: 1712–1717, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Chun TY, Pratt JH. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol Cell Endocrinol 239: 55–61, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Dahly AJ, Hoagland KM, Flasch AK, Jha S, Ledbetter SR, Roman RJ. Antihypertensive effects of chronic anti-TGF-β antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol 283: R757–R767, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Eren M, Painter CA, Gleaves LA, Schoenhard JA, Atkinson JB, Brown NJ, Vaughan DE. Tissue- and agonist-specific regulation of human and murine plasminogen activator inhibitor-1 promoters in transgenic mice. J Thromb Haemost 1: 2389–2396, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest 92: 1686–1696, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husted RF, Sigmund RD, Stokes JB. Mechanisms of inactivation of the action of aldosterone on collecting duct by TGF-β. Am J Physiol Renal Physiol 278: F425–F433, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda T, Shirasawa T, Esaki Y, Yoshiki S, Hirokawa K. Osteopontin mRNA is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta. J Clin Invest 92: 2814–2820, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keeton MR, Curriden SA, van Zonneveld AJ, Loskutoff DJ. Identification of regulatory sequences in the type 1 plasminogen activator inhibitor gene responsive to transforming growth factor beta. J Biol Chem 266: 23048–23052, 1991. [PubMed] [Google Scholar]
- 18.Khalil N TGF-beta: from latent to active. Microbes Infect 1: 1255–1263, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329: 1456–1462, 1993. [DOI] [PubMed] [Google Scholar]
- 20.Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol 13: 1464–1472, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Liaw L, Lombardi DM, Almeida MM, Schwartz SM, deBlois D, Giachelli CM. Neutralizing antibodies directed against osteopontin inhibit rat carotid neointimal thickening after endothelial denudation. Arterioscler Thromb Vasc Biol 17: 188–193, 1997. [DOI] [PubMed] [Google Scholar]
- 22.Lijnen HR Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 86: 324–333, 2001. [PubMed] [Google Scholar]
- 23.Lund LR, Riccio A, Andreasen PA, Nielsen LS, Kristensen P, Laiho M, Saksela O, Blasi F, Dano K. Transforming growth factor-beta is a strong and fast acting positive regulator of the level of type-1 plasminogen activator inhibitor mRNA in WI-38 human lung fibroblasts. EMBO J 6: 1281–1286, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma J, Weisberg A, Griffin JP, Vaughan DE, Fogo AB, Brown NJ. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int 69: 1064–1072, 2006. [DOI] [PubMed] [Google Scholar]
- 25.MacLeod AB, Vasdev S, Smeda JS. The role of blood pressure and aldosterone in the production of hemorrhagic stroke in captopril-treated hypertensive rats. Stroke 28: 1821–1828, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev 14: 627–644, 2000. [PubMed] [Google Scholar]
- 27.Okamoto H Osteopontin and cardiovascular system. Mol Cell Biochem 2006. [DOI] [PubMed]
- 28.Ridker PM, Gaboury CL, Conlin PR, Seely EW, Williams GH, Vaughan DE. Stimulation of plasminogen activator inhibitor in vivo by infusion of angiotensin II. Evidence of a potential interaction between the renin-angiotensin system and fibrinolytic function. Circulation 87: 1969–1973, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Rocha R, Chander PN, Khanna K, Zuckerman A, Stier CT Jr. Mineralocorticoid blockade reduces vascular injury in stroke-prone hypertensive rats. Hypertension 31: 451–458, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Rocha R, Chander PN, Zuckerman A, Stier CT Jr. Mineralocorticoid antagonism reduces ANG-II induced renal injury in stroke-prone hypertensive rats (Abstract). Am J Hypertens 11: 94, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Rocha R, Chander PN, Zuckerman A, Stier CT Jr. Role of aldosterone in renal vascular injury in stroke-prone hypertensive rats. Hypertension 33: 232–237, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Rocha R, Martin-Berger CL, Yang P, Scherrer R, Delyani J, McMahon E. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology 143: 4828–4836, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 111: 2509–2517, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Rossi GP, Bernini G, Desideri G, Fabris B, Ferri C, Giacchetti G, Letizia C, Maccario M, Mannelli M, Matterello MJ, Montemurro D, Palumbo G, Rizzoni D, Rossi E, Pessina AC, Mantero F. Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension 48: 232–238, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Sam F, Xie Z, Ooi H, Kerstetter DL, Colucci WS, Singh M, Singh K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am J Hypertens 17: 188–193, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Sawathiparnich P, Kumar S, Vaughan DE, Brown NJ. Spironolactone abolishes the relationship between aldosterone and plasminogen activator inhibitor-1 in humans. J Clin Endocrinol Metab 87: 448–452, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Stier CT, Mahboubi K, DiPippo VA, Levine S, Chander PN. The antiproteinuric action of enalapril in stroke-prone spontaneously hypertensive rats is unrelated to alterations in urinary prostaglandins. J Pharmacol Exp Ther 260: 1410–1415, 1992. [PubMed] [Google Scholar]
- 38.Sugiyama T, Yoshimoto T, Hirono Y, Suzuki N, Sakurada M, Tsuchiya K, Minami I, Iwashima F, Sakai H, Tateno T, Sato R, Hirata Y. Aldosterone increases osteopontin gene expression in rat endothelial cells. Biochem Biophys Res Commun 336: 163–167, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Zhang J, Zhang JQ, Ramires FJ. Local angiotensin II and transforming growth factor-beta1 in renal fibrosis of rats. Hypertension 35: 1078–1084, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res 88: 1080–1087, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest 95: 995–1001, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]