

Abstract
We report a new method called Metal Affinity Capture that when coupled with tandem mass spectromery (MAC-MSMS) allows for the selective detection and identification of phosphopeptides in complex mixtures. Phosphopeptides are captured as ternary complexes with GaIII or FeIII and Nα,Nα-bis-(carboxymethyl)lysine (LysNTA) in solution, and electrosprayed as doubly or triply charged positive ions. The gas-phase complexes uniformly dissociate to produce a common (LysNTA + H)+ ion that is used as a specific marker in precursor ion scans. The advantages of MAC-MSMS over the current methods of phosphopeptide detection are as follows. (1) MAC-MSMS uses metal complexes that self-assemble in solution at pH < 5, which is favorable for the production of positive ions by electrospray. (2) Phosphorylation at tyrosine, serine, and threonine is detected by MAC-MSMS. (3) The phosphopeptide peaks in the mass spectra are encoded with the 69Ga-71Ga isotope pattern for selective recognition in mixtures. Detection by MAC-MSMS of singly and multiply phosphorylated peptides in tryptic digests is demonstrated at low nanomolar protein concentrations.
Introduction
Phosphorylation is one of the most important post-translational modifications of proteins1,2 that plays a role in several cellular biochemical pathways, in particular in signaling.3 Thus, quantifying phosphorylated proteins and detecting phosphorylation sites on hydroxylated amino acids (Ser, Thr, and Tyr) has been of considerable interest in proteomic applications of mass spectrometry. Several methods have been developed to selectively analyze the phosphoprotein content in complex biological samples. A common feature of the current methods is that the protein in the sample is first purified by electrophoretic or reverse-phase chromatographic separation. The next step typically involves digestion with a cleavage enzyme, such as trypsin, although some methods include a phosphoprotein enrichment step prior to digestion. Phosphopeptide analysis relies on separation by liquid chromatography followed by tandem mass spectrometry (LC-MSn). This involves a neutral loss scan which identifies peptides that on collisional activation eliminate H3PO4 (98 Da) or HPO3 (80 Da) from phosphorylated Ser and Thr residues.4 Alternatively, a parent ion scan can be employed to search for precursors of the common product ions H2PO4− or PO3− in negative ionization mode.5,6 Infrared multiphoton dissociation of protonated phosphopeptides was found to show selectivity to the phosphate groups and can be potentially used in conjunction with LC separation of peptide mixtures.7
Many phosphate-specific enrichment methods have been developed to aid in the quantitation of relative phosphorylation levels. Some reported phosphopeptide enrichment techniques are variations of the Isotope-Coded Affinity Tags metod (ICAT)8 and involve base-induced phosphate elimination, followed by Michael addition of a biotin or mercaptoethanol handle to the dephosphorylated Ser or Thr residue. The chemically modified peptides can be separated by a specific capture/elution on bound streptavidin or by the use of sulfur specific antibodies.9-12 Since phosphotyrosine residues cannot undergo β-elimination of the phosphate moiety a modified approach, developed by Zhou et al,10 uses ethyl carbodiimide to catalyze the addition of cysteamine to the phosphorylated residue allowing for purification over glass beads. Disadvantages of these techniques stem from the excessive sample handling steps resulting in lost sample, thus limiting sensitivity.
Alternatively, immobilized metal-ion affinity chromatography (IMAC)13,14 can also be used to enrich for phosphopeptides without the inherent need for antibody based resins or sample derivatization. With this method the negatively charged phosphates are captured by a metal cation, such as FeIII or GaIII, that is immobilized on the surface of an agarose bead which is derivatized with a chelating agent such as nitrilotriacetic acid (NTA) or iminodiacetic acid (IDA).13,15 After the flow through portion is washed away, the captured phosphopeptides can be eluted with a phosphate-containing buffer, typically PBS. Since the immobilized metal ion can also capture the negatively charged carboxylates of both the peptide C-terminus and Asp and Glu side chains, methyl esterification of these carboxylate groups is often employed to ensure a more selective enrichment.16 An inherent limitation of the IMAC method is non-specific binding of hydrophobic peptides to the gel beads. The interactions leading to this capture are difficult to alleviate and are actually enhanced by methyl esterification of carboxylic groups. Thus, ongoing efforts exist to develop a more specific and comprehensive phosphopeptide analysis. For example, TiO2 and ZrO2 columns have been recently used for phosphopeptide enrichment.17 Many such efforts involve combining two or more enrichment steps to ensure selective isolation of phosphopeptides.7,18
In this study we introduce a new approach to specific phosphopeptide detection that relies on the metal coordination properties of the phosphate group, but is independent of the amino acid residue that is phosphorylated and does not require LC separation of the peptide mixture. We call this method Metal Affinity Capture Tandem Mass Spectrometry, or MAC-MSMS for short. We present a study of the complexation of phosphopeptides with NTA-containing ligands via trivalent metal cations, FeIII and GaIII, in solution with the goal of determining their utility as an analytical tool in phosphopeptide analysis. The NTA-like lysine derivative, Nα,Nα-dicarboxymethyl lysine, was used as a metal chelating agent. NTA binds strongly to four of the six FeIII or GaIII coordination sites leaving two free sites for phosphopeptide binding. Upon ESI-MSn these ternary complexes show specific fragmentations that are promising for MAC-MSMS to be developed into a useful tool in phosphopeptide analysis.
Experimental
Methods
Electrospray ionization tandem mass spectra were acquired on either an API III Perkin-Elmer Sciex triple quadrupole, or an Applied Biosystems API 4000 Sciex triple quadrupole. In addition, a Bruker Esquire ion trap mass spectrometer was used for the initial characterization of the complexes. All these instruments were operated in positive ion mode. All peptides and peptide complexes were sampled as solutions in 1:1 methanol/water acidified with acetic acid to pH 3−5. Direct infusion via a syringe pump was used at flow rates of 1−10 μL/min.
Optimum electrospray ionization was achieved at a capillary voltage of 4000 V and a nitrogen drying gas flow rate of 5−6 mL/min. Measurements on the Bruker Esquire ion trap instrument were performed with a 30 V skimmer potential and a 70 V capillary exit offset. Optimal trap drive settings were sample dependent falling within the range of 55 to 65 of the instrument units. MSMS measurements on the API 4000 triple quadrupole instrument were performed at collision gas setting of 5 psi and collision energy of 30−40 eV (for singly charged ions). The nebulizer gas (GS1) and curtain gas (CUR) settings were 20 and 10 psi, respectively. A 50 V declustering potential (DP), 10 V collision cell entrance potential (EP), and 15 V collision cell exit potential (CXP) were found to provide optimum phosphopeptide detection. MSn spectra on the ion trap were obtained for confirmation and initial characterization of phosphopeptide complexes. MSMS data were acquired on a tandem quadrupole instrument in Q1 scan, neutral loss scan, or parent ion scan mode.
Materials
Nα,Nα-dicarboxymethyl lysine was synthesized by the method of Schmitt et al.19 Briefly, Nε-carboxybenzyl lysine (H-Lys(Z)-OH, Fluka), 4 mmol, was dissolved in 5 mL of 2 M NaOH, and the solution was added dropwise with stirring at 0°C to bromoacetic acid (Fluka, 8 mmol) dissolved in 5 mL of 2 M NaOH. The reaction was stirred for 2 h at which point the cooling bath was removed, and the reaction was stirred at room temperature overnight. Following heating to 50°C for 2 h, 20 mL of 1 M HCl was added and the product crystallized after 4 h of cooling in ice. The solid precipitate was redissolved in 7 mL of 1 M NaOH, and recrystallized from the same volume of 1 M HCl. The product Nα,Nα–Bis[carboxymethyl]–Nε–[benzyloxycarbonyl]-L-lysine (Nε-Z-NTA) was confirmed by melting point (170−175°C)15 and ESI-MS (MH+ m/z 397).
The Nε-protected dicarboxymethyl lysine was dissolved in 10:1 CH3OH/H2O and hydrogenated over a Pd-on-charcoal catalyst at room temperature and 1 atm of H2 for 2 h. Hydrogenation was monitored by TLC in 4:1 CH3CN/H2O. The Pd catalyst was filtered off, and the solvent was removed under vacuum. The nitrilotriacetic acid product (LysNTA) was purified by recrystallization from 10:1 C2H5OH:H2O and characterized by electrospray mass spectrometry. Collision-induced dissociation (CID) of (LysNTA + H)+ precursor ions at m/z 263 leads to common losses of NH3, (CO + H2O), and (CO + 2H2O) giving rise to fragments at m/z 246, 217, and 199, respectively (Figure S1, Supporting Information). Furthermore, the spectrum showed a major fragment ion at m/z 130 that corresponds to an elimination of iminodiacetic acid by a cleavage of the N—Cα bond leaving (Lys + H − NH3)+, which is also the major fragment observed in the CID spectrum of protonated lysine.
LysNTA metal-ion complexes were formed with FeCl3 or GaCl3 (Aldrich). The formation constants for LysNTA-GaIII and LysNTA-FeIII complexes of 1:1 ligand-metal ion stoichiometry are on the order of 1013 and 1015 mol−1, respectively.20 Thus, a large excess of metal is not required to drive the complex formation to completion. A 2:1 ratio of metal to LysNTA was sufficient to facilitate complete complex formation, as determined by the absence of the free (LysNTA + H)+ ion (m/z 263) in the ESI-MS spectrum of metal-LysNTA solutions (Figure S2, Supporting Information).
The peptides used to characterize the ternary LysNTA-metal-peptide complexes include NQLLpTPLR, QLLpTPLR, MpSGIFR, MSGIFR, and pYWQAFR that were purchased from Genscript Corporation, Piscataway, NJ. Peptides from the tryptic digestion of αS1-casein were also used to characterize the formation of ternary complexes with LysNTA and metal cations. Sequence grade trypsin and bovine α-casein (>70% of the S1 variant) were purchased from Aldrich. Tryptic digestion of α-casein was performed at a 1:100 enzyme to substrate weight ratio in 20 mM NH4HCO3 and incubated overnight at 37° C. Peptide aliquots were purified by C-18 ziptip (Millipore, Eugene, OR) in a process involving a 10% HOAc binding and washing solution and 5 μL of 20% AcCN elution solution. The flow-through portions from this purification were also examined as these were found to contain significant amounts of tryptic phosphopeptides.
Results and Discussion
Phosphoester Binding in Solution
Mass spectra of the LysNTA-metal ion ternary complexes were initially acquired using phosphorylated serine, threonine, and tyrosine. In order to quantitatively characterize phosphoester binding to LysNTA−GaIII in solution, formation constants were evaluated for the LysNTA−GaIII−pSer ternary complex by monitoring the intensity of the precursor ion at m/z 514 (the 69Ga isotope) as a function of pSer concentration at constant LysNTA−GaIII concentrations. The ternary formation constants are represented as β values20 which define the equilibrium constant for the formation of the LysNTA−GaIII−pSer complex from free LysNTA, GaIII and pSer (eq 1).
| (1) |
The equilibrium concentration of phosphoserine is equivalent to the initial concentration of pSer minus the equilibrium concentration of the ternary complex. Furthermore, if excess LysNTA and GaIII are used then we can assume [LysNTA] ≅ [LysNTA]0 and [GaIII] ≅ [GaIII]0 and the expression can be rewritten as (eq 2):
| (2) |
To determine the equilibrium concentration of the complex precursor we assume that the ion intensity for the ternary complex, Im/z 514, is proportional to its concentration scaled by a response factor, αm/z 514.21 Substituting in eq 2 and rearranging it gives an expression for Im/z 514 (eq 3) that shows that Im/z 514 depends linearly on [pSer]0.
| (3) |
The slope, m (eq 4), still contains two unknowns, β and αm/z 514, the latter of which is eliminated by constructing the linear regression at two values of [LysNTA]0[GaIII]0.
| (4) |
From the m1 and m2 slopes thus obtained, and assuming that α1 = α2, β can be expressed by eq 5:
| (5) |
The values of β were measured at several solution pH, as summarized in Figure S3 (Supporting Information). The data suggest that, in accordance with previous literature14,22 the ternary complex with pSer is most stable at pH < 5, where log β ≤ 8.9. The pH dependence can be explained by the phosphoserine becoming less able to bind the metal center as the concentration of OH− competitor ions increases. The acidic environment is necessary to provide a bare, LysNTA-bound metal ion for phosphate binding. A beneficial corollary of this result is that the optimal pH for complex formation is also a desirable one for ESI-MS detection in positive ion mode. Thus, detection of these complexes by ESI-MS was performed in solutions of 1:1 methanol/water acidified to pH 3−4 with acetic acid. The values of β ≤ 109 mol−2 were further used to estimate the concentrations of LysNTA (5−100 μM) and GaIII (10−200 μM) for the efficient formation of phoshopeptide ternary complexes.
MSMS of Gas-Phase Complexes
Electrospraying the LysNTA−GaIII−pSer complex produces mainly singly charged gas-phase cations at m/z 514. The m/z value indicates that the ligands are overall doubly deprotonated, but the deprotonation positions (any of the four COOH groups or the phosphate) are unknown. Therefore, we denote the ligands generically as LysNTA and pSer without specifying their deprotonation in the complexes. The CID spectrum of the m/z 514 ion is shown in Figure 1. The fragment ions all retain the LysNTA−GaIII moiety and correspond to losses of CH2=C(NH2)COOH from the pSer moiety (m/z 427), serine (m/z 409), and phosphoserine (m/z 329). In contrast, CID of the LysNTA−GaIII complexes with phosphopeptides lead to a different fragmentation forming galliated phosphopeptide ions and a common (LysNTA + H)+ ion (m/z 263) as major products. Figure 2 illustrates the fragmentations of LysNTA−GaIII and LysNTA−FeIII complexes with the synthetic phosphopeptide MpSGIFR. Complexes of NQLLpTPLR and pYWQAFR gave quite analogous CID spectra that are given as Supporting Information (Figures S4 and S5, respectively). Major fragments result from cleavage at the LysNTA−metal bond resulting in the (LysNTA + H)+ ion and a metallated peptide ion whose charge is equal to the precursor ion charge minus one.
Figure 1.
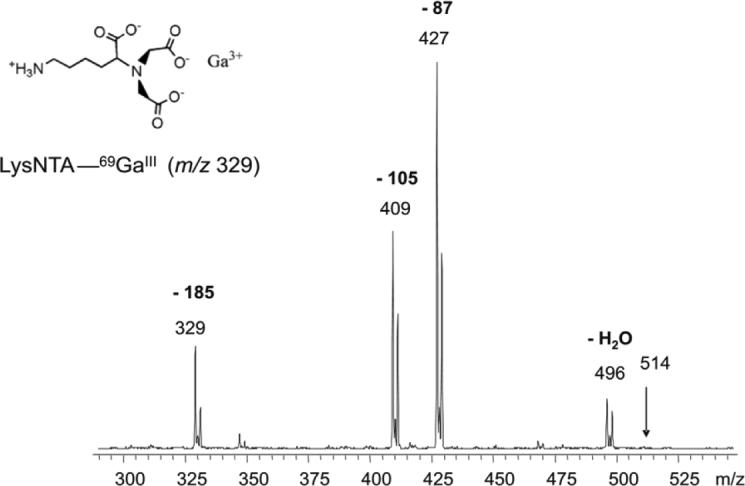
CID mass spectrum of the m/z 514 ion from ESI of LysNTA-GaIII-pSer in 1% HOAc.
Figure 2.

CID mass spectrum of the LysNTA-GaIII (top) and LysNTA-FeIII complexes with MpSGIFR.
Gallium III and iron III were both investigated as metal centers in this study. The MSMS spectra of LysNTA−GaIII complexes with each of the three synthetic phosphopeptides provide significantly more b and y type gallium-free peptide fragment ions than do the spectra of LysNTA−FeIII complexes with the same peptides (Figure 2). The cause of variation in fragmentation with metal ion was not studied in depth, but the observation might be attributed to the relative stabilities of the galliated and ferrated-peptide fragment ions. The observed result is consistent with the relative phosphopeptide-binding characteristics of iron and gallium. Specifically, it has been noted that iron III binds to phosphopeptides more strongly than gallium III.22
One advantage of gallium-containing fragments is that they can be more readily identified than ferrated ones due to the well resolved isotope doublets stemming from the existence of two naturally abundant gallium isotopes of 69 and 71 Da present in a 3:2 ratio. For example, in the CID spectrum of the singly charged, LysNTA−GaIII−pSer ion, the galliated fragments can be identified by the abundant ions at m/z 427, 409 and 329 (Figure 1). Similar assignments can be made for galliated ions from the phosphopeptides in the CID and precursor scan spectra (vide infra).
In another series of experiments, the molar ratio of LysNTA-GaIII to the phosphopeptide was varied from 1000:1 to 30:1, while the peptide concentration was kept at 300 nM. At the lowest LysNTA-GaIII : phosphopeptide ratio we observed about 50% of the peptide as the LysNTA-GaIII ternary complex and the other 50% as protonated ions. Thus, phosphopeptide coordination to LysNTA-GaIII to form ternary complexes efficiently competes with protonation, and the formation of the ternary complex can be forced by increasing the concentration of LysNTA-GaIII.
As mentioned above, the location of protons in the gas-phase phosphopeptide complexes was not directly investigated in this study, but a hypothesis can be made based on correlating the parent and product ion m/z values. The LysNTA−GaIII−peptide complexes, containing each of the three synthetic peptides, acquire a net charge of +2 on electrospray. If we assume that the ε-NH2 group in LysNTA is protonated while the three NTA COOH groups are deprotonated then a net charge of +1 is added by the LysNTA−GaIII component to the phosphopeptide. This leaves the possibility that the phosphopeptide ligand is singly deprotonated at the phosphate and doubly protonated along the peptide backbone, probably at both the N-terminus and the basic side-chain of the C-terminal Arg. Thus, cleavage at the LysNTA−metal bond to eliminate the (LysNTA + H)+ fragment at m/z 263 must be accompanied by the transfer of three protons from the peptide moiety to the LysNTA carboxylates. Alternately, only two protons would necessarily be transferred during fragmentation if the precursor ion were instead deprotonated at only two LysNTA COOH groups and deprotonated twice at the phosphate group of the phosphopeptide. The fact that GaIII always remains coordinated to the phosphopeptide following CID seems to favor structures with doubly deprotonated phosphate groups, because those would provide stronger binding to the metal ion.
Scheme 1 illustrates the likely protonation sites for both the precursor and product ions in the dissociation of the ternary complex (LysNTA−GaIII−MpSGIFR)2+. The product ion resulting from the loss of (LysNTA + H)+ (m/z 273) from the Ga complex corresponds to a galliated peptide with a charge that is one less than the charge of the precursor ion. In the case of the synthetic peptide complexes this means that doubly charged precursors fragment into a singly charged galliated peptide ion. In this case, two mobile protons are transferred from the peptide to the LysNTA carboxylates in a fashion described by Scheme 1. This protonation scheme can also explain why CID of the LysNTA−GaIII−pSer complex (Figure 1) does not produce the (LysNTA + H)+ fragment, because phosphoserine does not contain the required two mobile protons.
Scheme 1.

The mobile proton assumption received support from our study of phosphopeptide methyl esters in which the carboxylic protons were replaced by methyl groups.16 ESI of methyl esters of NQLLpTPLR, QLLpTPLR, MpSGIFR, and pYWQAFR showed formation of ternary complexes with GaIII-LysNTA, although with efficiencies that were 5−10% of those of the corresponding phosphopeptides with free carboxylic groups. However, upon collisional activation, the complexes did not produce the diagnostic (LysNTA + H)+ ion at m/z 273. This indicates that the peptide carboxyl proton plays a role in the dissociation. It appears plausible that the phosphopeptide carboxylate participates in displacing the LysNTA ligand and is coordinated to the metal cation in the GaIII-contaning fragment ions.
As a further characterization of phosphopeptide complexes with LysNTA−GaIII, an αS1-casein tryptic digest was analyzed by ESI-MSMS. A 10 μM solution of this tryptic digest, without added LysNTA or GaCl3, in 1:1 CH3OH/H2O in 1% HOAc, was analyzed on a Bruker Esquire ion-trap ESI-MS. The doubly charged, singly phosphorylated peptide, YKVPQLEIVPNpSAEER (m/z 976.7) was selected for MSMS characterization Figure 3, top). CID of this ion results in loss of H3PO4 giving the major product ion at m/z 927.8. Several fragments of the b and y ion series also appear in the spectra, confirming the identity of the peptide. In contrast, the MSMS spectrum of the triply-charged, LysNTA−GaIII complex with the phosphorylated α-Cas tryptic peptide at m/z 761 (Figure 3, bottom) shows major product ions that include a doubly-charged galliated peptide fragment, m/z 1010, and a singly charged (LysNTA + H)+ ion at m/z 263. A few b and y ions are also produced by CID of this ternary complex, allowing for further confirmation of the peptide identity. As anticipated, the galliated peptide product ion maintains a charge that is one less that the complex precursor ion charge. Thus, the triply charged complex produces a doubly charged galliated product ion.
Figure 3.
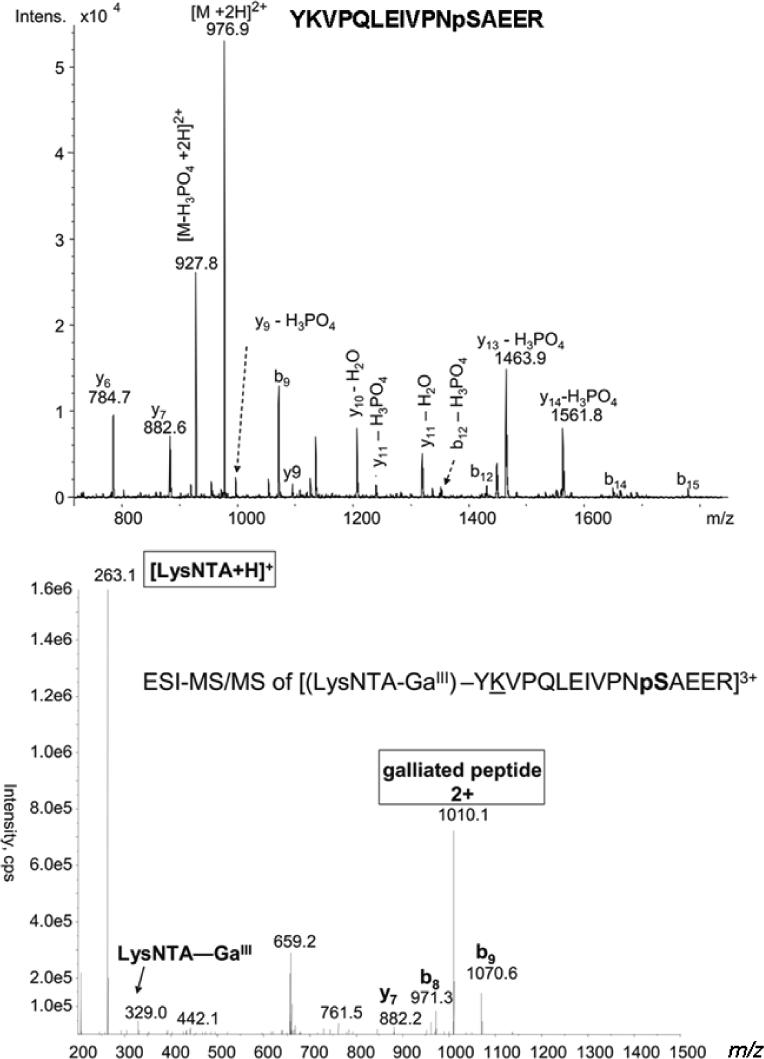
CID mass spectrum of the LysNTA-GaIII ternary complex with α-casein tryptic phosphopeptide YKVPQLEIVPNpSAEER.
Several other LysNTA−GaIII complexes with α-Cas phosphopeptides were characterized by this ESI-MSMS approach. The precursors at m/z 1173.7 (Figure S6) and at m/z 1119.5 (Figure S7) correspond, respectively, to the triply charged complexes of (LysNTA−GaIII)2−EKVNELSKDIGpSEpSTEDQAMEDIK (αS1-Casein residues 50−73) and (LysNTA)2−(GaIII)4−QMEAESIpSpSpSEEIVPNSVEQK (αS1-Casein residues 74−94). Note that each phosphoserine residue coordinates one GaIII ion. When selected for MSMS in the tandem quadrupole instrument, the triply charged precursor ion at m/z 1173.7 dissociates into a doubly-charged galliated peptide, m/z 1629.0, and the (LysNTA + H)+ ion (Figure S6). Although two LysNTA-GaIII are apparently coordinated to the two peptide phosphorylation sites, only one (LysNTA + H)+ is lost through CID. It is possible that the close proximity of the two LysNTA−GaIII bound phosphorylation sites allows for the vacated GaIII to bind the remaining LysNTA forming a stabilized, bridged structure, preventing loss of this second LysNTA in CID. The stability of such a GaIII−LysNTA−GaIII bridge might also be responsible for the observed composition of the complex ion at m/z 1119.5. The peptide in this complex contains four phosphorylated serine residues and a free threonine residue. Since the four pSer residues fall within a five-residue segment of each other, the four bound GaIII ions are likely in close proximity allowing binding to only the observed two LysNTA ligands, which create two bridged structures disallowing further LysNTA binding. Indeed, each of the LysNTA−GaIII peptide complexes detected contains no more than two gallium bound LysNTA moieties, possibly due to the close proximity of these phosphorylation sites in the casein motif. The role of the multiple glutamic acid residues near the phosphorylated segment in preventing further LysNTA binding in the complex is unclear. It cannot be ruled out that the three-dimensional structures of these peptides also allow the Glu side-chain carboxylates to occupy GaIII coordination sites, and further prevent LysNTA binding. Thus, according to these results the bridging of LysNTA species to multiple GaIII creates a stabilized structure which prevents both the loss of multiple LysNTA in CID and the binding of more than two LysNTA to the GaIII bound, multiply phosphorylated casein motif.
Parent-Ion Scanning of Phosphopeptide Complexes
In light of this data, the common product ion, (LysNTA + H)+ at m/z 263 was targeted as a possible phosphopeptide marker from the CID of phosphopeptide complexes with LysNTA−GaIII. The prescribed method involves parent-ion scanning on a triple quadrupole mass spectrometer, whereby precursor ions are selected in a scanning mode by Q1 for CID fragmentation in Q2. Subsequently Q3 selects only fragment ions at or near m/z 263 for detection, thus identifying only precursors that are associated with phosphopeptide complexes. Figure 4 (bottom) illustrates the data collected from the parent ion scan analysis of phosphopeptide GaIII complexes. A mixture of four synthetic peptides, NQLLpTPLR, pYWQAFR, MpSGIFR and MSGIFR, each at 300 nM, was combined with excess LysNTA−GaIII as described above. The three precursors appearing at m/z 559.7, 639.7, 681.2, and their 71Ga satellites correspond to doubly charged ternary complexes of LysNTA−GaIII with each synthetic phosphopeptide. The precursor at m/z 672.8 and its 71Ga satellite correspond to the (LysNTA−GaIII−NQLLpTPLR)2+ complex that eliminated NH3 on CID. Detection of the pYWQAFR peptide is especially promising since pY species cannot be detected by neutral loss scans. Figure 4 (top) shows a neutral loss scan for the 80 Da difference corresponding to the elimination of metaphosphoric acid (HPO3) from singly protonated phosphopeptide precursor ions. While peaks are detected for MpSGIFR (m/z 790.3) and NQLLpTPLR (m/z 1034.4), the peak for pYWQAFR that would be expected at m/z 950 is absent. Apparently, HPO3 is not easily eliminated from the phenylphosphate group of tyrosine. The [LysNTA−GaIII−pYWQAFR]2+ precursor ion is detected with a somewhat lower intensity than those for the phosphoserine and phosphothreonine peptides, perhaps reflecting the decreased ionization efficiency of the more hydrophobic pYWQAFR peptide.
Figure 4.
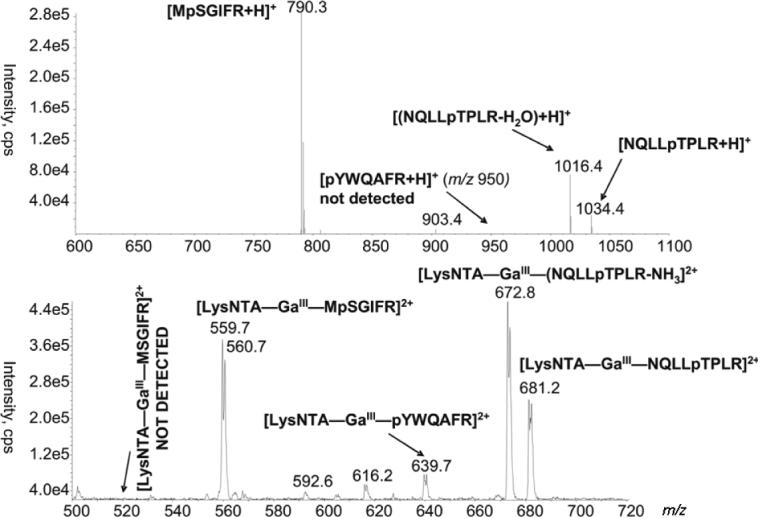
Top spectrum: Neutral loss (80 Da difference) scan of a mixture of singly protonated peptides. Bottom spectrum: Precursor scan for the m/z 263 common fragment ion from NTA-GaIII complexes of the same peptide mixture.
The non-phosphopeptide, MSGIFR, was added to the mixture above to test the selectivity of the parent scanning method. As shown in Figure 4 (bottom), the doubly charged complex with this non-phosphorylated peptide (theoretical m/z 519.7) is not detected while its phosphorylated analogue, MpSGIFR is detected as the 69Ga-71Ga doublet at m/z 559.7−560.7. The selectivity of LysNTA−GaIII binding to phosphopeptides and hydrophilic peptides that contain several acidic Asp or Glu residues was examined with an α-casein tryptic digest, as presented below.
Detection of phosphopeptides from an α-casein (α-Cas) tryptic digest by parent scanning of phosphopeptide complexes was compared to detection of these phosphopeptides by neutral loss scanning on a triple quadrupole mass spectrometer. For direct comparison of these detection techniques, the α-Cas tryptic phosphopeptide, YKVPQLEIVPNpSAEER, residues 119−134, was targeted for analysis by both neutral loss scanning of the peptide digest and parent ion scanning of the peptide complexes with LysNTA−GaIII. The neutral loss scanning method was optimized for losses of orthophosphoric acid, H3PO4, from the triply (32.6 Da), doubly (49 Da) and singly (98 Da) charged precursors. Since the doubly charged precursor at m/z 976.7 of the targeted phosphopeptide is detected with the greatest intensity relative to the other charge states, the neutral loss of 49 Da was targeted for detection of this phosphopeptide at digest concentrations of 400 nM (Figure 5a) and 40 nM (Figure S8).
Figure 5.
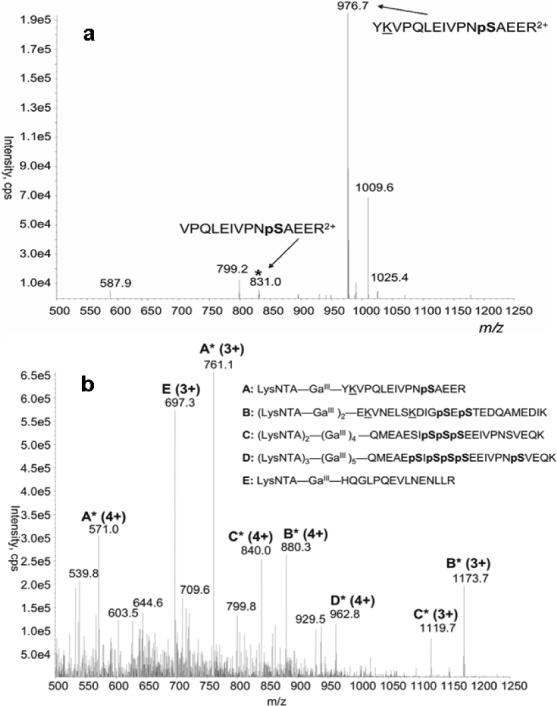
Top spectrum: (a) Neutral scan (49 Da difference) of a phosphopeptide mixture from trypsinolysis of α-casein at 400 nM. Bottom spectrum: (b) Precursor scan or the m/z 263 common fragment ion from LysNTA-GaIII complexes of the same peptide mixture.
For comparison, solutions consisting of 100 μM NTA, 200 μM GaCl3 and either 400 nM (Figure 5b) or 40 nM (Figure S9) of an α-Cas tryptic digest were analyzed by parent ion scanning for the common product ion at m/z 263. Both the neutral loss scan and parent ion scanning methods were able to detect the targeted phosphopeptide, YKVPQLEIVPNpSAEER, at these digest concentrations. However, the neutral loss scan was unable to detect several multiply phosphorylated peptides that the parent ion scan identifies. This is a common drawback of neutral loss scanning, since multiply phosphorylated peptides maintain a larger number of negatively charged components and their detection by ESI-MS in positive ion mode is suppressed. Furthermore, this data illustrates an inherent advantage of the parent ion scan over the neutral loss scan, that is the ability to detect multiple charge states of the same peptide in a single experiment. For example, both the triply charged, m/z 761, and quadruply charged, m/z 571, precursors of the targeted phosphopeptide complex, LysNTA−GaIII−YKVPQLEIVPNpSAEER, are detected by parent ion scanning. Thus, the precursor ion scan is able to detect a greater number of phosphorylated species than scanning for any single neutral loss, 32.5, 49, or 98 Da. While the advantages of precursor ion scanning over neutral loss scanning in detecting multiply phosphorylated species have not been investigated, two hypotheses might be offered. First, since complexation of the negatively charged phosphate moiety to the LysNTA−GaIII seems to result in a net gain of one positive charge to the peptide, ionization of these complex precursors in positive-ion mode might be enhanced relative to the free phosphopeptides. This enhancement is similar to that achieved with copper ternary complexes for the detection of amino acids and other coordinating analytes.23,24 Secondly, since the product ion m/z does not depend on the precursor charge state, we can identify phosphopeptides in several charge states with a single scanning acquisition. This is not possible with neutral loss scanning since elimination of phosphoric acid will result in different apparent neutral losses depending on the precursor ion charge. Thus, precursor ion scan of LysNTA-GaIII complexes has the definite advantage of being a universal, charge independent detection method for phosphopeptides in positive ion mode.
Figure 5b shows additional peaks to those of the phosphopeptide complexes. In particular, the acidic tryptic peptide HQGLPQEVLNENLLR that has three COOH groups forms a triply charged ternary complex with GaIII-LysNTA (ion E) that fragments to produce the (LysNTA + H)+ ion at m/z 263. In contrast, peaks of GaIII-LysNTA complexes for other nonphosphorylated acidic tryptic fragments, e.g., FFVPFPEVFGK, EDVPSER, VPQLEIVPNS AEER, YLGYLEQLLR, and EPMIGVNQELAYFYPELFR, are not present at above-background relative intensities. This indicates that while complexation to GaIII-LysNTA is not completely selective for phosphopeptides, only a few peptides containing acidic residues (D and E) are picked up by the precursor ion scan.
One feature that distinguishes the potentially interfering HQGLPQEVLNENLLR peptide from the phosphopeptides is the much diminished intensity of its higher charge states. In particular, whereas the A, B, and C phosphopeptides give abundant precursors for the +3 and +4 states (Figure 5b), the E peptide gives only a weak peak for the 4+ precursor at m/z 523. In spite of the less than 100% selectivity for phosphopeptides, the presence of a small number of acidic tryptic peptides in the precursor scan does not impair the multiplex detection of phosphopeptides, especially if the latter is based on more than one charge state. In most proteomic applications, acidic peptides can be identified from the known protein sequence and distinguished from tryptic peptides carrying phosphorylated S, T, and Y residues.
Conclusions
Ternary complexes of LysNTA−GaIII/FeIII with phosphopeptide were formed in aqueous solution and, when subjected to ESI-MSMS analysis, were found to yield a common (LysNTA + H)+ product ion that could be used as a marker in phosphopeptide analysis. Complexes of GaIII gave a greater amount of spectral information without a significant loss in signal relative to FeIII complexes. A triple quadrupole, parent-ion scanning method was developed to detect several phosphopeptides through their LysNTA−GaIII−phosphopeptide complexes in peptide mixtures. Detection of phosphopeptides from an αS1-Cas digest was performed by both neutral loss scanning for the common loss of phosphoric acid and by parent ion scanning for the common (LysNTA + H)+ product ion from peptide complexes with LysNTA−GaIII. The parent scanning method provides the ability to detect phosphopeptides in several charge states in a single scanning data acquisition. Furthermore, complexation to LysNTA−GaIII enhanced detection of multiply phosphorylated peptides by ESI-MSMS in positive ion mode. The advantages of this parent scan method will undoubtedly prove to be sample dependent. Of course, there are inherent advantages to precursor scanning in positive ion mode as opposed to precursor scanning in negative ion mode or neutral loss scanning. Precursor scanning in positive ion mode allows for fragmentation data, and thus sequence information, to be acquired. In contrast, scanning in negative ion mode (for loss of PO3− or H2PO3−) requires a second run at reversed polarity to acquire such information. Similarly fragmentation data cannot be obtained during a neutral loss scan. Instead, the neutral loss scan must trigger a product ion scan in a data dependent manner. Furthermore, precursor scanning allows for the simultaneous detection of several charge states of the same peptide whereas a separate scan is required for each charge state to detect peptides in neutral loss scanning. Thus, only parent ion scanning allows detection of multiple charge states of the same component. For these reasons parent ion scanning in positive ion mode offers definite advantages over parent ion scanning in negative ion mode or neutral loss scanning, when applied to peptide analysis.
Supplementary Material
Acknowledgement
Funding of this research by NIH/NDDK (Grant DK67859) is gratefully acknowledged. We also thank Dr. Martin Sadilek for technical assistance with mass spectrometric measurements.
Footnotes
Supporting Information Available. Figures S1-S9 of ESI and CID mass spectra of peptide complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Fischer EH, Krebs EG. Phosphorylase activity of skeletal muscle. J. Biol. Chem. 1955;216:113–120. [PubMed] [Google Scholar]
- 2.Cohen P. The origins of protein phosphorylation. Nat. Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 3.Hunter T. Signaling -- 2000 and Beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 4.Carr SA, Huddleston MJ, Annan RS. Selective detection and sequencing of phosphopeptides at the femtomole level by mass spectrometry. Anal. Biochem. 1996;239:180–192. doi: 10.1006/abio.1996.0313. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JC. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 6.Chang EJ, Archambault V, McLachlin DT, Krutchinsky AN, Chait BT. Analysis of protein phosphorylation by hypothesis-driven multiple-stage mass spectrometry. Anal. Chem. 2004;76:4472–4483. doi: 10.1021/ac049637h. [DOI] [PubMed] [Google Scholar]
- 7.a Flora FW, Muddiman DC. Gas-Phase Ion Unimolecular Dissociation for Rapid Phosphopeptide Mapping by IRMPD in a Penning Ion Trap: An Energetically Favored Process. J. Am. Chem. Soc. 2002;124:6546–6547. doi: 10.1021/ja0261170. [DOI] [PubMed] [Google Scholar]; b Crowe MC, Brodbelt JS. Differentiation of Phosphorylated and Unphosphorylated Peptides by High-Performance Liquid Chromatography-Electrospray Ionization-Infrared Multiphoton Dissociation in a Quadrupole Ion Trap. Anal. Chem. 2005;77:5726–5734. doi: 10.1021/ac0509410. [DOI] [PubMed] [Google Scholar]
- 8.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 9.Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. Phosphoprotein isotope-coded affinity tag for isolating and quantitating phosphopeptides in proteome-wide analyses. Anal. Chem. 2001;73:2578–2586. doi: 10.1021/ac010081x. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nat. Biotech. 2001;19:375–378. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 11.Oda Y, Nagasu T, Chait BT. Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nat. Biotech. 2001;19:379–382. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 12.Vosseller K, Hansen KC, Chalkley RJ, Trinidad JC, Wells L, Hart GW, Burlingame AL. Quantitative analysis of both protein expression and serine/threonine posttranslational modifications through stable isotope labeling with dithiothreitol. Proteomics. 2005;5:388–398. doi: 10.1002/pmic.200401066. [DOI] [PubMed] [Google Scholar]
- 13.Porath J, Carlsson J, Olsson I, Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975;258:598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- 14.Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+ affinity chromatography. Anal. Biochem. 1986;154:250–254. doi: 10.1016/0003-2697(86)90523-3. [DOI] [PubMed] [Google Scholar]
- 15.Hochuli E, Dobeli H, Schacher A. New metal chelate adsorbent selective for proteins and peptides containing neighboring histidine residues. J. Chromatography. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 16.Ficarro S, McCleland M, Stukenberg P, Burke D, Ross M, Shabanowitz J, Hunt D, White F. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 17.a Pinkse JWH, Uitto PM, Hilhorst MJ, Ooms B, Heck AJR. Selective Isolation at the Femtomole Level of Phosphopeptides from Proteolytic Digests Using 2D-NanoLC-ESI-MS/MS and Titanium Oxide Precolumns. Anal. Chem. 2004;76:3935–3942. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]; b Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJD. Highly Selective Enrichment of Phosphorylated Peptides from Peptide Mixtures Using Titanium Dioxide Microcolumns. Mol. Cell. Proteomics. 2005;4:873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]; c Kweon HK, Hakansson K. Selective Zirconium Dioxide-Based Enrichment of Phosphorylated Peptides for Mass Spectrometric Analysis. Anal. Chem. 2006;78:1743–1749. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
- 18.Tao W, Wollscheid B, O'Brien R, Eng J, Li X, Bodenmiller B, Watts JD, Hood L, Aebersold R. Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat. Methods. 2005;8:591–598. doi: 10.1038/nmeth776. [DOI] [PubMed] [Google Scholar]
- 19.Schmitt L, Dietrich C, Tampé R. Synthesis and characterization of chelator lipids for reversible immobilization of engineered proteins at self-assembled lipid interfaces. J. Am. Chem. Soc. 1994;116:8485–8491. [Google Scholar]
- 20.Martell AE, Smith RM. Critical Stability Constants, Vol. 1: Amino Acids. Plenum Press; New York: 1974. pp. 116–118.pp. 142–143. [Google Scholar]
- 21.a Gatlin CL, Tureček F. Acidity Determination in Droplets Formed by Electrospraying Methanol-Water Solutions. Anal. Chem. 1994;66:712–718. [Google Scholar]; b Di Marco VB, Bombi GG. Electrospray mass spectrometry (ESI-MS) in the study of metal-ligand solution equilibria. Mass Spectrom. Rev. 2006;25:347–379. doi: 10.1002/mas.20070. [DOI] [PubMed] [Google Scholar]
- 22.Posewitz MC, Tempst P. Immobilized gallium (III) affinity chromatography of phosphopeptides. Anal. Chem. 1999;71:2883–2992. doi: 10.1021/ac981409y. [DOI] [PubMed] [Google Scholar]
- 23.Gatlin CL, Tureček F, Vaisar T. Copper(II) Amino Acid Complexes in the Gas Phase. J. Am. Chem. Soc. 1995;117:3637–3638. [Google Scholar]
- 24.Gatlin CL, Tureček F. Quantitative Electrospray Ionization Mass Spectrometric Studies of Ternary Complexes of Amino Acids with Cu2+ and Phenanthroline. J. Mass Spectrom. 2000;35:172–177. doi: 10.1002/(SICI)1096-9888(200002)35:2<172::AID-JMS926>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.