

Abstract
Treatment of an N-terminal containing His6 tagged insulysin (His6-IDE) with proteinase K led to the initial cleavage of the His tag and linker region. This was followed by C-terminal cleavages resulting in intermediate fragments of ~95 kDa and ~76 kDa and finally a relatively stable ~56 kDa fragment. The ~76 kDa and ~56 kDa fragments exhibited a low level of catalytic activity but retained the ability to bind substrate with a similar affinity as the native enzyme. The kinetics of reaction of the IDE ~76 and ~56 kDa proteolytic fragments with a synthetic fluorogenic substrate produced hyperbolic substrate versus velocity curves, rather than the sigmoidal curve obtained with His6-IDE. The ~76 and ~56 kDa IDE proteolytic fragments were active toward the physiological peptides β-endorphin, insulin, and amyloid β peptide 1–40. Although activity was reduced by a factor of ~103 to 104 with these substrates, the relative activity and the cleavage sites were unchanged. Both the ~76 and ~56 kDa fragments retained the regulatory cationic binding site that binds ATP. Thus the two proteinase K cleavage fragments of IDE retain the substrate and ATP binding sites, but have low catalytic activity, and lose the allosteric kinetic behavior of IDE. These data suggest a role of the C-terminal region of IDE in allosteric regulation.
Insulysin (IDE, insulin degrading enzyme, EC 3.4.22.11) is a 110-kDa zinc metalloendopeptidase first described on the basis of its ability to degrade insulin. The enzyme exhibits an oligomeric structure existing in a dimer-tetramer equilibrium, with the dimer being the predominant species [1]. IDE cleaves insulin on both the A and B chains with major cleavage sites on the A chain between Leu13-Tyr14 and Tyr14-Gln15. Major cleavage sites on the B chain are between Ser9-His10, His10-Leu11, Glu13-Ala14, Try15-Leu16, and Phe25-Try26 [2]. The importance of IDE in regulating insulin levels is apparent from studies of the GK rat model of type II diabetes mellitus. In the GK rat naturally occurring missense mutations in the IDE gene that reduce its enzymatic activity result an elevation in insulin levels [3]. IDE is predominantly localized to the cytosol and peroxisomes, although secreted and plasma membrane forms have been reported [4]. The substrate specificity of IDE is somewhat complex with the enzyme cleaving peptides preferentially at hydrophobic and basic residues [5]. Although it has been suggested that IDE has a preference for peptides that can form β-pleated sheet structures [6], in vitro it effectively cleaves β-endorphin and a number of dynorphin related peptides [7] that do not form such structures. Recent interest in IDE comes from the initial reports of Kurochkin and Goto [8] and that of McDermott and Gibson [9] that IDE can cleave amyloid β-peptides in vitro. It was found that IDE is a major amyloid β-peptide degrading activity in a number of cell lines [10,11].
The importance of IDE in the degradation of amyloid β-peptides in vivo was solidified by studies with IDE deficient mice in which the IDE gene was disrupted [12,13]. Homozygous IDE deficient mice exhibit a statistically significant increase in brain amyloid β-peptide levels, with heterozygous mice having an intermediately level. In addition a number of genetic studies have implicated IDE as being linked to late onset Alzheimer disease [14–18]. However, this link has yet to be unequivocally established and not all genetic studies support this linkage [19,20].
We recently reported that IDE exhibits allosteric kinetic behavior [1]. With the synthetic substrate Abz-GGFLRKHGQ-EDDnp plots of initial velocity versus [S] were sigmoidal exhibiting a Hill coefficient greater than 2.0. In addition when IDE substrates were tested as alternate substrate inhibitors activation rather than inhibition was observed. Interestingly, although the peptide substrate dynorphin B-9 increased the rate of amyloid β-peptide hydrolysis by IDE, no such effect was seen with insulin as a substrate. This makes IDE a potential target for drugs that would selectively increase amyloid β-peptide clearance without affecting insulin levels.
Another form of regulation of IDE activity was first reported by Camberos et al. [21] in which nucleotide triphosphates were found to inhibit insulin hydrolysis by IDE. We showed that the effect of nucleotide triphosphates could be attributed to the triphosphate moiety and result from the binding of the polyanion to a cationic site on the enzyme distinct from the active site [22]. Although polyanions did not effect the hydrolysis of insulin or amyloid β-peptides, they increased the rate of cleavage of smaller dynorphin peptides and thus shift the specificity of the enzyme toward these smaller peptides.
In order to gain an insight into the structure of IDE we have utilized limited proteolysis to determine if structural domains exist. We now report that IDE can be cleaved into a relatively stable fragment of ~56 kDa that retains substrate binding and the polyanion binding site but not homotropic or heterotropic interactions, and has significantly reduced catalytic efficiency.
Methods
β-endorphin was obtained from Multiple Peptide Systems through the National Institute on Drug Abuse Research Tools program. Insulin was purchased from Bachem, USA, while amyloid β-peptide 1–40 was obtained from California Research Peptide Inc. 3′ (or 2′)-O-(2,4,6-trinitrophenyl)adenosine triphosphate (TNP-ATP) was obtained from Molecular Probes (Eugene, OR). Trypsin, chymotrypsin, soybean trypsin inhibitor, and diisopropylfluorophosphate were purchased from Sigma Chemical Co., St. Louis, MO. Staphylococcus aureus protease V8 (proteinase V8) was purchased from Worthington Biochemical Corp, Freehold, NJ. Proteinase K was obtained from Invitrogen. The fluorogenic substrate Abz-GGFLRKHGQ-EDDnp was synthesized as previously described [23].
A rat IDE cDNA, (pECE-IDE), kindly provided by Dr. Richard Roth of Stanford University (Stanford, CA), was subcloned into the baculovirus-derived vector pFASTBAC (Invitogen) through BamHI and XhoI restriction sites such that a His6-affinity tag and a linker region became fused to the N terminus of the protein. The IDE portion of the fusion protein started at methionine 42, which corresponds to the second putative start site in the coding region. Generation of recombinant baculovirus and expression of the recombinant IDE in Sf9 cells was performed according to the manufacturer’s instructions. Cells were harvested after three days and frozen. For the purification of recombinant IDE, a 1:10 (w/v) suspension of frozen Sf9 cells was prepared in 20 mM Tri-HCl, pH 7.4, containing 1 mM dithiothreitol. The suspension was sonicated 12 times, each burst for 1 sec, using a Branson sonifier (setting 3 at 30%) and then centrifuged at 15,000 rpm for 45 min to pellet cell debris and membranes. The supernatant containing recombinant rat IDE was loaded onto a 2 ml His-Select HC nickel affinity gel (Sigma) that had been equilibrated with 20 mM Tris-HCl, pH 7.4, containing 1 mM dithiothreitol. After extensive washing of the column with starting buffer, and then with 20 mM imidazole buffer, pH 7.4, the enzyme was eluted with 100 mM imidazole buffer, pH 7.4. The enzyme was then dialyzed against 50 mM Tris-HCl, pH 7.4 containing 20% glycerol and stored at −80°C until use. Under these conditions the enzyme is stable for at least 12 months. It was previously established that the hexa-histidine and linker region did not affect IDE activity [1] and therefore was not removed in these studies.
The proteinase K digestion fragments of IDE were expressed in insect Sf9 cells as N-terminal hexa-histidine fusion proteins. To accomplish this the cDNA for rat IDE in the vector pFastBacHTb [21] was used as a template for PCR to generate the appropriate cDNA. For the expression of the 56 kDa form of IDE (amino acids 42 to 534) PCR primers:
| 5-gtgtctagagactcgacccaaccaag-3′ and 5′-gtgctcgagtcaatttgtaggaatgaatt-3′ | |
| Xba1 | Xho1 stop |
were used to generate an ~1.1 kb fragment which replaced a 2.4 kb Xba1-Xho1 fragment from the rat IDE cDNA in pFastBacHTb. Similarly, for the expression of the 76 kDa form of IDE (amino acids 42 to 705) PCR primers:
| 5-tctgctagcaggcctgagctatgatc-3′ and 5′-gtcctcgagtcaatcgagggcttctttta-3′ | |
| Nhe1 | Xho1 stop |
were used to generate an ~280 bp Nhe1-Xho1 fragment containing a stop codon which was used to replace a 1.2 kb Nhe1-Xho1 fragment from the rat IDE cDNA in pFastBacHTb.
We also generated two forms of His6-IDE in which we inserted a second TEV protease site (ENLYFQ/G) to produce the 76 and 56 kDa forms in situ while retaining a stoichiometric amount of the C-terminal fragment.
Proteolysis by trypsin and proteinase V8 was performed in 20 mM potassium phosphate buffer, pH 7.3. With the trypsin reaction 1 mM CaCl2 was included, and adding a 10-fold molar excess of soybean trypsin inhibitor terminated the reaction. Proteolysis by proteinase V8 was terminated with 1 mM diisopropylfluorophosphate. Chymotrypsin hydrolysis was conducted in 100 mM potassium phosphate buffer, pH 7.3. The reaction was terminated by addition of PMSF to 0.5 mM plus TLCK to 0.05 mM. The proteinase K digestion reaction was terminated by addition of PMSF to 1 mM.
The N-terminal sequence of the stable proteinase K generated IDE fragments were determined by automated Edman degradation performed at the Protein Structure Core at the University of Nebraska Medical Center. The molecular weights of proteinase K generated IDE fragments were determined using a gold target with a Ciphergen PBSIIc linear MALDI-TOF mass spectrometer calibrated with peptide standards. This analysis was done at the Proteomics Core of the University of Kentucky Center on Structural Biology, which is supported in part by grant P20RR20171 from the NIH/NCRR.
IDE activity assays were conducted in reaction mixtures containing 10 μM Abz-GGFLRKHGQ-EDDnp in 50 mM Tris-HCl buffer, pH 7.4. Peptide bond cleavage resulted in an increase in fluorescence that was followed on a SpectraMax Gemini XS fluorescence plate reader at an excitation wavelength of 318 nm and an emission wavelength of 419 nm.
The rate of cleavage of insulin, β-endorphin, and amyloid β-peptide 1–40 was measured in reaction mixtures containing 10 μM peptide in 50 mM Tris-HCl buffer, pH 7.4. The rate of hydrolysis was quantified by following the decrease in the peak area of the substrate by HPLC. A C4 reverse phase column and a linear gradient from 0.1% trifluoroacetic acid in 95% water/5% acetonitrile to 0.1% trifluoroacetic acid in 50% water/50% acetonitrile was employed. Peptides were followed by their absorbance at 214 nm and quantified from their peak area.
Analytical ultracentrifugation was performed at 4.0 ± 0.1°C in a Beckman XL-A centrifuge using an AN 60 Ti rotor as previously described [24]. Scans were obtained at 280 nm with a step size of 0.001 cm. When scans made 6 h apart were indistinguishable (~20 hr) the approach to equilibrium was considered to be complete. Five scans were averaged for each sample at each rotor speed and analyzed as previously described [24].
The binding of TNP-ATP to IDE and its proteolytic fragments was measured in 50 mM Tris-HCl buffer, pH 7.4 using a Perkin Elmer LS55 Luminescence spectrometer.
Results
In an attempt to provide insights into the domain structure of IDE we conducted limited proteolysis experiments. Although an N terminally His6-tagged IDE was not cleaved by trypsin, proteinase V8, or chymotrypsin at molar ratios of up to 1:10 protease to IDE, it was cleaved by proteinase K into discreet bands. As shown in Figure 1A, over a 24 hr period proteinase K at a 1/200 molar ratio produced the sequential cleavage of the ~110 kDa His6-IDE to produce intermediate fragments of ~105 kDa and ~95 kDa, followed by the formation of a ~73–76 kDa fragment and finally a relatively stable ~54–57 kDa fragment. The activity declined over this time period to ~1% of the initial enzyme activity, figure 1B. We determined the kinetics of cleavage of the synthetic substrate Abz-GGFLRKHGQ-EDDnp at 0 and 24 hrs of digestion and found the substrate versus velocity curves changed from sigmoidal at zero time (Hill coefficient = 2.27) to nearly hyperbolic at 24 hrs (Hill coefficient = 1.38).
Figure 1. Proteinase K digestion of IDE.
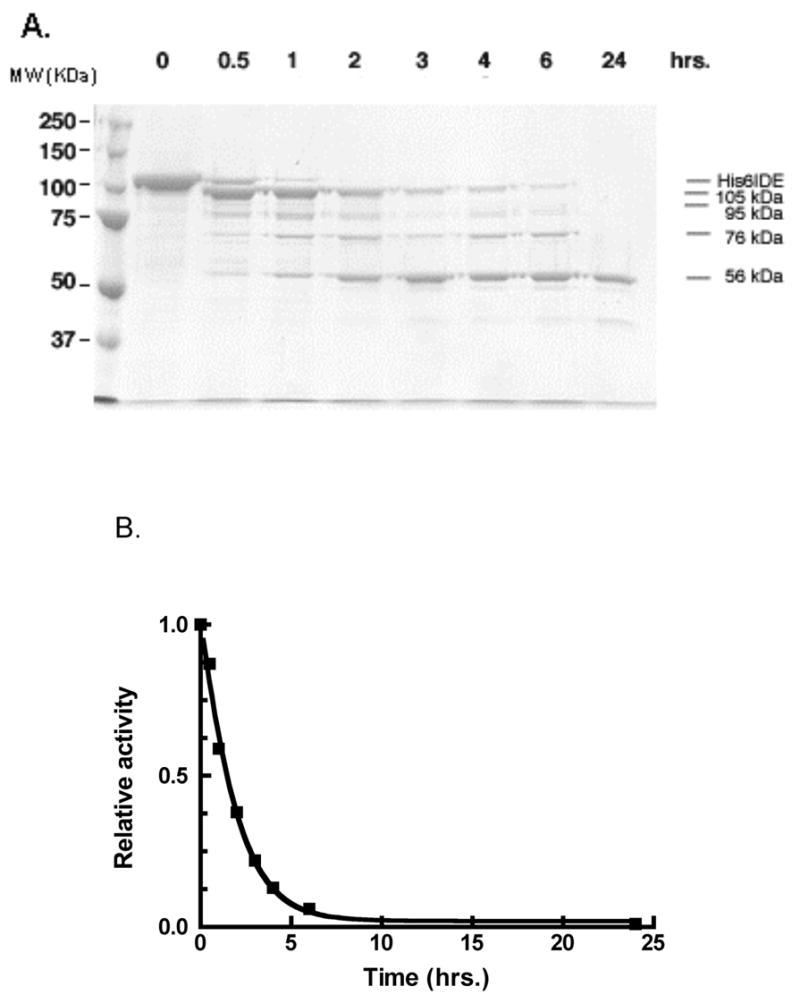
A. IDE (100 μg) in 20 mM potassium phosphate buffer, pH 7.3, was treated with 0.3 μg of proteinase K in a 250 μL reaction mixture. At the time periods indicated an aliquot was withdrawn to which was added PMSF to a final concentration of 0.8 mM to terminate digestion. This aliquot was subjected to SDS-PAGE.
B. IDE activity measurements with 10 μM Abz-GGFLRKHGQ-EDDnp as substrate measured as a function of time of protease K digestion.
To identify where in the IDE molecule cleavage occurred, the 54–57 and 73–76 kDa proteinase K digestion fragments were excised from a gel and subjected to N-terminal sequencing. This procedure yielded the sequence X-N-P-A-I-Q-K for the 54–57 kDa fragment and N-N-P-A-I for the 70–73 kDa fragment. These sequences correspond to cleavage at the methionine that forms the junction between the linker region of the hexahistidine fusion protein and the N-terminal sequence of IDE, Figure 2. It should be noted that we used methionine 42 as the first amino acid for IDE since it appears that this methionine represents the major start of translation for the enzyme [5]. Thus both the stable 54–57 kDa and the intermediate 73–76 kDa proteinase K digestion products resulted from cleavages toward the C-terminal part of the enzyme. In order to obtain a more precise measure of the proteinase K cleavage sites, the IDE fragments were isolated by molecular sieve chromatography and their molecular weight determined by mass spectrometry. A value of 56,601 Da was obtained for the smaller more stable fragment and a value of 76,719 Da was obtained for the larger intermediate fragment, thus we refer to these enzyme forms as IDE56 and IDE76, respectively.
Figure 2.
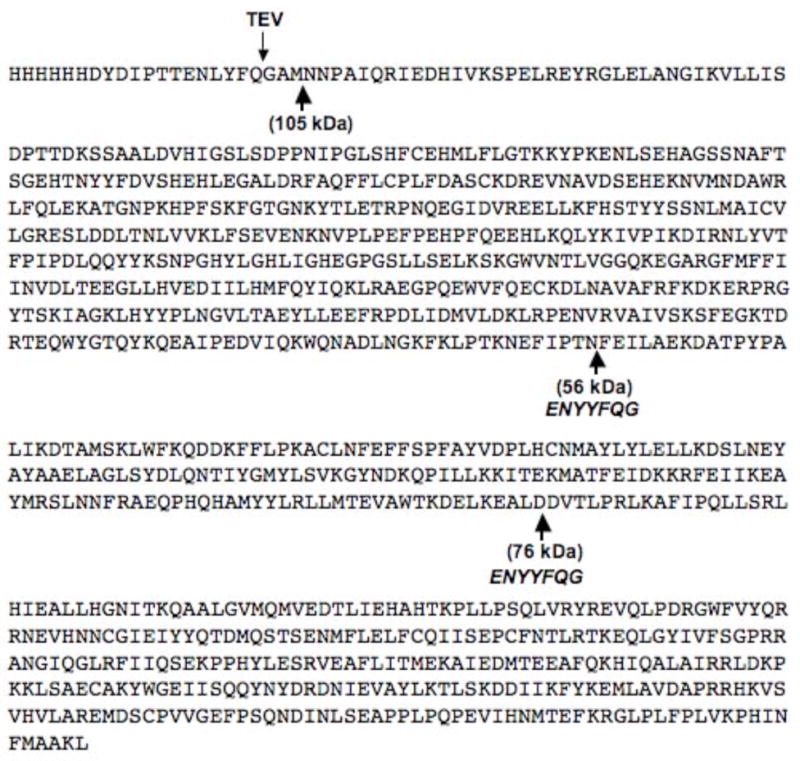
The sequence of His6-IDE showing the proteinase K cleavage sites under the sequence and the position and sequence where additional TEV cleavage sites were inserted. The arrow on the top shows where TEV cleavage occurs.
Using the mass spectral molecular weights we generated truncated forms of IDE that corresponded to IDE56 and IDE76 (Figure 2) as hexahistidine fusion proteins in Sf9 cells using the baculovirus expression system. These were purified to homogeneity on a His-select nickel affinity resin. The activity of these IDE fragments was verified with the fluorogenic substrate Abz-GGFLRKHGQ-EDDnp. As noted during the analysis of the protease K digestion reaction at 24 hrs, the sigmoidal substrate versus velocity curve seen for His6-IDE, appeared as a hyperbolic curve for IDE56, Figure 3. Although not shown a hyperbolic curve was also obtained for IDE76, A summary of the kinetic data with Abz-GGFLRKHGQ-EDDnp as substrate is given in Table 1. It can be seen from this data that both IDE56 and IDE76 retained the ability to bind substrate with the same or increased affinity as the untreated enzyme. In contrast, both of the proteolytic fragments retained very low catalytic activity as evidenced by kcat values that were ~1% of that of the wild type enzyme. We also generated IDE56 (and IDE76) from a full-length construct in which we inserted TEV protease sites in the appropriate positions, Figure 2. Although the IDE56 (and IDE76) form was produced by TEV cleavage from full length IDE, this procedure did not produce a more active enzyme nor were the allosteric properties retained.
Figure 3. Comparison of the kinetics of wild type IDE to IDE56 and IDE76.
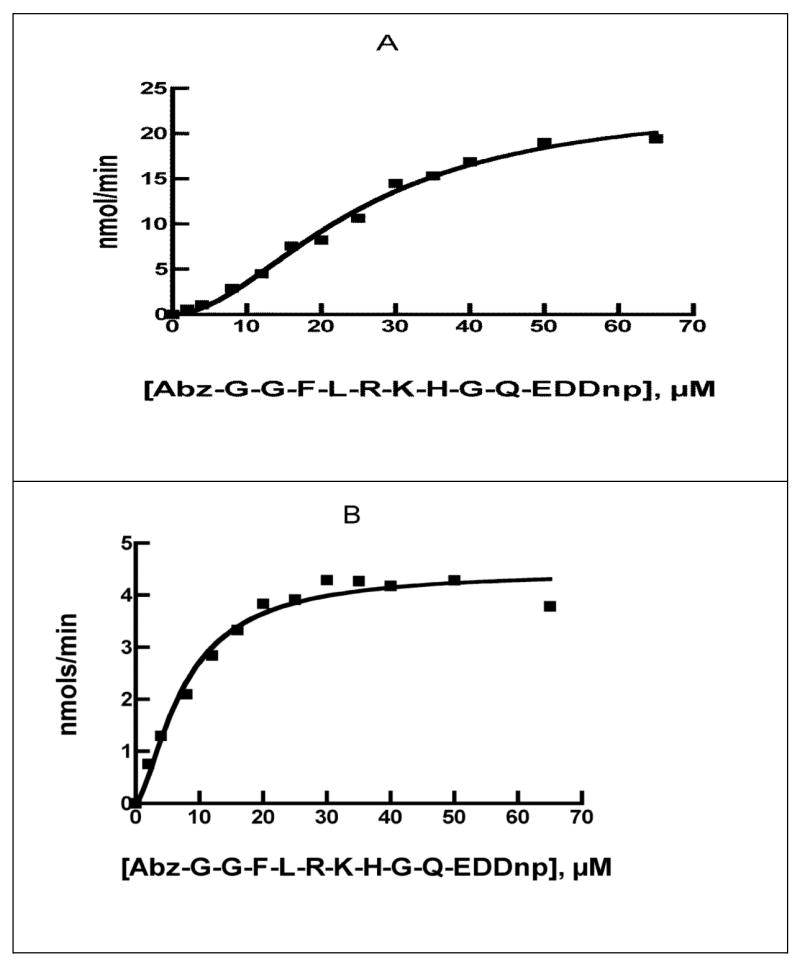
Kinetics of the reaction of 0.50 μg of wild-type IDE (A) or 10 μg of the 56 kDa form of IDE, IDE56 (B) with Abz-GGFLRKHGQ-EDDnp in 50 mM Tris-HCl buffer, pH7.4.
Table 1.
Kinetics for the reaction of IDE and its proteinase K fragments with Abz-GGFLRKHGQ-EDDnp.
| K 5 or Km (μM) | kcat (min−1) | Hill Coefficient | |
|---|---|---|---|
| His6-IDE | 25.4 ± 2.4 | 5,590 ± 402 | 1.9 ± 0.2 |
| IDE56 | 5.9 ± 0.4 | 43 ± 2 | 1.2 ± 0.1 |
| IDE76 | 7.5 ± 0.9 | 46 ± 1 | 1.2 ± 0.2 |
We further tested the activity of IDE56 and IDE76 with respect to the physiological peptides β-endorphin, insulin, and amyloid β peptide 1–40. The results of this analysis are summarized in Table 2. It can be seen that in each case the residual activity is reduced by a factor of ~103 to 104. However, the relative activity of the two IDE truncated forms toward the physiological peptides is unchanged. Although not shown the cleavage sites are also unchanged.
Table 2.
Comparison of the reaction of wild type IDE to IDE56 and IDE76 with physiological peptides.
| Rate of cleavage (nmols/hr/nmol IDE) | |||
|---|---|---|---|
| β-endorphin | amyloid β peptide 1–40 | Insulin | |
| His6-IDE | 18,374 | 4,105 | 396 |
| IDE56 | 3.54 | 0.62 | 0.10 |
| IDE76 | 0.4 | 0.27 | 0.06 |
| Ratio His6-IDE/IDE56 | 5.2×103 | 6.6×103 | 4.0×103 |
| Ratio His6-IDE/IDE76 | 45.9×103 | 15.2×103 | 6.6×103 |
Reactions were conducted in 0.15 ml reaction mixtures containing 50 mM Tris buffer, pH 7.4, 10 μM of peptide at 37°C. For the reaction of wild type IDE with β-endorphin 25 ng of protein was used, while 50 ng of protein was used for the reactions with amyloid β peptide 1–40 and insulin. Reactions were incubated for 15 to 30 min. For the reaction of IDE56, 10 μg of protein was used for all three peptides, with reaction times of 2 h to 17h. For the reaction of IDE76, 20 μg of protein was used for all three peptides, with reaction times of 5 h to 17 h. The cleavage of each peptide was followed by the disappearance of the substrate peak by HPLC. Data were quantified by the change in substrate peak area.
Substrate versus velocity curves for IDE56 (and IDE76), with Hill coefficients close to 1 indicates that IDE56 (and IDE76) do not retain the homotropic allosteric properties of His6-IDE (or native IDE). We further tested for the presence of heterotropic activation by measuring the effect of dynorphin B-9 on the hydrolysis of the fluorogenic substrate Abz-GGFLRKHGQ-EDDnp. As previously shown with wild type IDE, dynorphin B-9 acts as an activator of Abz-GGFLRKHGQ-EDDnp hydrolysis [1]. Rather than produce activation, dynorphin B-9 acted as a weak inhibitor of the IDE56 dependent hydrolysis of Abz-GGFLRKHGQ-EDDnp exhibiting a Ki value of 140 μM.
Since IDE exists predominantly as a dimer we determined whether the IDE56 form retained this subunit structure. Sedimentation equilibrium analysis showed that IDE56 exists predominantly as a monomer, with some tetrameric species present. However, no dimeric IDE56 was detected.
We have previously shown that IDE contains a cationic regulatory site distinct from the active site [22] that binds the triphosphate moiety of nucleotide triphosphates and free triphosphate. Binding at this site increases the rate of hydrolysis of small peptides of up to ~13 amino acids, but has no effect on the rate of hydrolysis of large peptide substrates such as insulin. Using Abz-GGFLRKHGQ-EDDnp as substrate we tested whether the IDE56 and IDE76 dependent reactions were affected by ATP. As shown in Figure 4, IDE56 retained the ability to be activated by ATP, although to a considerably lesser extent that native IDE. The activation constant for ATP, KAATP, was determined to be 1 mM for IDE56, which can be compared to a value of 1.4 mM obtained for native IDE [22]. ATP maximally increased the rate 6.6 fold, which is considerably less than the 40–80 fold increase seen with wild type IDE. In contrast to the effect of ATP on IDE56, we observed little if any effect of ATP on the IDE76 dependent cleavage of Abz-GGFLRKHGQ-EDDnp, Figure 4.
Figure 4. Activation of IDE56 and IDE76 by ATP.

Activity was determined in 50 mM Tris-HCl buffer, pH 7.4, with 10 μM Abz-GGFLRKHGQ-EDDnp as substrate and the indicated concentration of ATP. Closed squares is IDE56 and closed triangles is IDE76
In order to directly detect ATP binding, we used the fluorescent ATP analog 3′ (or 2′)-O-(2,4,6-trinitrophenyl)adenosine triphosphate (TNP-ATP). Binding of TNP-ATP to protein has been shown to result in an increase in its fluorescence due a change in environment [25]. We first tested TNP-ATP for its ability to increase the rate of hydrolysis of Abz-GGFLRKHGQ-EDDnp with wild type IDE and its two truncated forms. In agreement with the data using ATP, we found that 10 μM TNP-ATP increased the rate of hydrolysis of 10 μM Abz-GGFLRKHGQ-EDDnp ~125 fold (4 mM ATP increased the rate in this experiment 80 fold). With IDE56 the rate was increased 3.6 fold (ATP increased the rate in this experiment 3.9 fold). However there was no change in the IDE76 reaction with either ATP or TNP-ATP.
The effect of these IDE forms on TNP-ATP fluorescence is shown in Figure 5. It can be seen that TNP-ATP fluorescence is increased in the presence of 3 μM His6-IDE. Although TNP fluorescence is also increased in the presence of 3 μM IDE56, the magnitude of the change is considerably less. Quite surprising is the finding that the largest change in TNP-ATP fluorescence occurs in the presence of 3 μM IDE76 demonstrating that the anionic binding site is in fact retained in IDE76. It is also worth noting that in each case the binding of TNP-ATP shifts the fluorescence maxima to a lower wavelength indicating that the protein bound TNP-ATP is in a more hydrophobic environment [26].
Figure 5. Binding of 3′(2′)-o-trinitrophenyl-adenosin 5′-triphosphate (TNP-ATP) to wild type IDE and to IDE56 and IDE76.
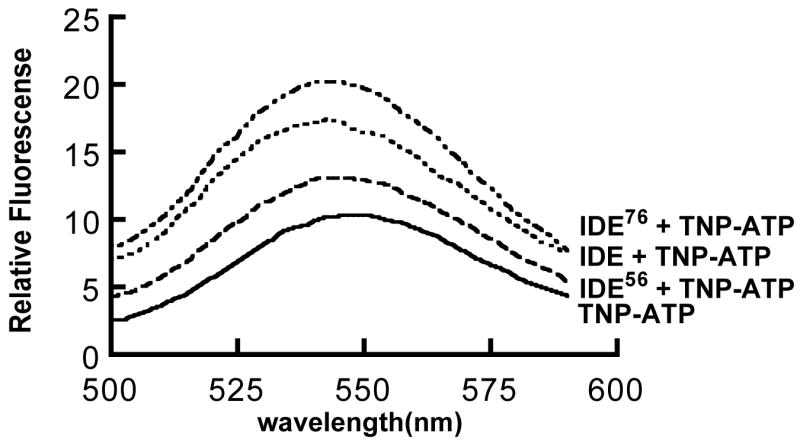
Reaction mixtures (4 ml) contained 50 mM Tris-HCl buffer, pH 7.4, 10 μM TNP-ATP, and 3 μM of wild type IDE or IDE56 or IDE76. Binding of TNP-ATP was measured by the quenching of TNP-ATP fluorescence using an excitation wavelength of 403 nm and an emission wavelength of 547 nm.
Discussion
Although IDE is fairly resistant to proteolysis by trypsin, chymotrypsin and proteinase V8, it can be cleaved by proteinase K. Initially the hexahistidine and linker region including the first methionine are cleaved leaving the N-terminus of IDE indicating that the junction between IDE and the linker region is unstructured. This is followed by C-terminal cleavages to generate two 95 and 76 kDa fragments and finally another C-terminal cleavage to produce a relatively stable 56 kDa species. Since proteinase K is a broad spectrum serine protease capable of cleaving IDE in a number of places, the finding of discreet proteolytic fragments suggests there are several unstructured regions of the enzyme within the C-terminal domains and a protease resistant ~56 kDa core N-terminal domain.
The intermediate 76 kDa and stable 56 kDa proteinase K cleavage fragments retain a low but detectable level of catalytic activity toward the synthetic fluorogenic substrate Abz-GGFLRKHGQ-EDDnp, but bind this substrate with the same or even higher affinity as native IDE. The activity of the proteinase K derived IDE fragments toward the physiological peptides β-endorphin, insulin, and amyloid β–peptide 1–40, are considerably lower than that towards the synthetic fluorogenic substrate. This might be interpreted to suggest that the loss of the C-terminal region may affect the ability of the enzyme to make extended binding interactions utilized in catalysis. It is likely that this results from conformational differences between the full-length enzyme and its proteolytic fragments. Thus the proteinase K fragments fold to produce a native-like substrate binding site, but lose catalytic efficiency.
The proteinase K cleavage fragments exhibit hyperbolic substrate-velocity response curves in contrast to the sigmoidal kinetics seen with native IDE. The ability of an alternate substrate such as dynorphin B-9 to serve as an activator is lost in the 56 and 76 kDa forms consistent with the change in kinetic properties from allosteric to classical Michaelis-Menton. In fact dynorphin B-9 acts as a typical, although weak, alternate substrate inhibitor with the proteinase K 56 kDa cleavage fragment as would be expected for an enzyme exhibiting classical Michaelis-Menton kinetics. This would suggest that the C-terminal part of the enzyme is required for its allosteric kinetic behavior and might be responsible for either transmitting conformational changes between subunits or for maintaining subunit-subunit interactions. Although native IDE exists primarily as a dimer [1], IDE56 exists predominantly as a monomer. This suggests that the C-terminal region of IDE contains its dimerization domain, and further suggest that the dimer is required for allosteric behavior.
Although the allosteric kinetic behavior of IDE is lost, the 56 and 76 kDa proteinase K cleavage fragments retain the allosteric cationic site, which binds polyanions. The 56 kDa proteinase K cleavage fragment retains the ability to bind ATP and to be activated by ATP binding, although the extent of activation appears considerably less than with native IDE. The affinity of ATP for this fragment is the same as for wild type IDE. Using the fluorescent ATP analog TNP-ATP, binding was demonstrated directly. This would suggest that activation by ATP and other polyanions may involve conformational changes within IDE, and does not require oligomerization. Surprisingly, the rate of substrate cleavage by the larger 76 kDa proteinase K cleavage fragment is unaffected by ATP, but this IDE form does bind ATP as shown by TNP-ATP fluorescence enhancement. Thus we might speculate that the ~20 kDa of C-terminal sequence found in the 76 kDa proteinase K cleavage fragment relative to the 56 kDa proteinase K cleavage fragment does not fold properly to permit transmittal of an ATP induced conformational change.
In summary, the 56 and 76 kDa proteinase K cleavage fragments of IDE retain an intact substrate binding site, but exhibit low catalytic activity. These IDE fragments lose the allosteric kinetic properties of IDE, but do retain the anion binding site. These results suggest that the C-terminal region of IDE is needed for dimerization, a structure that is likely linked to its allosteric behavior. These smaller IDE fragments could prove useful for determining the structure of the substrate and anion binding sites.
While this paper was in revision Li et al. [27] published a communication in which they reported that IDE expressed in E. coli could be cleaved into ~55 kDa and ~57 kDa fragments by trypsin. The trypsin cleavage site was near the site generating IDE56 in this study. From the analysis of the trypsin cleavage products Li et al. [27] concluded as we have, that the C-terminal region of IDE is involved in oligomerization.
Acknowledgments
We thank Dr. Carol Beach for performing the mass spectral analysis. This work was supported in part by grants DA 02243 from the National Institute on Drug Abuse, AG 24899 from the National Institute on Aging, NS 46517 from the National Institute on Neurological Disorders and Stroke, and P20 RR02017 from the NCRR.
Abbreviations
- IDE
insulin-degrading enzyme
- TNP-ATP
3′(2′)-O-(2,4,6-trinitrophenyl)-adenosine triphosphate
- Abz
2-aminobenzoyl
- EDDnp
ethylenediamine-2, 4-dinitrophenyl
References
- 1.Song ES, Juliano MA, Juliano L, Hersh LB. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. J Biol Chem. 2003;278:49789–49794. doi: 10.1074/jbc.M308983200. [DOI] [PubMed] [Google Scholar]
- 2.Duckworth WC. Insulin degradation: mechanisms, products, and significance. Endocrinol Rev. 1988;9:319–345. doi: 10.1210/edrv-9-3-319. [DOI] [PubMed] [Google Scholar]
- 3.Fakhrai-Rad H, Nikoshkov A, Kamel A, Fernstrom M, Zierath JR, Norgren S, Luthman H, Galli J. Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Hum Mol Genet. 2000;9:2149–2158. doi: 10.1093/hmg/9.14.2149. [DOI] [PubMed] [Google Scholar]
- 4.Hersh LB. Peptidases, proteases and amyloid beta-peptide catabolism. Curr Pharm Des. 2003;9:449–454. doi: 10.2174/1381612033391676. [DOI] [PubMed] [Google Scholar]
- 5.Song ES, Mukherjee A, Juliano MA, Pyrek JS, Goodman JP, Jr, Juliano L, Hersh LB. Peptidases, proteases and amyloid beta-peptide catabolism. J Biol Chem. 2001;276:1152–1155. doi: 10.1074/jbc.M008702200. [DOI] [PubMed] [Google Scholar]
- 6.Kurochkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci. 2001;26:421–425. doi: 10.1016/s0968-0004(01)01876-x. [DOI] [PubMed] [Google Scholar]
- 7.Safavi A, Miller BC, Cottam L, Hersh LB. Peptidases, proteases and amyloid beta-peptide catabolism. Biochemistry. 1996;35:14318–14325. [Google Scholar]
- 8.Kurochkin IV, Goto S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 9.McDermott JR, Gibson AM. Degradation of Alzheimer’s beta-amyloid protein by human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochem Res. 1997;22:49–56. doi: 10.1023/a:1027325304203. [DOI] [PubMed] [Google Scholar]
- 10.Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem. 1997;272:6641–6646. doi: 10.1074/jbc.272.10.6641. [DOI] [PubMed] [Google Scholar]
- 11.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny M, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 12.Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science. 2000;290:2302–2303. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- 15.Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM. Susceptibility locus for Alzheimer’s disease on chromosome 10. Science. 2000;290:2304–2305. doi: 10.1126/science.290.5500.2304. [DOI] [PubMed] [Google Scholar]
- 16.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science. 2000;290:2303–2304. doi: 10.1126/science.290.5500.2303. [DOI] [PubMed] [Google Scholar]
- 17.Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA, Jr, Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ait-Ghezala G, Abdullah L, Crescentini R, Crawford F, Town T, Singh S, Richards D, Duara R, Mullan M. Confirmation of association between D10S583 and Alzheimer’s disease in a case--control sample. Neurosci Lett. 2002;325:87–90. doi: 10.1016/s0304-3940(02)00243-4. [DOI] [PubMed] [Google Scholar]
- 19.Abraham R, Myers A, Wavrant-DeVrieze F, Hamshere ML, Thomas HV, Marshall H, Compton D, Spurlock G, Turic D, Hoogendoorn B, Kwon JM, Petersen RC, Tangalos E, Norton J, Morris JC, Bullock R, Liolitsa D, Lovestone S, Hardy J, Goate A, O’Donovan M, Williams J, Owen MJ, Jones L. Substantial linkage disequilibrium across the insulin-degrading enzyme locus but no association with late-onset Alzheimer’s disease. Hum Genet. 2001;109:646–652. doi: 10.1007/s00439-001-0614-1. [DOI] [PubMed] [Google Scholar]
- 20.Boussaha M, Hannequin D, Verpillat P, Brice A, Frebourg T, Campion D. Polymorphisms of insulin degrading enzyme gene are not associated with Alzheimer’s disease. Neurosci Lett. 2002;329:121–123. doi: 10.1016/s0304-3940(02)00586-4. [DOI] [PubMed] [Google Scholar]
- 21.Camberos MC, Perez AA, Udrisar DP, Wanderley MI, Cresto JC. ATP inhibits insulin-degrading enzyme activity. Exp Biol Med (Maywood) 2001;226:334–341. doi: 10.1177/153537020122600411. [DOI] [PubMed] [Google Scholar]
- 22.Song ES, Juliano MA, Juliano L, Fried MG, Wagner SL, Hersh LB. ATP effects on insulin-degrading enzyme are mediated primarily through its triphosphate moiety. J Biol Chem. 2004;279:54216–20. doi: 10.1074/jbc.M411177200. [DOI] [PubMed] [Google Scholar]
- 23.Csuhai E, Juliano MA, Pyrek JS, Harms AC, Juliano L, Hersh LB. New fluorogenic substrates for N-arginine dibasic convertase. Anal Biochem. 1999;269:149–154. doi: 10.1006/abio.1999.4033. [DOI] [PubMed] [Google Scholar]
- 24.Song ES, Daily A, Fried MG, Juliano MA, Juliano L, Hersh LB. Mutation of active site residues of insulin-degrading enzyme alters allosteric interactions. J Biol Chem. 2005;280:17701–17706. doi: 10.1074/jbc.M501896200. [DOI] [PubMed] [Google Scholar]
- 25.Hiratsuka T. Fluorescent and colored trinitrophenylated analogs of ATP and GTP. Eur J Biochem. 2003;270:3479–3485. doi: 10.1046/j.1432-1033.2003.03748.x. [DOI] [PubMed] [Google Scholar]
- 26.Huang SG, Weisshart K, Fanning E. Characterization of the nucleotide binding properties of SV40 T antigen using fluorescent 3′ (2′)-O-(2,4,6-trinitrophenyl)adenine nucleotide analogues. Biochemistry. 1998;37:15336–15344. doi: 10.1021/bi981094g. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Kuo WL, Yousef M, Rosner MR, Tang WJ. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochem Biophys Res Commun. 2006;343:1032–1037. doi: 10.1016/j.bbrc.2006.03.083. [DOI] [PubMed] [Google Scholar]