

Abstract
Due to their size, small molecules can not be simultaneously bound by two antibodies precluding their detection by noncompetitive two-site immunoassays, which are superior to competitive ones in terms of sensitivity, kinetics, and working range. This has prompted the development of anti-immune complex antibodies, but these are difficult to produce, and often exhibit high cross-reactivity with the unliganded primary antibody. This work demonstrates that anti-immune complex antibodies can be substituted by phage particles isolated from phage display peptide libraries. Phages bearing specific small peptide loops allowed to focus the recognition to changes in the binding area of the immune complex. The concept was tested using environmental and drug analytes; with improved sensitivity and ready adaptation into onsite formats. Peptides specific for different immune complexes can be isolated from different peptide libraries in a simple and systematic fashion allowing the rapid development of noncompetitive assays for small molecules
Keywords: pesticides, monoclonal antibodies, molinate, atrazine
INTRODUCTION
Owing to their excellent sensitivity and specificity, immunodetection techniques, which use the fine specificity of antibodies to detect trace amounts of target compounds, are particularly suitable for a wide range of applications in biomedical and environmental analysis. Depending on the format, the assays fall into two main categories, noncompetitive and competitive immunoassays. Noncompetitive assays, mostly used for large molecules and proteins, are based on two different antibodies (or other binding molecules) able to bind simultaneously to two independent epitopes on the analyte (two-site assays). Analysis of low-molecular-mass analytes such as drugs, hormones, toxins, pesticides, explosives, etc, not large enough to bind two antibodies simultaneously, requires a competitive immunoassay format. In this format, the analyte competes with a labelled (or immobilized) analyte analogue (hapten), and the measured signal is inversely proportional to the concentration of analyte in the sample. In this way, the formation of the analyte-antibody immune complex is indirectly quantitated by measuring the empty binding sites of the unreacted antibody. Mathematical modeling of immunoassay performance, shows that competitive assays are inferior to noncompetitive ones in terms of sensitivity, precision, kinetics, and working range 1. In addition, noncompetitive immunoassays are more easily adapted into rapid ‘on-site’ formats such as dipstick or immunochromatography, as well as to microfluidics and biosensor approaches. For this reason, several attempts have been made to implement small-molecule noncompetitive assays, but they have usually been limited to certain chemical structures or require analyte labelling 2, 3.
A method based on the blockage of the unreacted sites of the antibody with a polydentate ligand has been described 4, and successfully applied to the detection of cortisol, but a major drawback appears to be the difficulty in obtaining a stable blockage of the unoccupied antibody binding sites. An elegant alternative is the development of the so called open-sandwich ELISA, an immunoassay based on antigen-dependent stabilization of antibody variable regions (V(H) and V(L) domains). In this format, the analyte-dependent association of the antibody domains is used to bring together a tracer enzymatic activity, such as by joining the N- and C-terminal domains of beta-galactosidase 5. The open-sandwich ELISA is quick and highly sensitive, but the technology is laborious and requires a stringent control of the background association of the recombinant V(H) and V(L) domains in the absence of analyte, which is again case specific.
The most common approach for the development of noncompetitive ELISAs for small-molecules relies in the use of anti-immune complex antibodies 6–11, however these antibodies are difficult to obtain. In general, when antibodies are elicited against the antigenic determinant of an antibody’s combining site (idiotope), the interface of the idiotope-antiiditope antibody complex buries a large surface that involves most of the complementarity determining regions (CDRs) of both antibodies 12. When the idiotope under consideration corresponds to that of an anti-hapten antibody, it is important to consider that its modification upon reaction with the hapten will be modest and restricted to the binding pocket. In these antibodies, up to 85% of the accessible surface of the hapten can be buried after binding 13, 14, and therefore the small portion of the hapten that remains exposed to the solvent has little contribution to the formation of the new idiotope. This imposes a serious limitation to the preparation of anti-immune complex antibodies for two-site assay development, because the residual affinity of the anti-immune complex antibody for the free site of the anti-hapten antibody can be significant, and a major cause of high background noise in the assay.
In order to overcome this limitation, we sought “to focus” the recognition of the immune complex to the region of the idiotope where major changes are produced after binding of the hapten. For this, we substituted the large surface of the antiidiotope antibody binding site by short peptide loops that specifically react with the exposed region of the hapten and the conformational changes caused by its binding. For the selection of these peptides, we used phage display peptide libraries, reviewed by Smith and Petrenko 15, which have shown to be an excellent source of peptide ligands for a large number of selector molecules. We demonstrate the potential of this approach using the herbicides molinate (MW=187 Da) and atrazine (MW=215 Da) as small model analytes, and further test the concept with the drugs cyclosporin A (MW=1203 Da) and digoxin (MW=780 Da). Due to the control that can be exerted during the selection process, the isolation of these peptides is more systematic and much less laborious than the trial-and-error process of anti-immune complex antibody preparation.
EXPERIMENTAL SECTION
Antibodies, herbicides and other compounds
Molinate and the thiocarbamate compounds used in the cross-reactivity assays were gifts from Stauffer Chemical Co. Thiobencarb was a gift from Chevron Chemical Co. Development of the monoclonal anti-molinate antibody (MoAb 14D7) has been described in detail previously 16. Atrazine and related triazines were a gift from Dr. Shirley Gee, and the anti-atrazine antibody was a gift form Dr. T. Giersch 17. Anti-cyclosporine (cat. G86313M), anti-digoxin (cat. G45330M) antibodies and digoxin-BSA conjugate (cat. Z6D53-3405) were purchased from BioDesign International. Cyclosporine and digoxin standards were purchased from Sigma.
Phage display peptide libraries
The phage display peptide library used for initial panning experiments was a kind gift from Dr. Peter Schatz (Affymax Research Institute, Palo Alto, CA). This is a constrained library constructed in the phagemid vector p8V2 18, expressing peptides containing 7 or 8 random amino acids flanked by two cysteine residues and linked to the N-terminus of the major pVIII phage coat protein through a long glycine-rich spacer (GG-C(X)7/8C(GGGGS)3-). The library size is 2.4 × 109 independent clones. For mutagenesis library construction, 200 μg of p8V2 were digested with the BstxI restriction enzyme and the digested vector was purified by ultracentrifugation on a potassium acetate gradient at 48,000 rpm. The band visualized with ethidium bromide corresponding to linearized p8V2 vector was separated, the dye was removed with butanol, and the DNA was precipitated with ethanol. Double stranded DNA coding for the randomized eight amino acid constrained peptides was constructed by annealing of 100 picomoles of oligonucleotide mol mut 1 (5′-GGCCCAGTGCTCACGCAGGAGGCTGTNNKNNKTGGGAHACNNNKNNKNNK TGTGGAGGCGGGGGTAGC-3′) and 100 picomoles of oligonucleotide ON-2 (5′-TAGGGCCCACCTTGCTGGGATCGTCACTTCCC CCACCGCCGCTACCCC CGCCTCC-3′) in 40 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 50 mM NaCl in a final volume of 45μl. After heating for 5 min at 70°C and slowly cooling to room temperature to allow annealing, a fill-in reaction was performed by adding 80 units of sequenase V 2.0 (Stratagene) and 200 μM of dNTPs. After incubation at 37°C for 1.5 h, excess dNTPs were removed by gel filtration using Microspin S-200 HR column (Pharmacia). The purified double stranded DNA was digested with BstXI and BsiHKAI by incubating ON at 55°C and the digested fragment was purified by gel filtration using Microspin S-300 HR columns (Pharmacia). Twenty micrograms of digested p8V2 were ligated with 1.25 μg of the dsDNA insert using T4 DNA Ligase (InVitrogen). The ligated DNA was precipitated with ethanol and dissolved in 20 μl of water. ARI 236 cells (Affymax Research Institute, Palo Alto, CA) were electroporated, transferred to SOC (2% Bacto tryptone, 5% Bacto yeast extract, 0.05% NaCl, 2.5 mM KCl, pH 7.0 with NaOH) medium and incubated without selection for one hour at 37°C. Dilutions of a small aliquot of cells were plated on LB (Luria-Bertani medium) agar petri dishes with ampicillin, allowing an estimation of library diversity at 108 independent clones. Cells were diluted in 500 ml of SOP medium (2% Bacto tryptone, 1% Bacto yeast extract, 100 mM NaCl, 15 mM K2HPO4, 5 mM MgSO4, pH 7.2 with NaOH) containing 2.5 ml of 20% glucose, and 1.5 ml of 50 mg/ml ampicillin, and grown at 37°C with vigorous shaking. After the culture reached an OD600= 0.5, 1 × 10 12 transducing units of M13KO7 helper phage (Pharmacia Biotech) were added and incubated without shaking for 30 minutes to allow phage infection. After that, 0.6 ml of 20 mg/ml kanamycin and 6 ml of 20% arabinose were added to the culture and phages were cleared the next day as described below.
Panning experiments for isolation of peptides specific for digoxin and cyclosporin immune complexes were performed with a disulfide constrained 10-mer peptide library fused to the phage coat protein pVIII (a kind gift from Dr. Peter Schatz Affymax Research Institute, Palo Alto, CA). The general structure of this peptide library was the same as that of the 8 mer library and has a size of 5.8 × 1010 independent clones.
Biopanning
For the selection procedure, Nunc-Immuno™ plates were coated with 100 μl of MoAb 14D7 (2 μg/ml) in phosphate-buffered saline (PBS) overnight at 4°C. After a blocking step of 1 hour at 37°C with 5% skimmed milk in PBS, 100 μl of 1000 ng/ml of molinate were added to form the molinate-MoAb 14D7 immune complex. After 20 min incubation, the wells were rinsed twice with cold PBS, then 100–1000 library equivalents were diluted in 600 μl of 5% skimmed milk in PBS containing 100 ng/ml of molinate, dispensed into 6 microtiter wells and incubated for 2 hours at 4°C. Unbound phages were removed by 20 washes with cold PBS containing 0.05% Tween 20 (PBST). Bound phages were eluted by a 10 min incubation with 0.1 N glycine-HCl, pH 2,5, 0.1% BSA, and immediately neutralized with 2 M Tris base. The eluted phage was directly amplified as described below; or alternatively, a post-selection-adsorption step was included in some panning experiments to deplete the eluted phage of clones with affinity for the uncombined antibody. For this step, the eluted phages were incubated for 30 min in ELISA wells coated with MoAb 14D7 in the absence of molinate. This phage preparations (600 μl) were added to 10 ml of log-phase E. coli ARI 292 cells (Affymax Research Institute, Palo Alto, CA) and amplified in LB media containing 0.25% K2HPO4, 0.1% MgSO4, 0.1% glucose and 100 μg/ml ampicillin to an OD600 = 0.4. M13KO7 helper phage at a multiplicity of infection 10:1 was added. After a period of 30 min at 37°C without shaking, arabinose and kanamycin were added to a final concentration of 0.02 % and 40 μg/ml respectively, and the cultures incubated overnight at 37°C with vigorous shaking. Phage from liquid cultures were obtained by clearing the supernatants by centrifugation at 12,000 × g for 15 min, precipitated with 0.2 volumes of 20 % polyethylene glycol 8000-2.5M NaCl, (PEG-NaCl) on ice during 1 hour, and centrifuged as above. Phage pellets were resuspended in 2 ml of sterile PBS and titrated in ARI 292. For the next round of selection, 1010 transducing units were used. The same procedure of panning was used for the isolation of phage clones specific for the atrazine immune complex. The panning experiments performed with digoxin and cyclosporin were essentially the same, except that in these cases a high concentration (5μg/ml) of the analytes was added during the adsorption step on the immune complexes, and after elution and neutralization phages were post adsorbed on uncombined antibody.
After three to four rounds of panning, serial dilutions of individual amplified phage clones were tested for their ability to bind to the analyte-antibody immune complex by phage ELISA. Positive clones were further selected by checkerboard titration and submitted for DNA sequencing using the primer ON891 (TGAGGCTTGCAGGGGTC) (Division of Biological Sciences, Automated DNA Sequencing Facility, UCDavis).
Phage ELISA
Nunc-Immuno™ Maxi-Sorp™ plates were coated with the corresponding antibody or the analyte-antibody immune complex as described before. The wells were blocked with 5% skimmed milk PBS, and washed three times with PBST. One hundred μl/well of an overnight culture of individual amplified phage clones were dispensed into the wells. The microtiter plates where then incubated for 1 h at room temperature with gentle rocking. After three washes with PBST, 100 μl of a 1/5000 dilution of horse radish peroxidase (HRP) labeled anti-M13 monoclonal antibody (Pharmacia, Uppsala) in PBST was added to each well. One hour later, the plates were thoroughly washed and 100 μl of peroxidase substrate (25 ml of 0.1 M citrate acetate buffer pH 5.5, 0.4 ml of 6 mg/ml DMSO solution of 3,3′,5,5′-tetramethylbenzidine and 0.1 ml of 1% H2O2) were dispensed into each well. The enzymatic reaction was stopped after 15–20 min by the addition of 50 μl of 2 M H2SO4, and the absorbance at 450 nm (corrected at 600 nm) was read in a microtiter plate reader (Multiskan MS, Labsystems). The supernatants showing high readings in wells coated with the immune complex and low response in antibody coated wells were prepared as stabilized phage suspensions (see below), and used for further analysis. For checkerboard titration, 100 μl of various concentrations of the antibody (0.3–2 μg/ml) was used for coating.
Stabilization of phage suspensions
Individual amplified phage clones were obtained as described above. After two steps of precipitation with PEG-NaCl, the phage particles were suspended in 1/50 volume of the original culture volume in PBS, which was supplemented with the Complete Protease Inhibitor Cocktail of Roche Diagnostics and sodium azide 0.05%. The preparations were filtered through a 0.22 μm filter and stored in aliquots at 4°C and −20°C.
Noncompetitive ELISA, PHAIA
Fifty μl per well of serial dilutions of analyte standard in PBST were dispensed into microtiter plates coated with the corresponding antibody, followed by addition of 50 μl/well of the adequate dilution of the stabilized phage suspension. The antibody concentration used for coating and the proper dilution of the phage suspension had been previously optimized by checkerboard experiments. After 1 hour of incubation at room temperature, the plates were washed and developed as previously described. Absorbance values were fitted to a four-parameter logistic equation using the Genesis Lite 3.03 (Life Sciences (UK) Ltd.) package software. The lower detection limit of the assays was estimated as the analyte concentration causing a 10% increase in signal when compared to the zero analyte signal.
Assay cross-reactivity
The specificity of the noncompetitive assay set up with the phage borne peptides was characterized by determining the cross-reactivity with related pesticides. Analyte concentrations in the 0–10,000 ng/ml range were used in the noncompetitive ELISAs. After the data were normalized, the molar compound concentration corresponding to the midpoint of the curve, which corresponds to the concentration of analyte producing 50% saturation of the signal (SC50), was used to express the cross-reactivity of the assay according to the equation:
Matrix effect analysis
For the analysis of matrix effect, runoff water samples from rural areas of Uruguay, with no register use of molinate or atrazine, were spiked with known amounts of analyte and assayed by PHAIA in the 0–100 ng/ml range, using 100 μl of undiluted water samples plus 10 μl of 10 × PBST.
Dipstick assay
One and a half microliters of serial dilutions of MoAb 14D7 in PBS (7.5–30 ng/spot) were spotted onto nitrocellulose membranes (0.45 micrometers) (Biorad Laboratories Inc., CA). Immediately after drying, the membranes were quenched by soaking in 5% skim milk in PBS for 30 min; after rinsing in PBST, the membranes were cut into strips, dried and kept at 4°C until used. The assay was performed by dipping the membrane strips for 15 min into PBST spiked with different amounts of molinate and containing the appropriate dilution of specific phage (about 1010 phage particles/ml), washed under tap water, incubated for 10 min in a 1/5000 dilution of HRP-labeled anti-M13 monoclonal antibody, and developed using diaminobenzidine with metal ion enhancement substrate (6 mg of 3, 3′-diaminobenzidine in 9 ml of 50 mM Tris pH 7.6, plus 1 ml 0.3% NiCl2 plus 10μl of 30% H2O2. The reaction was stopped by washing the membrane in tap water.
RESULTS
Antibody-analyte immune complex specific peptides can be isolated from phage display peptide libraries
A general scheme of the procedure for isolation of anti-immune complex peptides is shown in Figure 1. Our first attempts to select anti-immune complex peptides were done using the anti-molinate monoclonal antibody (MoAb)14D7 19 and cyclic (disulfide constrained) peptide libraries consisting of 7 and 8-mer length peptides. This was performed on plates coated with the molinate-MoAb 14D7 complex in the presence of 100 ng/ml of molinate. After three rounds of selection (panning) individual phage clones were grown and assayed by ELISA for differential reactivity with the immune complex and the unreacted antibody, using wild type phage as a control. Four different sequences were obtained, three of them from the 8-mer library (insert in Figure 2b). All clones had the consensus sequence WDT, shown in bold. To test whether the selection of anti-immune complex peptides could be extended to other analyte-antibody systems we also explored its applicability to the competitive immunoassay for the herbicide atrazine described by Giersch et al. 17. This is a highly sensitive assay (IC50 (analyte concentration producing 50% signal inhibition) = 0.5 ng/ml) that utilizes the MoAb K4E7. Using the selection strategy described above, several peptide sequences highly specific for the atrazine-MoAb K4E7 immune complex were isolated (insert in Figure 2c). All of these clones exhibited strong reactivity with the immune complex, showing that the isolation of peptides that bind to the molinate-14D7 antibody complex is not a specific phenomenon associated with this system. Again there was an evident consensus motif, WFD which occurred in position 2 or 5 of the random 8 mer peptide. The only exception was clone 12A which had a three residue stretch that resembles the WFD consensus sequence, with His substituted for Phe, and Glu, another acidic residue, substituting for the aspartic acid residue of the consensus.
Figure 1. Scheme of the procedure for the selection of anti-immune complex phage and set up of Phage Anti-immune complex Assay (PHAIA).
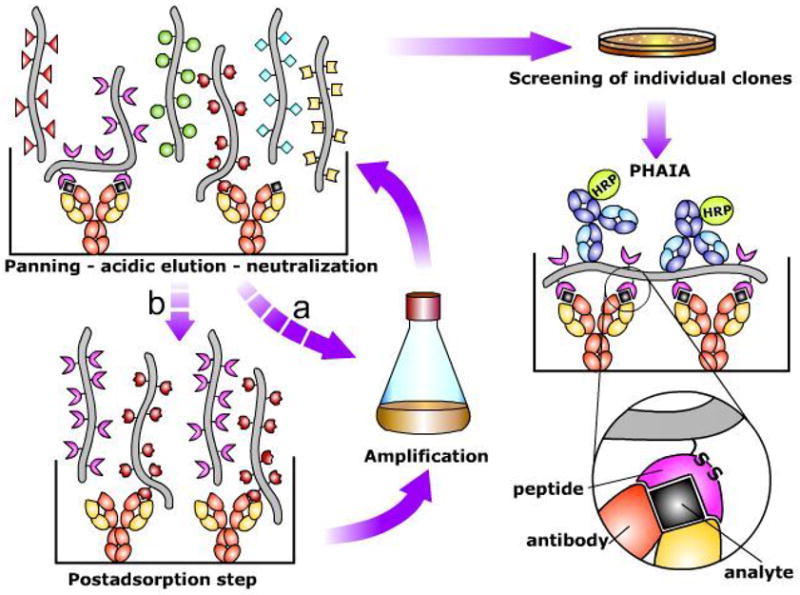
An anti-analyte antibody adsorbed onto the solid phase of an ELISA plate is used to capture the analyte (black square). A phage display peptide library is then added, and after several washing steps, specific phages recognizing the immune complex are eluted by addition of low pH buffer. The supernatant is neutralized, and the selected subpopulation of phages can be used directly for E. coli infection and phage amplification (a), or submitted to an optional post adsorption step to eliminate phages with strong affinity for the uncombined antibody (b). After several rounds of selection (panning) individual clones are tested for differential binding to the immune complex and the free antibody by phage ELISA. Phage clones showing low reactivity with the empty antibody are selected for development of PHAIA. In this assay, the immune complex formation is detected by its reaction with the phage borne peptide, followed by an anti-pVIII phage coat protein antibody coupled to horse radish peroxidase (HRP). A possible scheme of the antibody-analyte-peptide trivalent interaction that allows development of PHAIA is shown in the lower right part of the figure. Note that the overall affinity of the peptide for the immune complex is postulated to be the result of their interaction with the analyte and the antibody. The relative role of these contributions is further discussed in the text.
Figure 2. PHAIA development: noncompetitive ELISAs for molinate and atrazine.

a) Effect of phage concentration in assay performance. Different phage concentrations of clone 1M were used to detect binding of molinate on plates coated with 50 ng/well of MoAb 14D7. Phage concentrations (phage particles/ml) were: 1010 (white circles), 5 × 109 (diamonds), 2.5 × 109 (white triangles), 1.25 × 109 (stars), 6.3 × 108 (black circles), and 3.1 × 108 (black triangles). b) Noncompetitive ELISA for molinate (structure shown in the insert) performed with different phage borne peptides. Clones sequences and their corresponding midpoint values (SC50) are shown in the insert. c) Noncompetitive ELISA for atrazine (structure shown in the insert) performed with different phage borne peptides. Clones sequences and their corresponding midpoint values (SC50) are shown in the insert. d) Peptides isolated from the mutagenesis library with the molinate-MoAb 14D7 immune complex.
Phage bearing peptides can be used to set up noncompetitive Phage Anti-Immune complex Assay (PHAIA)
Checker-board titration analysis was used to optimize the amount of coating antibody in the presence of a fixed amount of analyte, in order to set up conditions for noncompetitive PHAIA. Once the amount of antibody was fixed, it was possible to develop a noncompetitive format to measure the analyte-bound sites using the set up shown in Figure 1. As expected, the sensitivity of PHAIA increased with the concentration of phage particles used in the assay, however after a certain point, the additional increase of phage concentration resulted in a high background signal due to direct binding of the peptide to the uncombined antibody (Figure 2a). The molinate assay performance was greatly influenced by the sequences flanking the conserved WDT motif of the phage borne peptides used for immune complex detection (Figure 2b). The most sensitive test for molinate was attained with clone 1M. With this phage borne peptide, the detection limit was 0.73 ng/ml, and the concentration of analyte producing 50% of signal saturation (SC50) was 5.0 ± 0.4 ng/ml. This sensitivity was about 30 folds better than that of the competitive ELISA set up with the same antibody 19 (detection limit = 22 ng/ml and IC50 = 69.2 ± 1.4 ng/ml). In a similar way, peptides isolated with the atrazine immune complex also allowed the set up of sensitive noncompetitive assays Figure 2c. As found for the molinate system, there was a marked influence of the residues flanking the WFD motif in the sensitivity of the assay. Clone 13A showed the best performance with a detection limit of 0.016 ng/ml and SC50 = 0.051 ± 0.002 ng/ml. Once more, this represented a significant improvement with regard to the competitive assay set up with the same antibody 17, which exhibited a detection limit of 0.18 ng/ml and IC50 = 0.64 ± 0.06 ng/ml.
The role of the flanking residues was further explored for the molinate-MoAb 14D7 system by building the mutagenesis library CXXwdtXXXC, where X indicates a fully random position, and lowercase letters indicate 50% probability of finding this residue in that position. Different strategies were used to pan this library, including extensive washing and post adsorption steps on fresh uncombined antibody, in addition, the screening of individual clones was biased towards phage exhibiting low binding affinity for the unreacted antibody. Ten new peptides were isolated using this strategy (Figure 2d), with the consensus sequence (S/N)XWDT(T/S)GW, where in addition to the WDT motif, Gly and Trp were also essential in position 7 and 8, respectively. While Thr in position 6 appeared to improve the assay sensitive, the role of Ser and Asn at position 1 was less clear. As expected due to our biased selection, most clones showed negligible cross-reactivity with the free antibody, particularly clone mA (see below in Figure 4b). However, this intuitive advantage of clone mA was not directly translated into a more sensitive assay, clone mA had a detection limit of 2 ng/ml and SC50 = 8.5 ± 0.5 ng/ml versus a detection limit of 0.73 ng/ml and SC50 = 5.0 ± 0.4 ng/ml for clone 1M. This highlights the relevance of the overall affinity of the peptide for the immune complex as a critical feature that determines the sensitivity of the assay.
Figure 4. Adaptation of the molinate PHAIA into dipstick format.
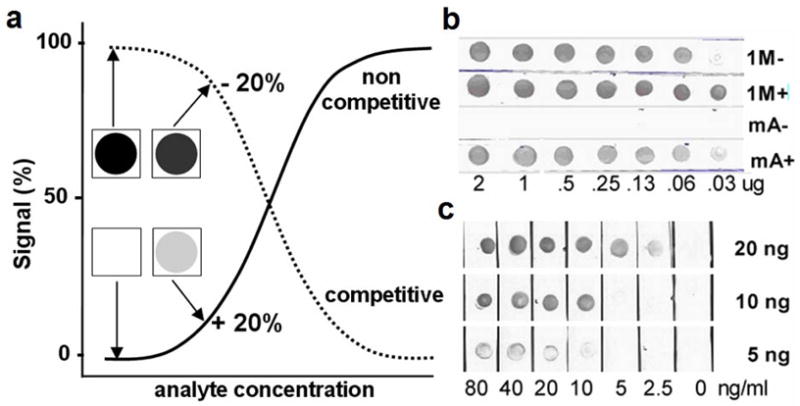
Dipstick assay for molinate using phage borne peptides. a) Theoretical representation of competitive (dotted line) and noncompetitive (solid line) immunoassays, and their adaptation into dipstick format. Notice that even in the case that both assays have equal sensitivity, the change in signal obtained in the noncompetitive format is much more convenient to detect trace amounts of analyte by naked eye visualization. b) Capture antibody titration. Different amounts of MoAb 14D7 (ranging from 2–0.03 μg) spotted onto nitrocellulose strips were incubated with 1010 phage particles/ml bearing the peptides 1M or mA in the presence (+) or absence (−) of 50 ng/ml of molinate. c) Dipstick assay set up with 20, 10 or 5 ng of MoAb 14D7 and clone 1M (1010 phage particles/ml) in the range of 0 to 80 ng/ml of molinate.
PHAIA cross-reactivitites and matrix effect
The simultaneous recognition of the analyte by the antibody and the peptide has a great potential for discriminating haptens improving the sensitivity of the assay. This was observed, particularly for atrazine, when the cross-reactivity of PHAIA and the competitive assays set up with the same antibodies were examined using a panel of molinate- and atrazine-related herbicides (table 1). PHAIA was further validated by performing molinate standard curves in runoff water obtained from representative agricultural areas of Uruguay. No relevant matrix effect was observed in the molinate PHAIA (Figure 3) when undiluted samples were analyzed. Similar results were obtained for the atrazine PHAIA (data not shown).
Table 1.
Cross reactivity (%) of molinate and atrazine PHAIA
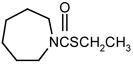
| Compound | Structure | Chemical hapten 19 | Clone 1M CSTWDTTGWC | Clone 2EM CRSHWDTWC | |
|---|---|---|---|---|---|
| molinate |
|
100 | 100 | 100 | |
| thiobencarb |
|
1 | 0 | 0 | |
| butylate |
|
1 | 0 | 0 | |
| EPTC |
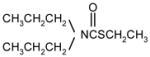
|
1 | 5 | 15 | |
| cycloate |
|
1 | 9 | 0 | |
| pebulate |
|
4 | 7 | 13 | |
| Vernolate |
|
4 | 4 | 5 | |
| Compound | Structure | Chemical hapten 17 | Clone 13A CTPVRWFDMC | ||
|
| |||||
|
R1 | R2 | R3 | ||
| atrazine | Cl | C2H5 | CH(CH3)2 | 100 | 100 |
| simazine | Cl | C2H5 | C2H5 | 48 | 0 |
| propazine | Cl | CH(CH3)2 | CH(CH3)2 | 116 | 144 |
| cyanazine | Cl | C2H5 | C2N(CH3)2 | 91 | 0 |
| ametryn | SCH3 | C2H5 | CH(CH3)2 | 0 | 0 |
| simetryn | SCH3 | C2H5 | C2H5 | 0 | 0 |
| prometryn | SCH3 | CH(CH3)2 | CH(CH3)2 | 0 | 0 |
| terbutryn | SCH3 | C2H5 | C(CH3)3 | 0 | 0 |
All data are the mean of two independent experiments. A value of 0 means that there was no observable cross reactivity with the highest concentration tested 104 ng/ml
Figure 3. Matrix effect of PHAIA.
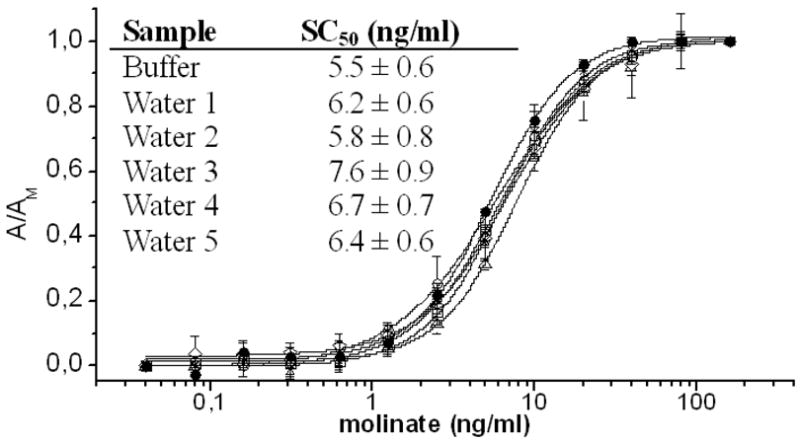
Molinate standard curves performed in assay buffer (black squares) and similar curves performed with undiluted water from agricultural runoffs.
PHAIA can be easily adapted into dipstick formats
The most attractive advantage of noncompetitive assays is the possibility of measuring near-zero signals at low analyte concentrations with much more ease than a competitive format. This advantageous property makes them the method of choice when rapid onsite test formats are needed, particularly, when the presence of the analyte needs to be visualized in a yes or no fashion (Figure 4a). To illustrate this concept we adapted the molinate assay into a dipstick format where the antibody immobilized onto a nitrocellulose strip is dipped into a molinate solution and the formation of the immune complex is revealed by its reaction with the phage borne peptide. Two clones were evaluated for this purpose, 1M and mA. The latter was chosen on the basis of its high specificity for the immune complex, and as shown in Figure 4b, clone mA did not react with the empty antibody even a the highest coating density. Conversely, clone 1M showed intense cross-reactivity with the uncombined antibody over a wide range of coating densities. However, once the proper conditions were set, clone 1M performed with higher sensitivity in the dipstick assay, allowing the detection by visual examination of up to 2.5 ng/ml of molinate (Figure 4c). This is an interesting result considering that the WHO guideline value for the concentration of this compound in drinking water is 6 ng/ml 20, and 20 ng/ml in the State of California 21.
The isolation of anti-immune complex peptides is of general applicability
To further demonstrate the concept we selected two additional target analytes, cyclosporin and digoxin. These are larger molecules that need careful monitoring during medication. We performed our panning experiments using two commercial MoAbs, cyclosporin (Kd 1 × 10−8 M) and digoxin (Kd 1.4 × 10−12 M). The selection process was done as previously, but using a 10 mer library and including post adsorption steps. In the case of both analytes, after three rounds of panning, most clones showed strong binding to the immune complex with negligible reactivity with the uncombined antibody. Out of 15 clones sequenced, three different sequences were obtained for cyclosporin and four for digoxin (inserts in Figure S-1). As observed in the case of molinate and atrazine, a three amino acid consensus sequences, FPD, was present in the family of peptides isolated for cyclosporin, but no consensus sequence was found in the case of digoxin. Sensitive PHAIAs could be set up with selected phage borne peptides (Figure S-1). Cyclosporin PHAIA using the peptide CELFPDLIHVWC attained a detection limit of 0.62 ng/ml and SC50 = 3.4 ± 0.2 ng/ml, while the digoxin PHAIA set with the peptide CMVLPELSPCTC performed with a detection limit of 0.070 ng/ml and SC50=0.22 ± 0.01 ng/ml. This represented a 10 fold improvement when compared with the competitive assay performed with the same antibody (detection limit = 0.72 ng/ml, SC50=2.1 ± 0.3 ng/ml). Because of the lack of commercially available coating antigens for cyclosporin the comparison between the direct and competitive formats using the same antibody could not be done.
DISCUSSION
Despite the many advantages of two-site immunoassays, to date, no systematic and simple methods exist for the development of these assays when the analyte is a low molecular weight molecule. The small size of these analytes is the critical obstacle that precludes the simultaneous binding of the capture and detecting antibody. The closest approach to overcome this situation has consisted in the use of antiidiotype antibodies that show differential reactivity with the antibody binding site upon binding of the analyte. However, as discussed before, due to the large surface buried in the antiidiotype immune complex interface, these antibodies constitute a rare event in the polyclonal immune response against immune complex antigens, and therefore are difficult to produce. Here we demonstrated that it is possible to “focus” the recognition of the immune complex using short peptide loops isolated from phage display libraries, allowing the trivalent detection of small analytes in a noncompetitive format.
In the different models examined, the transition from detection of unreacted antibody binding sites (competitive assay) towards detection of the analyte-antibody reaction boosted the sensitivity of the tests. The formation of the trivalent complex appears to be the result of the combined interactions of the peptide with the exposed regions of the analyte plus additional bonding to residues in the antibody. While the latter is an undesirable source of cross-reactivity with the free antibody, it may also work as a key contribution to the detection of trace amounts of immune complex in the PHAIA system. Thus, the proper balance of this characteristic appears to be a critical feature of the peptide, and care should be taken in the design of the panning and screening experiments in order to select convenient candidate phage borne peptides for PHAIA. In addition, the shift to a positive or noncompetitive readout facilitates the detection of near zero signals, and therefore makes easier the adaptation of the PHAIA into rapid yet sensitive dip-stick formats for ‘point of need’ testing, as shown here for the molinate assay.
Another important aspect of PHAIA is its general applicability and simplicity. The availability of commercial phage display peptide libraries makes this technology accessible to non specialized laboratories, and therefore prone to be applied in biomedical, agricultural and environmental sciences. In addition to the four examples presented here, the principle has also worked for other analytes such as phenoxibenzoic acid, a pyrethroid metabolite. In this case the peptides were isolated utilizing affinity purified polyclonal antibodies and the immunoassay performed with the total IgG fraction (manuscript in preparation). Moreover, we have found a great flexibility with regard to the type of library that can be used. Anti-immune complex peptides have been isolated from phage libraries expressing different lengths of random peptides, and also from libraries expressing the peptide on the phage minor coat protein pIII. Conversely to the challenging and laborious preparation of detection antibodies, the selection of phage borne peptides is systematic and can be accomplished in few days. Furthermore, the final product of this selection, namely the phage particle bearing the specific peptide sequence, can be directly used as convenient immunoassay reagent 22. It is worth noting that the robustness and filamentous polymeric structure of the phage particle opens many possibilities for its chemical modification using dyes, fluorescent compounds, acridinium esters, enzymes, etc. which could enable the automation of the noncompetitive PHAIA technology in microfluidic and biosensor platforms.
CONCLUSIONS
The principle described here represents a new concept for the development of noncompetitive immunoassays that is simple and of general applicability. In addition to providing a positive readout, in all cases examined, PHAIA resulted in an increased sensitivity (about ten fold). The trivalent detection principle possesses the potential to increase the specificity of the assay and is amenable to be adapted into rapid on site dipstick formats.
Supplementary Material
Acknowledgments
This work was supported in part by NIH Fogarty International Center Grant TW05718; PDT54-41 DINACYT, Uruguay; the NIEHS Superfund Basic Research program, P42 ES04699; and the NIEHS Center, P30 ES05707. We would like to thank Dr. Peter Shatz, Affymax Research Institute, Palo Alto, California, USA, for his generous donation of some of the phage libraries used in this study.
References
- 1.Jackson TM, Ekins RP. J Immunol Methods. 1986;87:13–20. doi: 10.1016/0022-1759(86)90338-8. [DOI] [PubMed] [Google Scholar]
- 2.Pradelles P, Grassi J, Creminon C, Boutten B, Mamas S. Anal Chem. 1994;66:16–22. doi: 10.1021/ac00073a005. [DOI] [PubMed] [Google Scholar]
- 3.Piran U, Riordan WJ, Livshin LA. Clin Chem. 1995;41:986–990. [PubMed] [Google Scholar]
- 4.Giraudi G, Anfossi L, Rosso I, Baggiani C, Giovannoli C, Tozzi C. Anal Chem. 1999;71:4697–4700. doi: 10.1021/ac981282c. [DOI] [PubMed] [Google Scholar]
- 5.Yokozeki T, Ueda H, Arai R, Mahoney W, Nagamune T. Anal Chem. 2002;74:2500–2504. doi: 10.1021/ac015743x. [DOI] [PubMed] [Google Scholar]
- 6.Towbin H, Motz J, Oroszlan P, Zingel O. J Immunol Methods. 1995;181:167–176. doi: 10.1016/0022-1759(94)00343-u. [DOI] [PubMed] [Google Scholar]
- 7.Nagata S, Tsutsumi T, Yoshida F, Ueno Y. Nat Toxins. 1999;7:49–55. [PubMed] [Google Scholar]
- 8.Kobayashi N, Shibusawa K, Kubota K, Hasegawa N, Sun P, Niwa T, Goto J. J Immunol Methods. 2003;274:63–75. doi: 10.1016/s0022-1759(02)00501-x. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi N, Kubota K, Oiwa H, Goto J, Niwa T, Kobayashi K. J Immunol Methods. 2003;272:1–10. doi: 10.1016/s0022-1759(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 10.Ullman EF, Milburn G, Jelesko J, Radika K, Pirio M, Kempe T, Skold C. Proc Natl Acad Sci U S A. 1993;90:1184–1189. doi: 10.1073/pnas.90.4.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Self CH, Dessi JL, Winger LA. Clin Chem. 1994;40:2035–2041. [PubMed] [Google Scholar]
- 12.Pan Y, Yuhasz SC, Amzel LM. Faseb J. 1995;9:43–49. doi: 10.1096/fasebj.9.1.7821758. [DOI] [PubMed] [Google Scholar]
- 13.Lamminmaki U, Kankare JA. J Biol Chem. 2001;276:36687–36694. doi: 10.1074/jbc.M102367200. [DOI] [PubMed] [Google Scholar]
- 14.Monnet C, Bettsworth F, Stura EA, Le Du MH, Menez R, Derrien L, Zinn-Justin S, Gilquin B, Sibai G, Battail-Poirot N, Jolivet M, Menez A, Arnaud M, Ducancel F, Charbonnier JB. J Mol Biol. 2002;315:699–712. doi: 10.1006/jmbi.2001.5284. [DOI] [PubMed] [Google Scholar]
- 15.Smith GP, Petrenko VA. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 16.Rufo C, Hammock BD, Gee SJ, Last JA, Gonzalez-Sapienza G. J Agric Food Chem. 2004;52:182–187. doi: 10.1021/jf034710a. [DOI] [PubMed] [Google Scholar]
- 17.Giersch T. Journal of Agriculture and Food Chemistry. 1993;41:1006–1011. [Google Scholar]
- 18.Wrighton NC, Farrell FX, Chang R, Kashyap AK, Barbone FP, Mulcahy LS, Johnson DL, Barrett RW, Jolliffe LK, Dower WJ. Science. 1996;273:458–464. doi: 10.1126/science.273.5274.458. [DOI] [PubMed] [Google Scholar]
- 19.Rufo C, Hammock BD, Gee SJ, Last JA, Gonzalez-Sapienza G. J Agric Food Chem. 2004;52:182–187. doi: 10.1021/jf034710a. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton D, AMbrus A, Dieterle R, Felsot S, Harris C, Holland P, Katamaya A, Kurihara N, Linders J, Unsworth J, Wong SS. Pure Appl Chem. 2003;75:1123–1155. [Google Scholar]
- 21.OEHHA. 1988 http://www.oehha.ca.gov/public_info/public/phg4.html.
- 22.Cardozo S, Gonzalez-Techera A, Last JA, Hammock BD, Kramer K, Gonzalez-Sapienza GG. Environ Sci Technol. 2005;39:4234–4241. doi: 10.1021/es047931l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.