

Abstract
An on-line sample concentration method using sample matrix switching and field amplification peak stacking has been developed. A microbore LC guard column is used to slightly retain the analytes in order to switch from a high ionic strength sample matrix (the physiological fluid) to a low ionic strength matrix (the LC mobile phase). The eluted LC peak is then trapped in a CE system and preconcentrated by field amplification peak stacking. The concentrated sample peak is then analyzed by CE. Compared to normal hydrodynamic injection, the sensitivity was increased by more than 500-fold without loss in resolution. A limit of detection of less than 10 nM for a physiological sample was achieved using UV adsorption detection. This method can be used for negatively or positively charged analytes.
A major limitation of capillary electrophoresis (CE) is the poor concentration detection limits achieved using spectrophotometric detectors. To achieve useable detection limits for most biological applications, special detectors such as laser-induced fluorescence or electrochemistry must be used. However, these are limited in the range of compounds that are detectable relative to UV absorbance. Therefore, approaches to improve the concentration detection limits achievable using spectrophotometric detection would dramatically increase the utility of CE.
To achieve useable detection limits with UV absorbance, a preconcentration step is typically performed prior to CE analysis. However, one of the strengths of CE is the small sample volume requirement of the technique. Traditional preconcentration approaches, such as solid-phase or liquid–liquid extractions, require sample volumes that eliminate this advantage. Therefore, several on-column approaches to sample preconcentration have been developed. These include field amplification, capillary isotachophoresis, and chromatographic preconcentration.
In field amplification stacking, sample analytes are concentrated into a narrow band at the edge of the sample zone.1-7 This is accomplished by generating a strong electric field across the sample zone by using a sample matrix of significantly lower conductivity than the running buffer. Analytes then migrate much faster in the sample zone than in the running buffer. This approach has been modified using polarity reversal to remove the low-conductivity sample matrix prior to the electrophoretic separation.8-10 Termed large-volume injection with polarity reversal, the sample is initially injected hydrodynamically; the matrix is electroosmotically removed under reverse polarity conditions while sample anions stack at the boundary of the sample zone and the running buffer. Once the sample matrix is largely removed, the separation is performed under normal polarity. On-column transient capillary isotachophoresis (CITP) can be viewed as a sophisticated form of stacking.11-14 For CITP, the sample zone is sandwiched between a leading electrolyte, with a mobility greater than any of the sample components, and a trailing electrolyte, with a mobility slower than any of the sample components. However, as field amplification and CITP are only applicable to low ionic strength samples, their utility for the analysis of high ionic strength physiological fluids is limited.
Chromatographic preconcentration has been accomplished by introducing a region of chromatographic media at the head of the CE capillary.15-23 Cai and Rassi developed an open tubular preconcentrator for use on-line with CE.15 On-line preconcentration with a C-18-coated capillary enhanced the analytical signal by a factor of 10. However, while this approach enabled the analysis of larger sample volumes compared to conventional CE, the coated capillary had a low capacity and was easily saturated. This approach also required 10–20 min to permit equilibration of the capillary with the binding electrolyte before each run. An alternative approach involved the insertion of a small bed of an appropriate adsorptive solid-phase material at the inlet of the CE capillary.16-23 The use of a packed-bed concentrator allowed introduction of increased sample volumes into a CE capillary for a variety of analyte types. Sensitivity increases of up to 700-fold have been reported.21 However, while this technique significantly improves the detection limit, the CE performance is frequently compromised. The use of solid-phase preconcentration-CE often results in reduced analyte resolution, column efficiency, and separation speed and substantial component tailing. The packed-bed concentrator must be loaded slowly, typically requiring 10–20 min. Furthermore, CE separation times are typically longer for this method than for normal CE.16,17,23 The increased back pressure induced in the CE capillary by the packing and frit material leads to a reduced hydrodynamic flow and other problems, such as formation of air bubbles. The combination of slow sample loading and longer separation times results in significantly lower sample through-put for solid-phase preconcentration-CE.
A hydrophobic adsorbent membrane, which acts rather like a reversed-phase chromatographic packing material, has been described as an alternative to the use of packed-bed concentrators.24,25 A typical configuration involves sandwiching the membrane between the ends of two capillaries, with a sheath tubing placed over the junction to form a mechanically stable structure. However, because of the limited amount of stationary phase in this microconcentrator, low capacity and overloading are problems, particularly with biological fluids. Furthermore, irreversible adsorption of endogenous components shortens the life of the preconcentrator. As with other adsorption-based preconcentration techniques, this approach required a rather long sample loading time (1 μL/10 min), separation time, and activation time of the adsorptive membrane. A supported liquid membrane device has recently been described for concentrating samples on-line prior to CE separation.26 This device incorporated field amplification stacking prior to the separation as a second concentrating step. On-line preconcentration techniques have recently been reviewed.27,28
In this report, an on-line matrix-switching/CE system enabling field amplification stacking of analytes injected in high ionic strength sample matrixes is described. Instead of packing the fused-silica capillary, a microbore LC guard column is connected to the capillary. In this system, the LC column is not used for concentrating the analyte but rather to switch the matrix from the high ionic strength biological fluid to a low ionic strength buffer. After this process, analytes could be concentrated by field amplification stacking because the sample matrix (the LC buffer) was of significantly lower ionic strength than the CE running buffer. The CE separation was then carried out in a separate capillary such that the stacking system did not compromise the separation. Compared to a normal CE method, the sensitivity was increased more than 500-fold without loss in resolution or efficiency. A detection limit of 10 nM was achieved for several analytes injected in biological samples. The utility of the system was ultimately demonstrated by determining bupivacaine in microdialysis samples.
EXPERIMENTAL SECTION
Materials and Reagents
Primaquine, doxepin, and bupivacaine were used as model compounds in this research. Bupivacaine hydrochloride, primaquine, and doxepin were purchased from Sigma Chemical (St. Louis, MO). All other chemicals were reagent grade or better and used as received. All water was purified by passing distilled water through a Barnstead water purification system.
Two buffers were used in this work. The first, the LC buffer, consisted of 25% (v/v) acetonitrile and 75% 7.5 mM lithium citrate buffer, pH 2.5. The second, the CE running buffer, consisted of 150 mM lithium acetate, pH 4.75. Ringer’s solution consisted of 155 mM NaCl, 5.5 mM KCl, and 2.3 mM CaCl2 in deionized water. All solutions were filtered through a 0.2-μm filter prior to use. Standard solutions were prepared by dissolving primaquine, doxepin, and bupivacaine in Ringer’s solution at pH 7.4.
Apparatus
The complete system consisted of a microbore LC system connected to a CE system as shown in Figure 1. The LC system, used for matrix switching, consisted of an Isco Laboratory Instruments (Lincoln, NE) model 2350 LC pump (P) connected to a Valco Instrument (Houston, TX) E045 electronic valve (S1) which was then connected to a Rheodyne 7125 injection valve (V). A 1 × 0.3 mm, 5-μm C-18 repackable guard column (LC Packings, San Francisco, CA) and a 14 × 1 mm, 5-μm C-18 guard cartridge (L) (Bioanalytical Systems, West Lafayette, IN) were used for buffer switching. Two-way valve S1 was used to divert the flow of the LC pump while the system was operating in the CE mode. This was found to be more efficient than turning the pump on and off. A length of restrictor tubing (R) (Bioanalytical Systems, Inc.) was connected to valve S1 to provide back pressure. Sample was loaded into the sample loop of the injection valve directly by syringe. The CE system consisted of an Isco model 3850 capillary electrophoresis system used in conjunction with a homemade safety box. Four fused-silica capillaries (C1, C2, C3, C4) (Polymicro Technology, Phoenix, AZ) were connected with a Valco cross microadapter (A). Capillaries C2 and C4 were connected to two-way switching valves (S2 and S4, Upchurch Scientific, Oak Harbor, WA). These switching valves had a 0.020-in. thru-hole and capillary tubing was connected using PEEK sleeves (Upchurch). These switching valves were used to prevent flow in the CE sections when in the LC mode. CE running buffer reservoirs were placed at the end of capillaries C2, C3, and C4. R2 was the high voltage end of the CE system while R3 and R4 were grounded. Buffer reservoir R4 was connected to a high-pressure argon system for hydrodynamic filling of the CE system. Two on-column UV detectors were used. The CE separation was monitored by a detector on capillary C2, while the matrix switching was monitored by a detector on capillary C3. Detection was performed at 210 nm. A Datajet integrator (model SP4600, Spectra-Physics, San Jose, CA) connected to a WINner/386 workstation was used for data acquisition.
Figure 1.
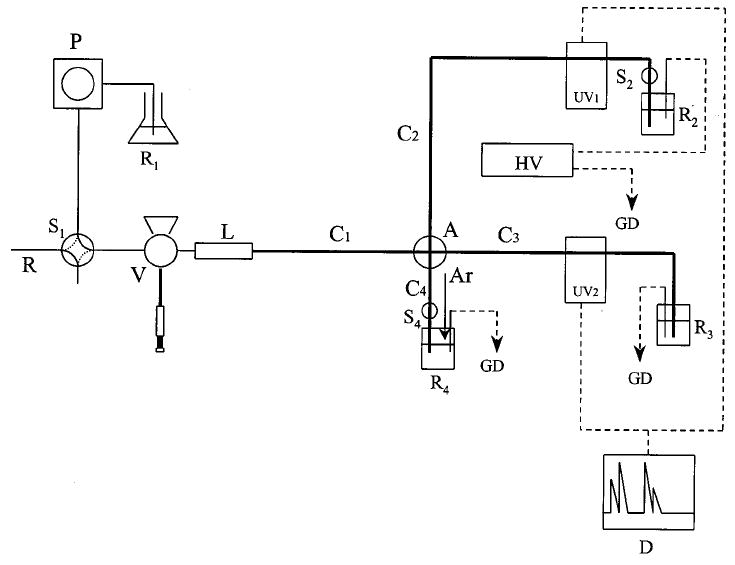
Schematic diagram of the matrix-switching LC–CE apparatus: P, LC pump; S1, S2, and S4, switching valves; V, LC injection valve; L, micro-LC column; C1 and C4, fused-silica capillary; C2, separation capillary; C3, stacking capillary; A, cross adapter; R1, LC mobile-phase reservoir; R2, R3, and R4, CE buffer reservoirs; UV1 and UV2, on-column UV detectors; R, restrictor tubing; D, data recording system; HV, high-voltage power supply; Ar, high-pressure argon; GD, ground.
Experimental Procedure
At the start of each day, the separation capillary was rinsed sequentially at 40 psi with 0.5 M EDTA (pH 13), water, and running buffer for 15, 5, and 10 min, respectively. All solutions were filtered through a 0.2-μm filter. Reservoir R1 contained the LC mobile phase while reservoirs R2, R3, and R4 contained the CE running buffer. Before sample injection, switching valve S1 was closed to ensure that mobile phase did not pass through the LC column, and switching valves S2 and S4 were open. In this position, the CE system (capillaries C2, C3, and C4) was filled with CE running buffer (150 mM acetate, pH 4.75) from reservoir R4 using argon pressure. Switching valves S2 and S4 were then closed, and S1 was opened to allow the mobile phase to pass through both the LC column and capillaries C1 and C3 but not capillaries C2 and C4. At the beginning of the day, the column was allowed to equilibrate with the mobile phase for 2 min after which samples were injected using the LC injection valve (V). A flow rate of 30 μL/min was used for all experiments. Elution of the analytes from the LC column was monitored by detector UV2. When the point of maximum absorbance was reached, switching valve S1 was closed to stop flow of the mobile phase through the capillary system. At this point, the analyte plug was in capillary C3 in the low ionic strength LC mobile phase while capillaries C2 and C4 were filled with the high ionic strength CE running buffer. Switching valve S2 was then opened and a potential of 22 kV applied between R2 and R3 for 80 s. This stacked the analytes at the head of capillary C2 by field amplification. Immediately following this, switching valve S2 was closed, switching valve S4 was opened, and the mobile phase in capillary C3 was replaced with running buffer by application of argon pressure in reservoir R4 for 10 s. The final step of the operation was to open switching valve S2 and apply 18 kV between R2 and R4 to achieve the CE separation. The separation was monitored with detector UV1. After each run, the system was flushed with running buffer by application of argon pressure in reservoir R4 for 20 s with both switching valves S2 and S4 opened. The system was then ready for the next sample injection.
RESULTS AND DISCUSSIONS
Sample Matrix-Switching and Field-Amplified Stacking Procedure
A schematic representation of the scheme for sample matrix switching and field amplification stacking is shown in Figure 2. Prior to sample injection, capillaries C2 and C4 are filled with the high ionic strength CE buffer and capillaries C1 and C3 are filled with the low ionic strength LC buffer. In the first step (Figure 2A), sample is injected onto the LC column. Analytes are slightly retained in the column while the high ionic strength sample matrix is flushed through the column. In the second step (Figure 2B), the analytes are eluted from the column into capillary C3 by the mobile phase. At this point, the analytes have been switched from the original high ionic strength sample matrix to the low ionic strength LC buffer. In the next step (Figure 2C), a high voltage is applied at the ends of C2 and C3, and the analytes are stacked at the interface between the high ionic strength CE buffer in C2 and the low ionic strength LC buffer in C3. This stacking occurs because there is a large difference in electric field strength between these two zones. While the analytes, stacked in this manner, are concentrated into a narrow zone, there is a small degree of movement of the analytes into the separation capillary C2, because of electroosmotic flow (EOF). In the fourth step (Figure 2D), the voltage is removed and the low ionic strength LC buffer in C3 is displaced with the CE buffer from reservoir R4 using a pressure-driven flush. Finally, the separation voltage is applied at the ends of capillaries C2 and C4 and the analytes are separated in capillary C2 by a normal CE process (Figure 2E).
Figure 2.
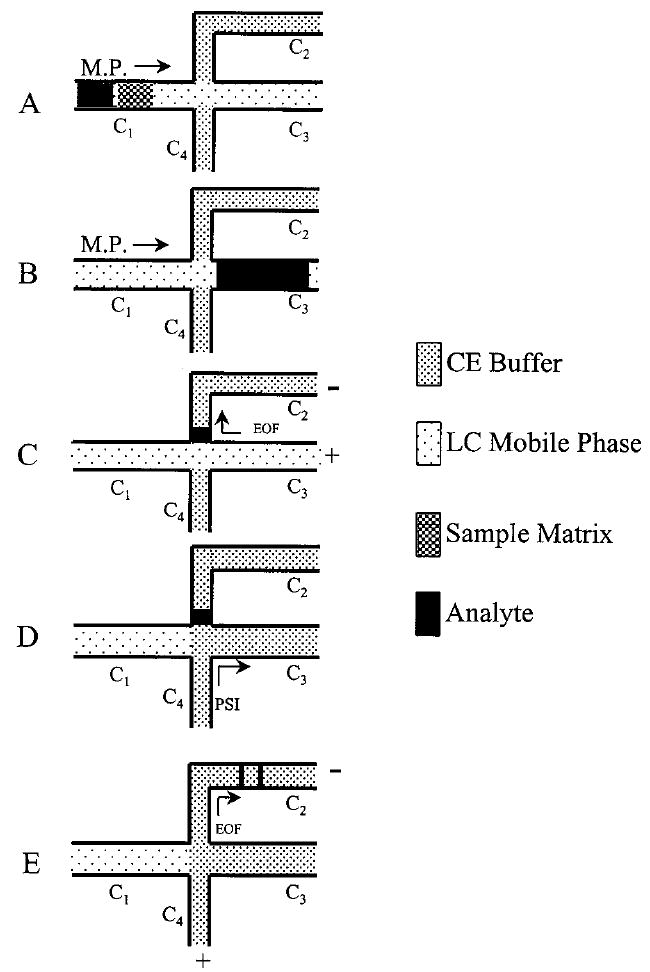
Schematic representation of the steps in matrix-switching LC–CE: (A) LC matrix switching, (B) trapping of analyte peak in the stacking capillary, (C) field amplification stacking of analyte, (D) flushing of LC mobile phase from the CE system, and (E) CE separation.
Sample Matrix Switching
The purpose of the LC column in this system is to separate the analytes from the sample matrix. This column is not intended for concentration of the analytes or separation of the analytes from each other. The ideal situation is one in which the analytes are completely separated from the sample matrix and are then immediately eluted from the LC column in a single band. To achieve this goal, a range of column dimensions, stationary-phase particle sizes, and chemistries was investigated (results not shown). A 1 × 14 mm short microbore column packed with 5-μm C-18 stationary phase was chosen to provide sufficient retention for matrix switching with a weak LC buffer. A repackable guard column (1 × 0.3 mm) was placed in front of the microbore column for protection. Figure 3 shows that bupivacaine and primaquine were just separated from the matrix (the large negative peak) and eluted as a single peak. If an analyte has too little retention on the short column, a longer column or smaller particle size may be selected. However, these changes lead to higher back pressure which may cause a problem when the mobile-phase flow is stopped for stacking and separation. While the C-18 phase worked well for the compounds used in this study, other stationary phase chemistries may be more suitable for other analytes.
Figure 3.
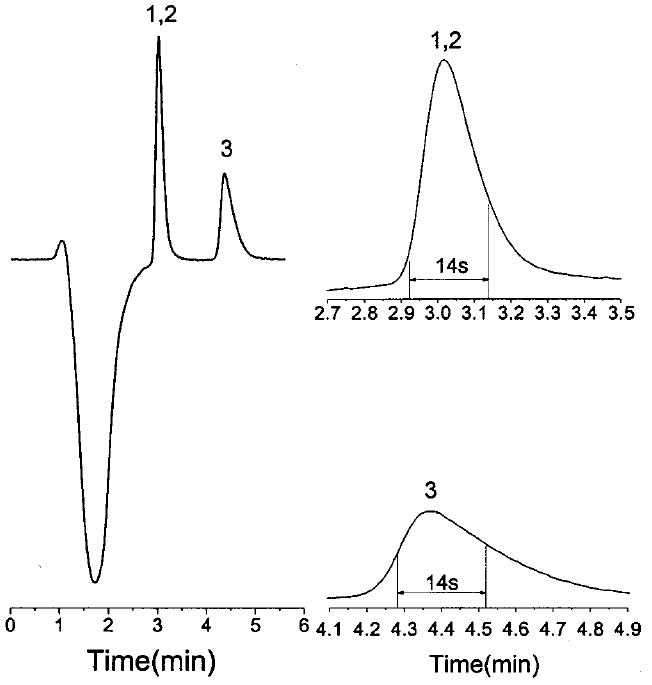
Matrix-switching chromatogram (obtained at detector UV2 in Figure 1) for a mixture bupivacaine, primaquine, and doxepin. Peak widths of the individual peaks are shown in the expanded chromatograms on the right: mobile phase, 10 mM lithium citrate, pH 2.5/acetonitrile (70:30 v/v); flow rate, 30 μL/min; sample, 5 μM each bupivacaine (1), primaquine (2), and doxepin (3) in Ringer’s solution. Other conditions as in Figure 3.
Proper selection of the dimensions of capillaries C1 and C3 is critical. The function of capillary C1 is to provide back pressure on the outlet of the LC column. This serves two purposes. First, the back pressure eliminates continued flow of mobile phase through the column after switch S1 is switched. Second, it prevents a sudden pressure drop at the end of the LC column. A sudden pressure drop will cause serious outgassing in the eluent, and the bubbles formed will consequently interfere with the CE operation. A 50-μm-i.d. capillary of 50-cm length was found to provide sufficient backpressure.
The choice of the dimensions for capillary C3 was based on the analyte peak width from the LC column and the requirements for field amplification stacking. This capillary must have sufficient volume to contain the analyte peak eluting from the LC column. However, a capillary with either too wide a bore or too long a length will result in a weak electric field and a long stacking time. The typical peak width was estimated as 20 s for the microbore guard cartridge used in this work. A 40-cm length of 150-μm-i.d. capillary for capillary C3 could contain a 14-s-wide peak (this corresponds to a volume of 7 μL). Increasing the volume of capillary C3 by either increasing the length or inner diameter of the capillary resulted in unacceptable stacking results.
While primaquine and bupivicaine coeluted from the LC column, addition of a more hydrophobic analyte, doxepin, resulted in a second peak eluting from the LC column (Figure 3). Variation of the mobile-phase ionic strength and pH did not result in coelution of doxepin with the other two analytes. The chromatogram shows that 80% of the first peak and 70% of the second peak can be kept in capillary C3. The reproducibility of the LC procedure based on elution time was 1.9% RSD. Therefore, once the elution time is known, this can be used as the switching time to stop the LC mobile phase for repetitive analysis of the same analyte. In this manner, detector UV2 is not necessary.
Removal of the LC Buffer from Capillary C3 Prior to CE Analysis
The low ionic strength LC buffer must be removed from capillary C3 after the stacking process before the CE separation begins. If the LC buffer remained in the stacking capillary C3 after stacking, leakage of the LC mobile phase from the capillary C3 into the separation capillary C2 was observed during the electrophoretic separation procedure. This is because the internal diameter of C3 is twice that of C2. Leakage of the LC mobile phase into the separation capillary results in a large void peak that interferes with the CE separation. Filling capillary C3 with the CE running buffer prior to separation circumvented this problem.
Effect of Buffer pH on Stacking and Separation
The pH of both the LC buffer and CE buffer affect the stacking and separation process with this system. A pH below 7 is necessary for both buffers to keep the analytes in their ionized form (i.e., protonated). In addition, a low pH is desirable for the LC buffer to suppress EOF in the stacking capillary. If the LC buffer pH is too high, the LC buffer moves into the separation capillary by EOF during the stacking process and deteriorates the separation. On the other hand, if the pH of the CE buffer is too low, the EOF in the separation capillary will be very slow and lead to poor separation efficiency as shown in Figure 4A. The optimal situation was to maintain the LC buffer at low pH (pH 2.5) and the CE buffer at higher pH (pH 4.75). These conditions provided both good stacking and separation efficiency (Figure 4B).
Figure 4.
Effect of the pH of the LC and CE buffers on the CE separation efficiency: (A) 10 mM lithium citrate, pH 2.5/acetonitrile (70:30 v/v) for the LC buffer and the CE buffer; (B) 10 mM lithium citrate, pH 2.5/acetonitrile (70:30 v/v) for the LC buffer and 100 mM lithium acetate, pH 4.75, for the CE buffer; sample, 10 μM bupivacaine and primaquine in Ringer’s solution; stacking conditions, 22 kV for 80 s; separation conditions, 18 kV. Peak identities as in Figure 3.
Effect of the Buffer Ionic Strengths
The larger the difference in conductivity between the separation capillary and the stacking capillary, the higher the stacking efficiency. There are two methods to change the conductivity. One is to change the ionic strength of the buffers. The other is to change the internal diameter of the capillaries. Ideally, the separation capillaries, C2 and C4, would have a larger inner diameter than the stacking capillary, C3, to provide a higher resistance in the stacking capillary. As described above, a 150-μm-i.d. capillary was optimal for the stacking capillary. Smaller inner diameter capillaries would provide better stacking conditions but have a much smaller sample capacity. Capillaries of 75-μm-i.d. were employed for the separation capillaries in this work as a larger inner diameter resulted in Joule heating and bubble formation. Therefore, only the ionic strengths of the LC and CE buffers could be manipulated to produce field amplification in the stacking capillary. Burgi and Chien8 indicated that at least a 10-fold difference in ionic strength is needed for efficient field amplification. Figure 5 shows the effect of using different ionic strength CE and LC buffers. The best result was achieved when the concentration of the CE buffer was 150 mM and that of the LC buffer was 7.5 mM. Increasing the concentration of the CE buffer further will cause Joule heating and bubble formation, whereas a further decrease in the concentration of the LC buffer provided too a low buffer capacity.
Figure 5.
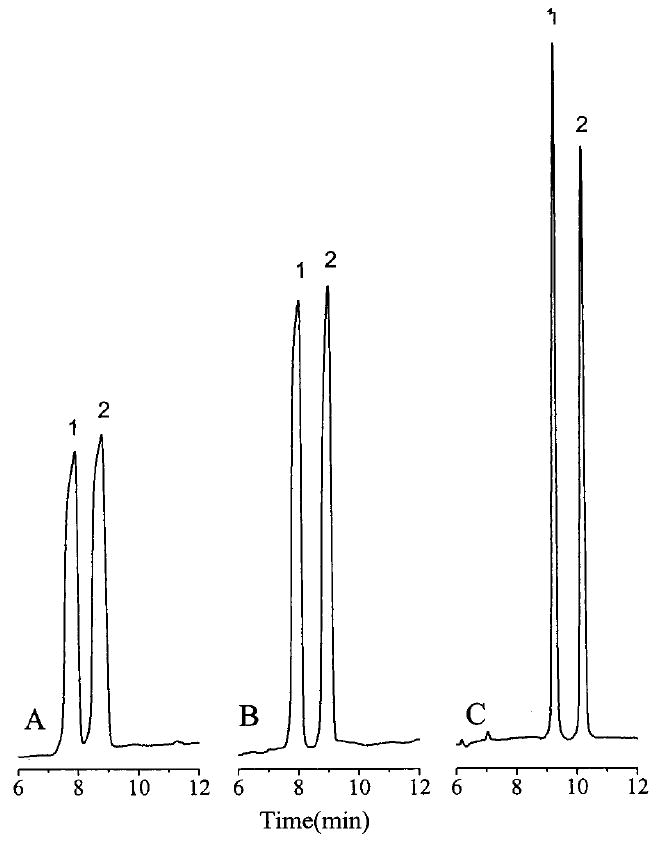
Effect of the ionic strength of the LC and CE buffers on the CE separation efficiency: (A) 10 mM LC buffer and 100 mM CE buffer; (B) 10 mM LC buffer and 150 mM CE buffer; (C) 7.5 mM LC buffer and 150 mM CE buffer, LC buffer, lithium citrate, pH 2.5/acetonitrile (70:30 v/v); CE buffer, lithium acetate, pH 4.75. Other conditions as in Figure 4.
Effect of the Stacking Time
During the field amplification stacking process, the charged analytes migrate rapidly through the low ionic strength zone (LC buffer) to the high ionic strength zone (CE buffer) because of the large electric field across the low ionic strength zone. On arrival at the interface between the two zones, they effectively stop because they now migrate very slowly in the high ionic strength zone with its weak electric field. However, it is important to note that the analytes do not completely stop but rather still migrate slowly into the separation capillary, C2. Therefore, when a long stacking time is used, this slow migration process leads to broadening of the stacked analyte zone. Too long of a stacking time consequently leads to lower efficiency and poor resolution. In addition, too long of a stacking time will lead to LC buffer transfer into the separation capillary and a resulting deterioration in the CE separation. This is shown in Figure 6A. On the other hand, too short of a stacking time will result in incomplete stacking. This leads to lower sensitivity (Figure 6C). A stacking time of 80 s was selected to provide the maximum sensitivity without significant loss in separation efficiency (Figure 6B).
Figure 6.
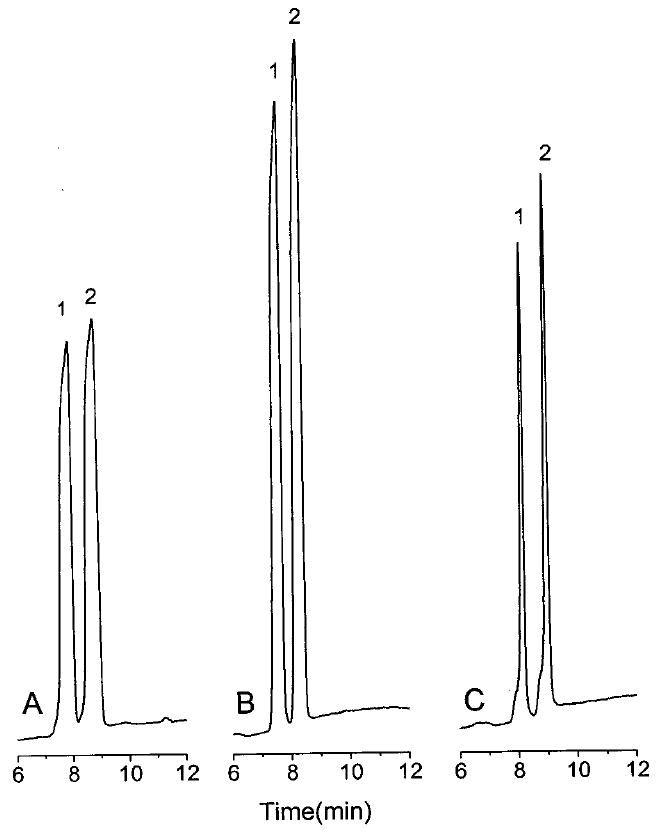
Effect of the stacking time on the CE separation efficiency: stacking time, (A) 90, (B) 80, and (C) 70 s; LC buffer, 10 mM lithium formate, pH 2.5/acetonitrile (70:30 v/v); CE buffer, 150 mM lithium acetate, pH 4.75. Other conditions and peaks identities as in Figure 4.
Optimized Conditions
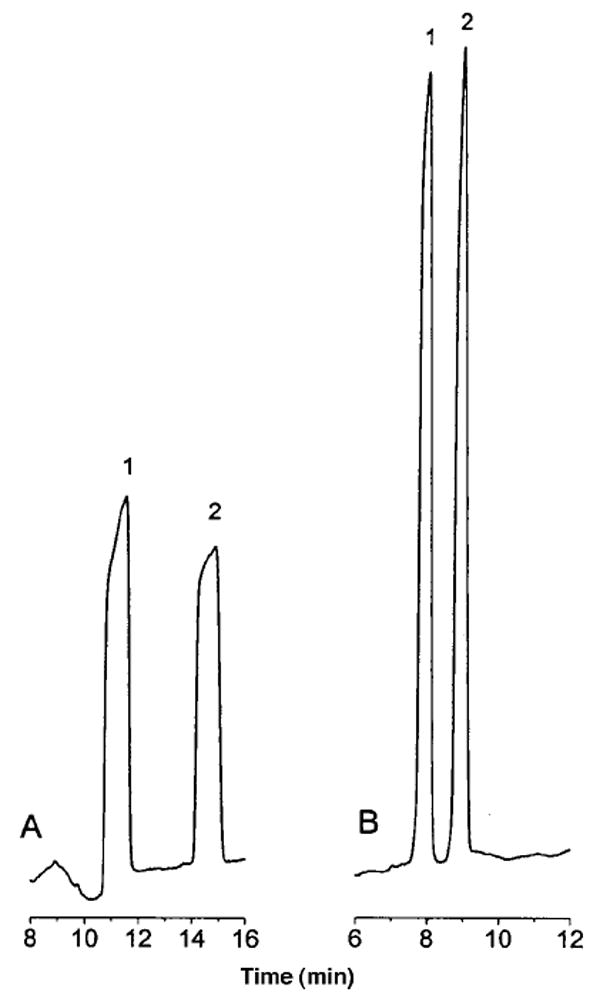
The optimized matrix switching, stacking, and separation conditions for the model compounds consisted of an LC mobile phase of 7.5 mM lithium citrate, pH 2.5/acetonitrile (70:30 v/v), a CE buffer of 150 mM lithium acetate, pH 4.75, a 60-cm-long (75-μm-i.d.) separation capillary with 40 cm to the detection window, and a 40-cm-long (150-μm-i.d.) stacking capillary. A flow rate of 30 μL/min was used for the LC elution process, and a 10-s flush of the CE buffer from reservoir R4 was used to remove the mobile phase after stacking. A stacking voltage of 22 kV was applied for 80 s. The separation voltage was 18 kV. A typical electropherogram obtained using these conditions is shown in Figure 7C. The sensitivity was increased more than 500-fold without loss in resolution compared to a normal hydrodynamic injection (Figure 7A).
Figure 7.
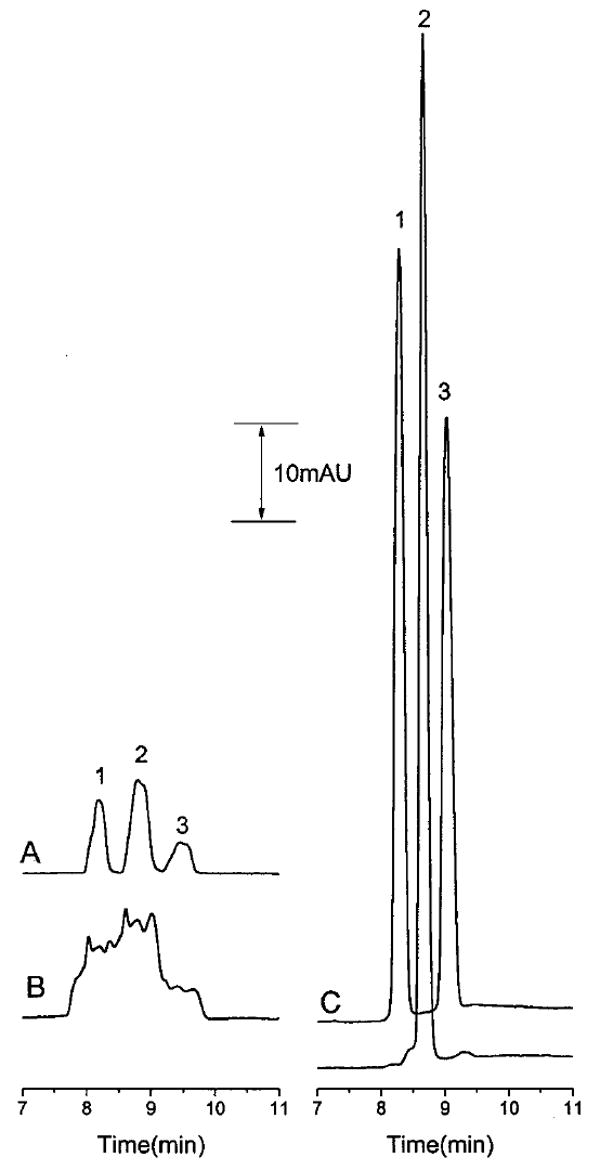
Comparison of hydrodynamic injection and the matrix-switching LC–CE method using two sample injections for multiple peaks: (A) hydrodynamic injection of 11-nL sample 100 μM in each analyte with CE separation; (B) hydrodynamic injection of 33-nL sample 100 μM in each analyte with CE separation; (C) injection of a 20-μL sample 5 μM in each analyte using matrix-switching LC–CE. Conditions for (A) and (B): running buffer, 100 mM lithium acetate, pH 4.75; separation voltage, 18 kV. Conditions for (C): LC buffer, 7.5 mM lithium citrate, pH 2.5/acetronitrile (70:30 v/v); CE buffer, 150 mM lithium acetate, pH 4.75; 80 s stacking time at 22 kV; separation voltage, 18 kV. Peak identities as in Figure 3.
The entire analysis time was less than 15 min with a detection limit of less than 10 nM (S/N) = 3) for all of the compounds. For the entire procedure, the reproducibility of migration time was less than 6% RSD; however, the reproducibility of the peak height was more than 10% RSD. The major source of variation at this time is that the procedure is performed manually. In particular, the timing of trapping the LC peak in the stacking capillary was determined by visual observation of the detector output and manually switching valve S1. Automating this system should lead to significant improvements in peak height reproducibility.
LC–CE Mode
In the system described, ideally, all of the analytes would elute from the LC column as a single peak. Unfortunately, this is not the case when the hydrophobicity of the analytes varies widely (Figure 3). To determine all of the analytes in this situation, two approaches can be taken. In the first approach, two injections of the sample are required. On the first injection, the first LC peak is trapped in the stacking column and subsequently analyzed by CE. The LC system is flushed, and a second injection of the same sample is made. For this second injection, the second LC peak is trapped and then analyzed by CE (Figure 7C). While this is not difficult for the simple sample used in this work, in general, the number of sample injections must be the same as the number of LC peaks. This could be a significant problem for complex samples with several analytes varying widely in hydrophobicity.
The second approach is to make a single injection into the LC system. The first LC peak is eluted and analyzed by CE while the rest of the analytes remain in the LC column. After CE analysis of the first peak, the LC mobile phase flow is restarted through the LC system and the second LC peak is eluted and subsequently analyzed by CE. This approach requires that the later eluting peaks from the LC remain in the LC column while the earlier peaks are analyzed by CE. Figure 8 shows the affect on the peak shape of the second LC peak of stopping the LC mobile phase for various periods of time. In this experiment, the mobile phase was stopped when the first peak had just passed the detector on capillary C3. Mobile-phase flow through the LC system was stopped by redirecting the flow by means of switching valve S1. After a period of time, the mobile phase was again directed through the LC column to elute the second peak. Holding the second peak in the LC column for 15-min resulted in some band broadening but not enough to significantly decrease the amount of sample captured in the stacking capillary C3. However, when the delay was 55 min or longer, less than 25% of the second peak could be retained in the stacking capillary because of significant band broadening in the LC column during the delay time. A 15 min delay is sufficient for the stacking and separation of the first LC peak (primaquine and bupivacaine). The second LC peak (doxepin) was then eluted into the stacking capillary C3 and the stacking and separation of the second LC peak carried out. Compared to the two injection method, the sensitivity of doxepin was reduced approximately 25% due to band broadening. However, this strategy gave an acceptable result as shown in Figure 8.
Figure 8.
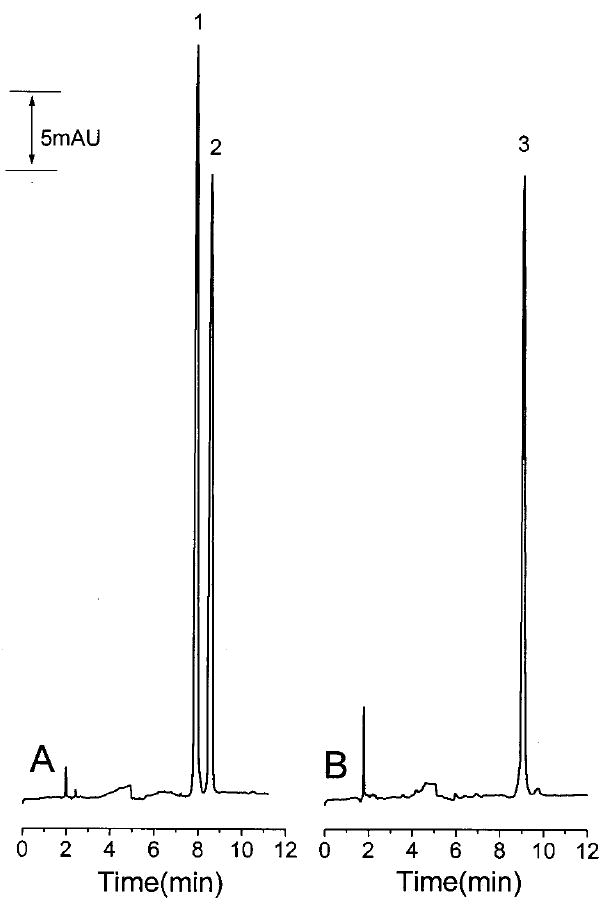
Electropherograms obtained using the single sample injection procedure. Conditions and peaks identities as in Figure 7 except that the LC flow was stopped after elution of the first LC peak and subsequently restarted after the CE separation shown in (A); the second LC peak was then trapped, stacked, and separated by CE for (B).
Application of the LC–CE Method to Microdialysis Samples
This method was used to analyze microdialysis sample. A 1.0-cm linear probe with a Cuprophan membrane was implanted into a hind leg muscle of a rat and 1-mL solution of bupivacaine (5 mg/mL) was injected im at a point approximately 0.5 cm from the probe. The probe was perfused with Ringer’s solution at 1 μL/min. The samples were diluted 5-fold with Ringer’s solution and injected directly into the system. Figure 9 shows the electropheromgram of a typical dialysate sample collected after administration of bupivacaine.
Figure 9.
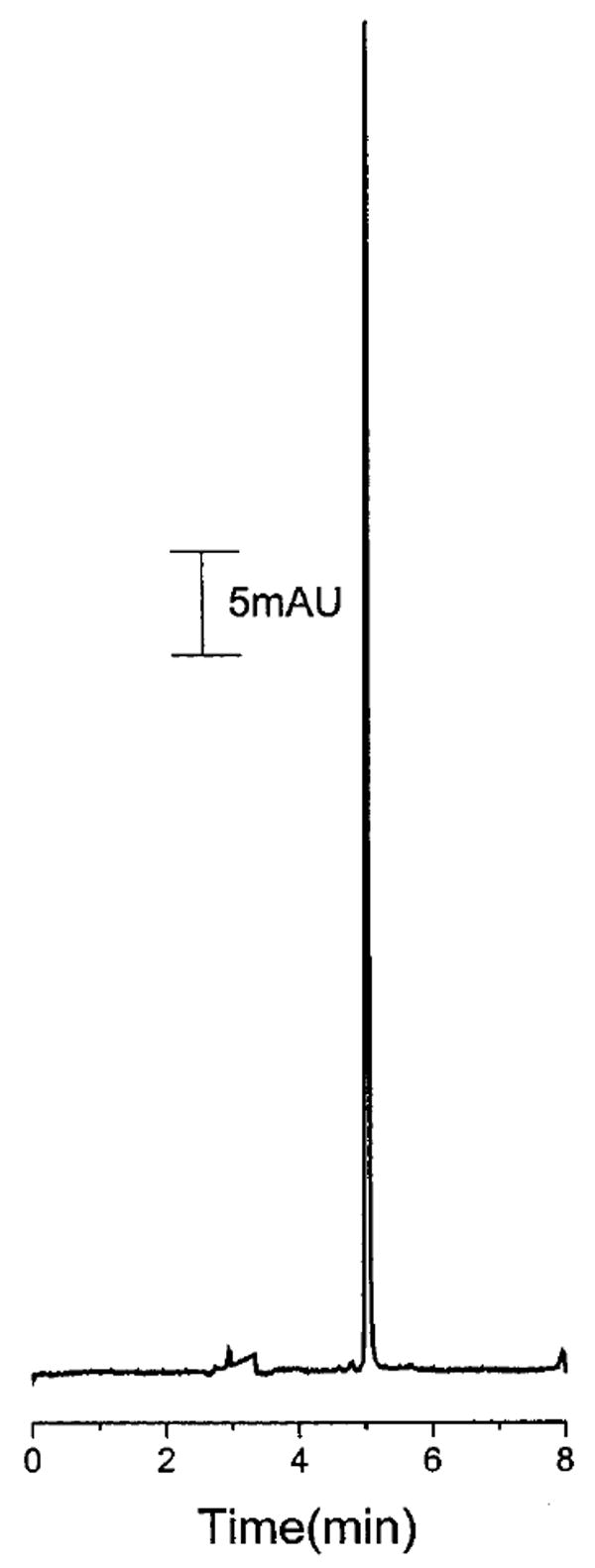
Electropherogram using matrix-switching LC–CE of a dialysis sample obtained after administration of bupivacaine im bolus. Experimental conditions as in Figure 7.
CONCLUSION
A LC–CE on-line sample concentration method with sample matrix-switching and stacking procedures has been developed. In this system, the LC is not used to separate the analytes but rather to switch the sample matrix from a high ionic strength solution to a low ionic strength solution. After this matrix switching, field amplification peak stacking is used to concentrate the sample prior to electrophoresis. Compared to a normal hydrodynamic injection, the sensitivity was increased by more than 500-fold without loss in resolution. While ideally the analytes elute from the chromatographic column as a single peak, multiple chromatographic peaks can be accommodated. Peaks can be sequentially eluted from the LC column, stacked, and analyzed by CE. This provides the capability of a two-dimensional separation for even higher resolution and peak capacity.
Acknowledgments
This work was supported by the National Institutes of Health (Grant GM44900-07).
References
- 1.Chien RL, Burgi DS. J Chromatogr. 1991;559:141–152. [Google Scholar]
- 2.Chien RL, Burgi DS. J Chromatogr. 1991;559:153–161. [Google Scholar]
- 3.Chien RL, Burgi DS. Anal Chem. 1992;64:1046–1050. [Google Scholar]
- 4.Liu Z, Sam P, Sirimanne SR, Mcclure PC, Grainger J, Patterson DG., Jr J Chromatogr. 1994;673:125–132. doi: 10.1016/0021-9673(94)87065-9. [DOI] [PubMed] [Google Scholar]
- 5.Chien RL, Burgi DS. Anal Chem. 1992;64:489A–496A. [Google Scholar]
- 6.Zhang C-X, Thormann W. Anal Chem. 1996;68:2523–2532. doi: 10.1021/ac951250e. [DOI] [PubMed] [Google Scholar]
- 7.Zhang C-X, Thormann W. Anal Chem. 1998;70:540–548. doi: 10.1021/ac9707085. [DOI] [PubMed] [Google Scholar]
- 8.Burgi DS, Chien RL. Anal Biochem. 1992;202:306–309. doi: 10.1016/0003-2697(92)90110-s. [DOI] [PubMed] [Google Scholar]
- 9.Albert M, Debusschere L, Demesmay C, Rocca JL. J Chromatogr A. 1997;757:281–289. doi: 10.1016/s0021-9673(97)00413-5. [DOI] [PubMed] [Google Scholar]
- 10.McGrath G, Smyth WF. J Chromatogr B. 1996;681:125–131. doi: 10.1016/0378-4347(95)00486-6. [DOI] [PubMed] [Google Scholar]
- 11.Weinberger R. Practical Capillary Electrophoresis. Academic Press; San Diego, CA: 1993. p. 215. [Google Scholar]
- 12.Demarest CW, Monnot-Chase EA, Jiu J, Weinberger R. In: Capillary Electrophoresis, Theory & Practice. Grossman PD, Colburn JC, editors. Academic Press, Inc.; San Diego, CA: 1992. p. 320. [Google Scholar]
- 13.Wang C-C, McCann WP, Beale SC. J Chromatogr B. 1996;676:19–28. doi: 10.1016/0378-4347(95)00416-5. [DOI] [PubMed] [Google Scholar]
- 14.Foret F, Szoko E, Karger BL. J Chromatogr. 1992;608:3–12. [Google Scholar]
- 15.Cai J, Rassi ZE. J Liq Chromatogr. 1992;15:1179–1192. [Google Scholar]
- 16.Swartz ME, Merion M. J Chromatogr. 1993;632:209–213. [Google Scholar]
- 17.Morita I, Sawada J. J Chromatogr. 1993;641:375–381. [Google Scholar]
- 18.Hoyt AM, Jr, Beale SC, Larmann JP, Jr, Jorgenson JW. J Microcolumn Sep. 1993;641:375–388. [Google Scholar]
- 19.Benson LM, Tomlinson AJ, Naylor S. J High Resolut Chromatogr. 1994;17:671–682. [Google Scholar]
- 20.Tomlinson AJ, Braddock WD, Benson LM, Oda RP, Naylor S. J Chromatogr B. 1995;669:67–78. doi: 10.1016/0378-4347(95)00127-5. [DOI] [PubMed] [Google Scholar]
- 21.Beattie JH, Self R, Richards MP. Electrophoresis. 1995;16:322–328. doi: 10.1002/elps.1150160153. [DOI] [PubMed] [Google Scholar]
- 22.Strausbauch MA, Madden BJ, Wettstein PJ, Landers JP. Electrophoresis. 1995;16:541–548. doi: 10.1002/elps.1150160189. [DOI] [PubMed] [Google Scholar]
- 23.Strausbauch MA, Landers JP, Wettestein PJ. Anal Chem. 1996;68:306–314. doi: 10.1021/ac9503217. [DOI] [PubMed] [Google Scholar]
- 24.Tomlinson AJ, Benson LM, Oda RP, Braddock WD, Riggs BL, Katzmann LA, Naylor S. J Capillary Electrophor. 1995;2:97–110. [Google Scholar]
- 25.Tomlinson AJ, Benson LM, Braddock WD, Oda RP, Naylor S. J High Resolut Chromatogr. 1995;18:381–392. [Google Scholar]
- 26.Pálmarsdóttir S, Thordarson E, Edholm L-E, Jönsson JÅ, Mathiasson L. Anal Chem. 1997;69:1732–1737. doi: 10.1021/ac960668p. [DOI] [PubMed] [Google Scholar]
- 27.Tomlinson AJ, Benson LM, Guzman NA, Naylor S. J Chromatogr A. 1996;744:3–15. [Google Scholar]
- 28.St Claire RL., III Anal Chem. 1996;68:569R–586R. [Google Scholar]