

Abstract
Accumulation of oxidative DNA damage in the human brain has been implicated in etiologies of post-traumatic and age associated declines in neuronal function. In neurons, because of high metabolic rates and prolonged life span, exposure to free radicals is intense and risk for accumulation of damaged DNA is amplified. While data indicate that the brain is equipped to repair nuclear and mitochondrial DNA, it is unclear whether repair is executed by distinct subsets of the DNA-repair machinery. Likewise, there are no firm assessments of brain capacity for accurate DNA repair under normal and more so compromised conditions. Consequently, the scope of DNA repair in the brain and the impact of resolution of oxidative lesions on neuronal survival and function remain largely unknown. This review considers evidence for brain levels and activities of the base excision repair (BER) pathway in the context of newly available, comprehensive in situ hybridization analyses of genes encoding repair enzymes. These analyses suggest that not all subsets of BER are equally represented in the brain. Because BER is the major repair process for oxidatively damaged DNA, to what extent parsimonious BER may contribute to development of neuronal dysfunction and brain injury under compromised conditions, is discussed.
Keywords: base excision repair, brain, neuron, oxidative DNA damage
INTRODUCTION
Generally robust DNA repair processes are associated with DNA replication in proliferating cells (Barnes and Lindahl, 2004). The long lived, highly metabolic neurons, however, lack the replication machinery, which is considered vital to renewal and maintenance of genetic information. For this reason, a specialized mode for genomic maintenance has been proposed for neuronal cells. This proposed mode, termed ‘differentiation associated repair’, posits that in neurons, both strands of transcribed genes would be preferentially repaired via the nucleotide excision repair pathway (Nouspikel and Hanawalt, 2002). Of note is also the recently reported, lesser accumulation of oxidative DNA damage in promoters of transcribed compared to non-transcribed genes in the aged human brain (Lu et al., 2004). Interestingly, new findings suggest that RNA polymerase II might be in fact, a primary sensor of DNA lesions via stalled transcription complexes (Derheimer et al., 2007; Lindsey-Boltz and Sancar, 2007). While such a scenario might be of major significance in postmitotic, highly metabolic neuronal cells, the actual in vivo spectrum of oxidative lesions in the brain is not well characterized. Therefore, it is still unclear to what extent predominant oxidative lesions and repair intermediates contribute to stalling of RNA pol II nucleated complexes, in vivo in the brain. Insofar however, that these notions await further experimental support, it is plausible that maintenance of neuronal genomes relies largely on the overall process of global genomic repair. It is conceivable therefore, that under compromised conditions and excessive oxidative stress the need for DNA repair exceeds neuronal capacity leading to mutagenic consequences and that in the long term, such ill-repaired DNA may predispose to neurodegenerative processes.
Since the brain is quite well protected from external insults by the cranium and blood brain barrier, endogenous DNA damage is arguably the most critical threat to genomic stability in neuronal cells. Endogenous DNA damage, which is incidental to normal cellular metabolism, consists of DNA lesions that are continually generated by spontaneous decay, depurination, depyrimidination, deamination, free radicals mediated oxidation and strand breaks and other DNA transactions including erroneous base incorporation, base methylation and alkylation (De Bont and van Larebeke, 2004; Lindahl, 1993). Accordingly, to sustain neuronal longevity, the brain is expected to be well equipped for repair of endogenous DNA damage, which in mammalian cells is carried out primarily, albeit not exclusively by the base excision repair (BER) pathway.
BER is a stepwise process initiated by DNA glycosylases, which release aberrant bases. The resultant abasic sites (AP) are cleaved and further processed to become substrates for the gap filling synthesis, which is carried primarily by DNA polymerase β, and nick ligation to complete the repair process (Dianov et al., 1998; Wilson and Kunkel, 2000). Several sub-pathways of mammalian BER have been characterized, initially based on the repair patch size; as a short (one-nucleotide) or long (few-nucleotides) gap filling synthesis (Dogliotti et al., 2001) and more recently on partition into apurinic/apyrimidinic endonuclease (APE1)-dependent and independent sub-pathways (Wiederhold et al., 2004). The latter paradigm follows the discovery of the NEIL (Nei-like) class of mammalian glycosylases that catalyze the βδ elimination reaction at AP sites generating a 3’ phosphate. In mammalian cells, removal of 3’ phosphate can be efficiently executed by the polynucleotide kinase-phosphatase and thus independently of APE1 (Karimi-Busheri et al., 1999; Wiederhold et al., 2004; Xu et al., 2003). Various DNA end-blocking modifications may form, either directly or as BER intermediates (Dianov and Parsons, 2007). Importantly, additional enzymes with ability to resolve DNA end-blocking modifications and thereby channel BER intermediates for DNA repair synthesis and ligation processes are being described (El-Khamisy and Caldecott, 2007; El-Khamisy et al., 2005; Katyal et al., 2007; Mani et al., 2007) gradually unraveling BER complexities.
Notwithstanding, how the BER process is orchestrated in vivo (Wilson and Kunkel, 2000) remains unclear. It is also not well understood how the different BER sub-pathways are selectively recruited at sites of damage in different types of mammalian cells and to what extent BER may cooperate with other repair pathways in the various cellular settings (Nussenzweig and Nussenzweig, 2007). These issues are even less clear in the context of mammalian brain, which likely also relies on additional pathways (Brooks, 2007; Brooks et al., 1996; Katyal et al., 2007) for genomic maintenance and thereby preservation of function. In this review, we utilize newly available gene expression data to evaluate likelihood for occurrence of the different BER sub-pathways in the brain.
Mammalian BER
Base excision repair (BER) is the major mammalian pathway for repair of oxidatively damaged nuclear and mitochondrial DNA (mtDNA). The details of the BER process have been extensively characterized in vitro, in reconstituted assays and cultured proliferating cells and to a much lesser degree in in vivo systems and in postmitotic cells. The BER process in nuclei and mitochondria is initiated by DNA glycosylases, which share but also differ in their targets and enzymatic properties. Key glycosylases for protection from mutagenic consequences of the frequent oxidative adduct, 7,8-dihydro-8-oxoguanine (8-oxoG) include OGG1 and MYH. OGG1 and MYH are orthologs of the E. coli MutM glycosylase that excises 8-oxoG and the E. coli MutY glycosylase, which cleaves adenine residues opposite 8-oxoG, guanine and other lesions. Removal of either 8-oxoG or mispaired adenines prevents mutagenic G:C to A:T transversions (McGoldrick et al., 1995). The NEIL (Nei-like) glycosylases are orthologs of the E. coli MutM/Nei enzymes and have affinity for 5-hydroxyuracil (5-OHU) (Dou et al., 2003; Hazra et al., 2002a; Hazra et al., 2002b), a product of oxidative deamination of cytosine. Interestingly, NEIL2, shows cell cycle independent expression pattern and preference for excision of 5-OHU incorporated into a bubble structure, suggesting that it might be specifically involved in repair of transcribed DNA and thereby have a role in sustaining the integrity of transcribed genes in post-mitotic cells (Englander and Ma, 2006). Similarly, the family of uracil DNA glycosylases (UDG) is strongly relevant to DNA modifications that are likely to occur in postmitotic cells, ie, deamination of cytosine or incorporation of uracil and generation of U:G or T:G mismatches. Interestingly, while the major mammalian uracil N-glycosylase 2 (UNG2) associates with replicating DNA, the single strand selective monofunctional UDG (SMUG1) is implicated in U:G mismatch repair in non proliferating cells (Pettersen et al., 2007). Similarly, the thymine DNA glycosylase (TDG) is eliminated during the S phase and restored when cells transition through G2 (Hardeland et al., 2007). Interestingly, both SMUG1 and TDG show significant expression in the brain, while the S phase-associated UNG2 appears to be absent (see below).
The abasic sites generated by the diverse glycosylases are integrated into the BER pathway by the apurinic/apyrimidinic endonuclease (APE1) or in the case of the NEIL1/2 by polynucleotide kinase phosphatase (PNKP) (Karimi-Busheri et al., 1999; Wiederhold et al., 2004). Subsequent gap-filling synthesis steps may proceed depending on the status of DNA ends, either through the short- or long-patch repair sub pathway by DNA polymerase β (pol-β) or by pol-β (pol-β, δ and ε), flap endonuclease 1 (FEN-1) and PCNA, respectively. The remaining nicks are then sealed by ligase III associated with the scaffold/chaperone protein XRCC1 or by ligase I to complete the repair process (Dianov et al., 1998; Mortusewicz et al., 2006; Sleeth et al., 2004). Here, the different BER sub-pathways are discussed in view of emerging data for expression and activities of repair proteins in the mammalian brain.
BER modulations during brain ontogeny in the context of physiological transition from proliferative to non-proliferative state
Since generally robust DNA repair is thought to be associated with DNA replication in proliferating cells, it was of interest to monitor BER during brain development, namely, in the course of a physiological transition from proliferative to non-proliferative state. We followed the status of cell proliferation during brain development by monitoring expression of the DNA replication associated protein Pcna (proliferating cell nuclear antigen). As expected, a sharp decline in density of Pcna positive cells was observed in the course of brain development, starting at the mid-late embryonic stage, E14, through early postnatal stages (Figure 1). The demarcated cortical regions, which are shown at low magnification in the upper panel were enlarged (lower panel) to visualize the dense PCNA staining in the cortical plate in E14–E16 embryos, with reduced staining in the corresponding regions in E21 and only sparsely distributed Pcna positive cells at postnatal stages P1 and P7. A pattern of decline in density of PCNA positive cells was observed also for the developing cerebellum (Lee et al., 2004). Declining Pcna staining was mirrored in the sharply reduced levels detected by western analyses (Figure 1). Interestingly, developmental decline in levels of BER enzymes was less pronounced, indicating that in the brain BER pathway is independent, at least in part, of DNA replication and cell proliferation.
Figure 1. (Top) Distribution of Pcna positive cells in the developing rat cerebral cortex and cerebellum.
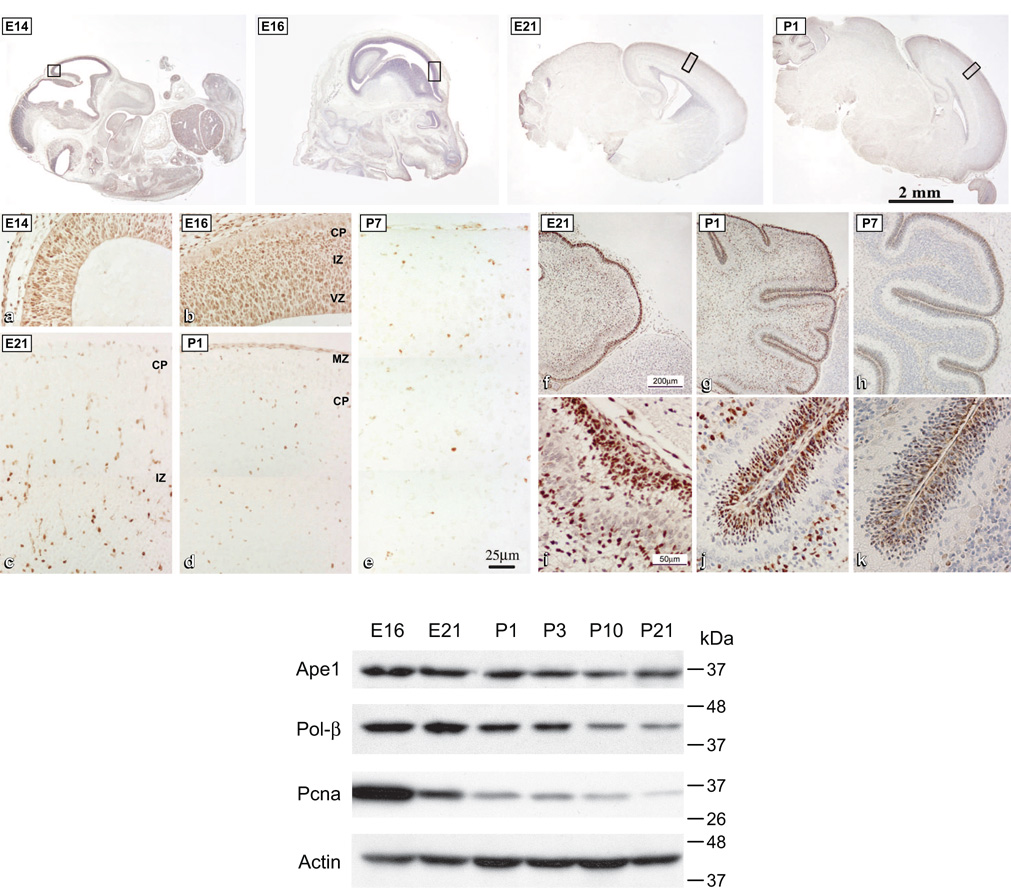
(Top) photomicrographs of sagittal sections of the whole rat embryo (E14), head (E16) and brain (E21 and P1). Demarcated regions are enlarged (a–e), stained for PCNA and counterstained with hematoxylin. CP, cortical plate; MZ, marginal zone; IZ, intermediate zone; VZ, ventricular zone. PCNA staining in the developing cerebellum (f–k), enlarged regions show external germinal layer where cells continue to proliferate postnatally (i–k). E, embryonic day; P, postnatal day. (Modified with permission from Lee HM, Hu Z, Ma H, et al. J Neurochem 2004;88(2):394–400). (Bottom) Developmental changes in levels of BER proteins and Pcna. Western blotting analyses of Ape1, pol-β and Pcna in the embryonic and postnatal brain. Total protein extracts were resolved by SDS-PAGE and probed as indicated; reprobing for actin served as a loading control. (Modified with permission from Englander EW, Ma H. Mech Age Dev 2006;127:64–69).
In the context of declining proliferation rates during brain ontogeny, the steady state mRNA measurements in the embryonic E21, neonatal P1, P5, P10, P21 and mature rat brain (Englander and Ma, 2006) revealed a pattern of decline for the 7,8-dihydro-8-oxoguanine DNA glycosylase (Ogg1), APE1 and DNA polymerase β. In all cases, a gradual decline to ~50% for the mature compared to embryonic rat brain, was recorded (Figure 2). In contrast, however, Neil1 and Neil2 mRNA levels did not diminish within this time frame. Remarkably, mRNA levels of the polynucleotide kinase-phosphatase (PNKP) (Figure 2) implicated in the Neil1/2 initiated, Ape1-independent BER sub-pathway remained unchanged in the course of brain ontogeny and in the mature brain (Englander and Ma, 2006).
Figure 2. Steady state mRNA levels of BER enzymes during brain ontogeny.

Real time RT-PCR analysis shows relative levels of mRNA expression in the embryonic (E16, E21), neonatal (P1, P5, P10, P21) and adult rat brain (three brains per group, duplicate assays). Graphs for Neil1, Neil2, Pnk (polynucleotide kinase-phosphatase), Ogg1 (7,8-dihydro-8-oxoguanine DNA glycosylase), Ape1 (apurinic/apyrimidinic endonuclease), pol-β (DNA polymerase beta) are shown. (Reprinted from Englander EW, Ma H. Mech Age Dev 2006;127:64–69, with permission).
To assess how well expression correlates with enzymatic activity, nuclear extracts from embryonic and adult brains were compared for their ability for handling the different types of DNA lesions. Both, embryonic and adult rat brains have capacity for excision of oxidatively modified bases, 8-oxoG and 5-OHU, and for incision of abasic sites (AP) (Englander and Ma, 2006). The decrease in Ogg1 and Ape1 mRNA levels in the adult compared to embryonic brain (E21), was accompanied by a similar decrease in the capacity to excise oxidized guanines from 8-oxoG:C containing synthetic substrates and the capacity to incise the AP(THF):A substrate, ie, Ogg1-like and AP endonuclease-like activity, respectively. In further agreement with mRNA levels, comparable activities were measured in the mature and embryonic rat brain, for excision of the 5-OHU incorporated into a bubble structured substrate (Figure 3).
Figure 3. Base excision/incision activities in embryonic E21 and adult rat brain.

(A) In vitro DNA cleavage assays. Nuclear extracts from cerebra of E21 and adult rats were assembled with substrates containing: i) 8-oxoG:C, targeted by Ogg1 and/or Ogg1-like activities. ii) AP(THF):A, an abasic site targeted by Ape1 and iii) 5-OHU:bubble, 5-OHU in a bubble structure targeted by Neil1 and Neil2. Reaction products were resolved in denaturing polyacrylamide gels and visualized by autoradiography. A) Time points are indicated for each substrate and negative and positive controls are included in external lanes. S, substrate; P, cleavage product. (B) Time courses: For each substrate percent cleavage with time was calculated by plotting Phosphorimager data using the ImageQuant software. Radioactivity values for at least 3 assays for each time point in the course of reaction for either E21 (n=3) or adult cerebrum (n=3) were averaged and plotted as a function of time. (Reprinted from Englander EW, Ma H. Mech Age Dev 2006;127:64–69, with permission).
BER enzymes expression analyses in the brain: in situ hybridization (ISH)
Brain expression profiles of mouse mRNAs for approximately 20,000 genes became recently available from the Allen Institute for Brain Science (Lein et al., 2007). These expression profiles were generated using automated high-throughput procedures and data acquisition analyses for in situ hybridization (ISH). The resultant output has been standardized to provide comprehensive information on distribution and localization of the individual mRNAs in the brain; it reveals regional enrichments and in some cases cell type-specific enriched gene expression. In general, large pools of data generated and analyzed in a uniform and systematic fashion are scarce, yet central for prediction of functional outcomes. In the case of DNA repair genes, availability of comprehensive expression analyses provides a gateway for assessments of the different repair pathways, sub-pathways and redundancy or the relative lack of redundancy of repair processes in the brain. Since this review is focused on the brain capacity for repair of oxidatively damaged DNA via the BER pathway, ISH images presented in Figure 4 are ordered by sequential steps of this process. Images of genes involved in BER, for which ISH failed to detect significant expression, are omitted from the composite figure; such genes are listed in the figure legend.
Figure 4. BER enzymes gene expression patterns in the brain.
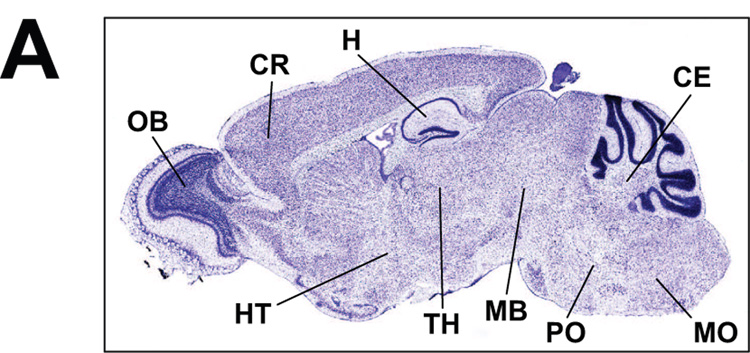

In Situ Hybridization (ISH) images of BER proteins: ISH images visualize distribution and relative levels of the individual BER proteins mRNAs in sagittal sections through the mouse brain. Sectioning is through the center plane and includes cerebrum with detailed central view of hippocampal formation, cerebral cortex, olfactory areas as well as central plane of the cerebellum. (A) Main brain structures are demarcated on a Nissl stained sagittal section: CR, cerebral cortex; H, hippocampus; CE, cerebellum; MO, medulla oblongata; PO, pons; MB, midbrain; TH, thalamus; HT, hypothalamus; OB, olfactory bulb; staining is indicative of neuronal density. (B) ISH images provide qualitative information on localization and relative levels of mRNA for each given gene (pixel intensity color scale ranges from blue/low to orange/high). Images include gene names and are ordered by sequential steps of the BER pathway: glycosylases, single strand break-binding proteins, endonucleases, DNA termini-processing enzymes, polymerases and ligases. Myh, MutY homolog; Tdg, thymine DNA glycosylase; Smug1, single strand selective monofunctional uracil DNA glycosylase; Neil, Nei like; Parp1, poly(ADP-ribose) polymerase 1; Ape1, abasic endonuclease 1; Pnkp, polynucleotide kinase/phosphatase; Pol-β, polymerase β; Fen-1, flap endonuclease 1; Pcna, proliferating cell nuclear antigen; Xrcc1, x-ray repair cross complementation group1. Canonical members of the BER pathway whose expression is undetectable by ISH analyses include: uracil DNA glycosylase (Ung2), N-methylpurine DNA glycosylase (Mpg), endonuclease III E. coli homolog 1 (Nth1), polymerases delta and epsilon, (as well as replicative polymerases alpha and iota). All images were downloaded from the Site, Allen Brain Atlas [Internet]. Allen Institute for Brain Science, © 2007, Seattle (WA).
Overall, ISH images for expression patterns of DNA repair genes in the mouse brain are consistent with data obtained by other methods for RNA, protein and enzymatic activities or immunohistochemical analyses. It must be noted, that occasionally steady state RNA levels captured by ISH are imperfect predictors of the levels of corresponding proteins. Our compilation revealed the strongest positive correlation with other types of assessments in the case of DNA polymerase beta (pol β), which is the major DNA polymerase in the brain and whose high expression in neurons has been documented early on (Waser et al., 1979). In excellent agreement, ISH shows high ubiquitous as well as hippocampal neuron-enriched expression of pol β in the mouse brain (Figure 4). Pol β is central to BER and although polymerases delta and epsilon are implicated in the long-patch gap-filling synthesis process, their brain expression is practically undetectable by ISH. As expected, expression of the replicative DNA polymerase α is also undetectable. On the other hand, lack of agreement with ISH data is noted in the case of the Ogg1, conflicting with evidence for Ogg1-like enzymatic activity in the brain (Englander and Ma, 2006; Imam et al., 2006; Verjat et al., 2000). Although Ogg1-like activity is clearly present in the adult rat brain (Englander and Ma, 2006), the ISH signal is very weak (not shown). Notwithstanding a technical problem or a lack of agreement between RNA and protein levels, excision of 8-oxoG in the brain is likely carried out also by enzymes other than Ogg1. This is consistent with the fact that Ogg1 knockout mouse models exhibit a range of subtle to mild phenotypes (Friedberg and Meira, 2006). ISH profiles of other BER glycosylases are more pronounced, with Myh and Smug1 showing quite ubiquitous expression patterns. We and others have previously reported significant Myh expression in rat and mouse brain, respectively (Englander et al., 2002; Ichinoe et al., 2004) and suggested that in postmitotic cells, Myh may be involved in repair of mtDNA. Both, Tdg and Neil1 on the other hand, exhibit a more localized expression pattern. In fact, in agreement with reported expression of NEIL1 in the S phase, the ISH analysis detects Neil1 in the olfactory region where cell proliferation continues also in the mature brain (Garcia-Verdugo et al., 1998). ISH data for Neil2 are not available at this time. Some examples of DNA glycosylases whose expression by ISH is undetectable include the Ung2, a uracil glycosylase that also shows preference for the S phase in proliferating cells and the N-methylpurine DNA glycosylase (Mpg).
The abasic sites generated by initiating glycosylases are integrated into the BER pathway by APE1 and in the case of NEIL1/2 glycosylases by PNKP. Interestingly Pnkp expression is high and broadly distributed in the brain as revealed by ISH, as well as by our earlier real time RT-PCR analyses (Englander and Ma, 2006). With respect to the subsequent gap filling DNA synthesis step, ISH data suggest that the in the brain gap filling synthesis proceeds mainly through the short patch sub-pathway, since for both Fen-1 and Pcna, ISH signals are very low. Likewise, no ISH signal is detected for polymerases δ and ε, which are associated with the long-patch BER pathway. In further support for predominance of the short patch sub-pathway, both ligase III and Xrcc1 show ubiquitous expression while ligase I, which is associated with the long-patch synthesis is low. Substantial uniform levels of XRCC1 mRNA have been previously reported in most regions of the human brain (Wilson and McNeill, 2007). Interestingly Ape2, which has weak endonuclease and a more significant 3’ exonuclease-phosphodiesterase activity (Burkovics et al., 2006), shows a non-uniform expression profile with enrichment in olfactory lobe and the wall of ventricle where cell proliferation does not cease in the mature brain (Garcia-Verdugo et al., 1998). ISH also reveals a lack of Pcna expression in the brain with signal which is limited only to a layer of proliferating cells in the wall of lateral ventricle. We have previously shown that Pcna positive cells become sparse early on in the neonatal rat brain (Lee et al., 2004).
Does parsimonious BER predispose to neuronal injury and dysfunction?
To what extent somatic mutations may preferentially accumulate in neurons because of limited capacity for timely and accurate repair of endogenous DNA damage is an open question. Likewise, it is not clear to what extent parsimonious repair may exacerbate injurious conditions to precipitate neuronal injury. Interestingly, while an early report suggested that somatic frameshift mutations occur at a high frequency in neurons (Evans et al., 1994), a follow-up work revealed that such frameshifts, which are a hallmark of certain neuropathologies, in fact occur at the level of mRNA while uncompromised wild-type DNA sequences are retained (Van Leeuwen et al., 2000).
Notably, neuronal dysfunction has been linked not only to under-repaired, but also to over-repaired DNA, as demonstrated in models of genetically engineered DNA repair deficiencies and excesses or in models of exogenously induced oxidative DNA damage. Examples include, increased postischemic brain injury in mice deficient in the uracil DNA glycosylase (Endres et al., 2004) and sensitization of dopamine neurons to manganese toxicity in Ogg1 knockout mice (Cardozo-Pelaez et al., 2005). In contrast, a salutary reduction of age-associated expansion of trinucleotide repeats in Huntington Disease model, was reported in mice lacking the Ogg1 protein (Kovtun et al., 2007). Furthermore, folic acid deficiency, which promotes generation of DNA damage (Blount et al., 1997) was reported to potentiate Abeta toxicity and predispose to Alzheimer disease (AD) (Kruman et al., 2002). Increased oxidative damage (Markesbery and Lovell, 2006; Wang et al., 2005) and increased levels of APE1 (Davydov et al., 2003) have been reported in AD brains. In contrast, reduction in brain APE1 protein was noted in a hypoxia/ischemia (Walton et al., 1997) as well as traumatic brain injuries (Lewen et al., 2001), suggesting that reduction in Ape1 might precede neuronal death in both the neonatal and mature brain. However, since the brain Ape1-like incision activity is quite robust (Englander and Ma, 2006), it is not clear to what extent this activity might be in fact significantly diminished in the different settings of injury. Although there is evidence for significant increases in levels of DNA strand breaks following traumatic brain injury (Clark et al., 2001), it is likely that formation of single strand breaks under these conditions does not necessarily result from APE1-mediated generation of intermediate products of the BER process. It is also possible that the noted association between the loss of the multifunctional protein APE1 and neuronal death in certain models of brain injury reflects loss of APE1 function other than that for incision of abasic sites.
With respect to the possibility that nontraditional members of BER may be involved in the repair process (Rass et al., 2007b), it is noteworthy that an inactivating mutation of the enzyme tyrosyl-DNA phosphodiesterase 1 (Tdp1), which generally repairs covalently bound topoisomerase l-DNA complexes, has been linked to spinocerebellar ataxia (Takashima et al., 2002). Defective Tdp1 protein was found to be sequestered into SSB-repair complexes, which may be handled by the BER process. This could link the mutated Tdp1 to impaired processing of SSBs in postmitotic neurons which may contribute to neuronal compromise underlying onset of spinocerebellar ataxia (El-Khamisy et al., 2005; Hirano et al., 2007; Katyal et al., 2007).
Similarly, recent studies identified a role in BER for aprataxin, a member of nucleotide hydrolases family expressed in the brain, which when mutated underlies a certain class of neurological disorders (Ahel et al., 2006; Rass et al., 2007a). Specifically, aprataxin was shown to function in BER by resolving abortive ligation reactions and thereby to have a ‘proof reading’ role at the penultimate step of the repair process. Since the Allen Brain Atlas data for canonical members of the long-patch BER sub-pathway suggest that the long-patch mode of BER is rather unlikely in the brain, aprataxin may compensate in part for the lack of FEN-1 mediated 5’-flap removal, which is integral to long-patch BER. This may occur in the brain when modified/oxidatively damaged DNA ends, which typically would be handled by the long patch sub-pathway, are formed. In support of this possibility, ISH analyses reveal quite a broad aprataxin expression in the brain with significant enrichment in hippocampal neurons.
Perspective
Redundancy of base excision activities at the first step of the mammalian BER process is underscored by the many knockout mice models of BER initiating DNA glycosylases, which confer subtle to mild phenotypes when generated singly or even doubly in some cases. In contrast, knockout models for enzymes involved in the subsequent integrative BER steps, including Ape1, pol-β, ligase 1 and ligase 3, are lethal. To what extent backup systems of mammalian BER might be restricted in postmitotic differentiated cells is not known; however, the new ISH data suggest that not all BER subsets are likely to be equally represented in the brain. Thus, it is plausible that parsimonious nuclear and mitochondrial BER may sensitize neuronal cells to oxidative injuries. Nonetheless, it is also possible that in the brain, limited redundancy of DNA repair processes and specifically of BER sub-pathways might be counterbalanced by specialized neuronal proteins whose roles in augmentation of repair of oxidatively damaged DNA remain to be uncovered.
Acknowledgment
Supported by NIH grants NS039449 and ES014613 and Shriners grant SHG8670 to EWE.
Abbreviations
- 8-oxoG
7,8-dihydro-8-oxoguanine
- OGG1
7,8-dihydro-8-oxoguanine DNA glycosylase
- APE1
apurinic/apyrimidinic endonuclease 1
- BER
base excision repair
- pol β
polymerase beta
- ISH
in situ hybridization
- MYH
MutY DNA glycosylase homolog
- PNKP
polynucleotide kinase phosphatase
- XRCC1
x-ray repair cross complementation group 1
- THF
tetrahydrofuran
- E
embryonic day
- P
postnatal day
- SSB
single strand break
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ. The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkovics P, Szukacsov V, Unk I, Haracska L. Human Ape2 protein has a 3'-5' exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006;34:2508–2515. doi: 10.1093/nar/gkl259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo-Pelaez F, Cox DP, Bolin C. Lack of the DNA repair enzyme OGG1 sensitizes dopamine neurons to manganese toxicity during development. Gene Expr. 2005;12:315–323. doi: 10.3727/000000005783992007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RSB, Chen M, Kochanek PM, Watkins SC, Jin KL, Draviam R, Nathaniel PD, Pinto R, Marion DW, Graham SH. Detection of single- and double-strand DNA breaks after traumatic brain injury in rats: comparison of in situ labeling techniques using DNA polymerase I, the Klenow fragment of DNA polymerase I, and terminal deoxynucleotidyl transferase. J Neurotrauma. 2001;18:675–689. doi: 10.1089/089771501750357627. [DOI] [PubMed] [Google Scholar]
- Davydov V, Hansen LA, Shackelford DA. Is DNA repair compromised in Alzheimer's disease? Neurobiol Aging. 2003;24:953–968. doi: 10.1016/s0197-4580(02)00229-4. [DOI] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, O'Hagan HM, Krueger HM, Hanasoge S, Paulsen MT, Ljungman M. RPA and ATR link transcriptional stress to p53. Proc Natl Acad Sci U S A. 2007;104:12778–12783. doi: 10.1073/pnas.0705317104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- Dianov GL, Parsons JL. Co-ordination of DNA single strand break repair. DNA Repair (Amst) 2007;6:454–460. doi: 10.1016/j.dnarep.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Dogliotti E, Fortini P, Pascucci B, Parlanti E. The mechanism of switching among multiple BER pathways. Prog Nucleic Acid Res Mol Biol. 2001;68:3–27. doi: 10.1016/s0079-6603(01)68086-3. [DOI] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia with axonal neuropathy-1. Neuroscience. 2007;145:1260–1266. doi: 10.1016/j.neuroscience.2006.08.048. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander EW, Hu Z, Sharma A, Lee HM, Wu ZH, Greeley GH. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J Neurochem. 2002;83:1471–1480. doi: 10.1046/j.1471-4159.2002.01259.x. [DOI] [PubMed] [Google Scholar]
- Englander EW, Ma H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech Ageing Dev. 2006;127:64–69. doi: 10.1016/j.mad.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Evans DA, van der Kleij AA, Sonnemans MA, Burbach JP, van Leeuwen FW. Frameshift mutations at two hotspots in vasopressin transcripts in post-mitotic neurons. Proc Natl Acad Sci U S A. 1994;91:6059–6063. doi: 10.1073/pnas.91.13.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair (Amst) 2006;5:189–209. doi: 10.1016/j.dnarep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Garcia-Verdugo JM, Doetsch F, Wichterle H, Lim DA, Alvarez-Buylla A. Architecture and cell types of the adult subventricular zone: in search of the stem cells. J Neurobiol. 1998;36:234–248. doi: 10.1002/(sici)1097-4695(199808)36:2<234::aid-neu10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Hardeland U, Kunz C, Focke F, Szadkowski M, Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007;35:3859–3867. doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002a;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002b;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007;26:4732–4743. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinoe A, Behmanesh M, Tominaga Y, Ushijima Y, Hirano S, Sakai Y, Tsuchimoto D, Sakumi K, Wake N, Nakabeppu Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004;32:477–487. doi: 10.1093/nar/gkh214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJ, Sgouros J, Miller GG, Fakhrai H, Davis EM, Le Beau MM, Weinfeld M. Molecular characterization of a human DNA kinase. J Biol Chem. 1999;274:24187–24194. doi: 10.1074/jbc.274.34.24187. [DOI] [PubMed] [Google Scholar]
- Katyal S, El-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Hu Z, Ma H, Greeley GH, Jr, Wang C, Englander EW. Developmental changes in expression and subcellular localization of the DNA repair glycosylase, MYH, in the rat brain. J Neurochem. 2004;88:394–400. doi: 10.1046/j.1471-4159.2003.02164.x. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lewen A, Sugawara T, Gasche Y, Fujimura M, Chan PH. Oxidative cellular damage and the reduction of APE/Ref-1 expression after experimental traumatic brain injury. Neurobiol Dis. 2001;8:380–390. doi: 10.1006/nbdi.2001.0396. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Lindsey-Boltz LA, Sancar A. RNA polymerase: The most specific damage recognition protein in cellular responses to DNA damage? Proc Natl Acad Sci U S A. 2007;104:13213–13214. doi: 10.1073/pnas.0706316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Mani RS, Fanta M, Karimi-Busheri F, Silver E, Virgen CA, Caldecott KW, Cass CE, Weinfeld M. XRCC1 Stimulates Polynucleotide Kinase by Enhancing Its Damage Discrimination and Displacement from DNA Repair Intermediates. J Biol Chem. 2007;282:28004–28013. doi: 10.1074/jbc.M704867200. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. DNA oxidation in Alzheimer's disease. Antioxid Redox Signal. 2006;8:2039–2045. doi: 10.1089/ars.2006.8.2039. [DOI] [PubMed] [Google Scholar]
- McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortusewicz O, Rothbauer U, Cardoso MC, Leonhardt H. Differential recruitment of DNA Ligase I and III to DNA repair sites. Nucleic Acids Res. 2006;34:3523–3532. doi: 10.1093/nar/gkl492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair (Amst) 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Pettersen HS, Sundheim O, Gilljam KM, Slupphaug G, Krokan HE, Kavli B. Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Res. 2007;35:3879–3892. doi: 10.1093/nar/gkm372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Actions of aprataxin in multiple DNA repair pathways. J Biol Chem. 2007a;282:9469–9474. doi: 10.1074/jbc.M611489200. [DOI] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007b;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- Sleeth KM, Robson RL, Dianov GL. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry. 2004;43:12924–12930. doi: 10.1021/bi0492612. [DOI] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen FW, Hol EM, Hermanussen RW, Sonnemans MA, Moraal E, Fischer DF, Evans DA, Chooi KF, Burbach JP, Murphy D. Molecular misreading in non-neuronal cells. Faseb J. 2000;14:1595–1602. doi: 10.1096/fj.14.11.1595. [DOI] [PubMed] [Google Scholar]
- Verjat T, Dhenaut A, Radicella JP, Araneda S. Detection of 8-oxoG DNA glycosylase activity and OGG1 transcripts in the rat CNS. Mutat Res. 2000;460:127–138. doi: 10.1016/s0921-8777(00)00022-7. [DOI] [PubMed] [Google Scholar]
- Walton M, Lawlor P, Sirimanne E, Williams C, Gluckman P, Dragunow M. Loss of Ref-1 protein expression precedes DNA fragmentation in apoptotic neurons. Brain Res Mol Brain Res. 1997;44:167–170. doi: 10.1016/s0169-328x(96)00291-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Waser J, Hubscher U, Kuenzle CC, Spadari S. DNA polymerase beta from brain neurons is a repair enzyme. Eur J Biochem. 1979;97:361–368. doi: 10.1111/j.1432-1033.1979.tb13122.x. [DOI] [PubMed] [Google Scholar]
- Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumix T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, McNeill DR. Base excision repair and the central nervous system. Neuroscience. 2007;145:1187–1200. doi: 10.1016/j.neuroscience.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- Xu YJ, DeMott MS, Hwang JT, Greenberg MM, Demple B. Action of human apurinic endonuclease (Ape1) on C1'-oxidized deoxyribose damage in DNA. DNA Repair (Amst) 2003;2:175–185. doi: 10.1016/s1568-7864(02)00194-5. [DOI] [PubMed] [Google Scholar]