

Abstract
The structural mechanisms by which proteins have evolved new functions are known only indirectly. We report x-ray crystal structures of a resurrected ancestral protein—the ∼450 million year-old precursor of vertebrate glucocorticoid (GR) and mineralocorticoid (MR) receptors. Using structural, phylogenetic, and functional analysis, we identify the specific set of historical mutations that recapitulate the evolution of GR’s hormone specificity from an MR-like ancestor. These substitutions repositioned crucial residues to create new receptor-ligand and intra-protein contacts. Strong epistatic interactions occur because one substitution changes the conformational position of another site. “Permissive” mutations—substitutions of no immediate consequence, which stabilize specific elements of the protein and allow it to tolerate subsequent function-switching changes—played a major role in determining GR’s evolutionary trajectory.
A central goal in molecular evolution is to understand the mechanisms and dynamics by which changes in gene sequence generate shifts in function and therefore phenotype (1, 2). A complete understanding of this process requires analysis of how changes in protein structure mediate the effects of mutations on function. Comparative analyses of extant proteins have provided indirect insights into the diversification of protein structure (3-6), and protein engineering studies have elucidated structure-function relations that shape the evolutionary process (7-11). To directly identify the mechanisms by which historical mutations generated new functions, however, it is necessary to compare proteins through evolutionary time.
Here we report the empirical structures of an ancient protein, which we “resurrected” (12) by phylogenetically determining its maximum likelihood sequence from a large database of extant sequences, biochemically synthesizing a gene coding for the inferred ancestral protein, expressing it in cultured cells, and determining the protein’s structure by x-ray crystallography. Specifically, we investigated the mechanistic basis for the functional evolution of the glucocorticoid receptor (GR), a hormone-regulated transcription factor present in all jawed vertebrates (13). GR and its sister gene, the mineralocorticoid receptor (MR), descend from duplication of a single ancient gene, the ancestral corticoid receptor (AncCR), deep in the vertebrate lineage ∼450 million years ago (Fig. 1A, ref. (13)). GR is activated by the adrenal steroid cortisol and regulates stress response, glucose homeostasis, and other functions (14). MR is activated by aldosterone in tetrapods and by deoxycorticosterone (DOC) in teleosts to control electrolyte homeostasis, kidney and colon function, and other processes (14). MR is also sensitive to cortisol, though considerably less so than to aldosterone and DOC (13,15). Previously, AncCR was resurrected and found to have MR-like sensitivity to aldosterone, DOC, and cortisol, indicating that GR’s cortisol specificity is evolutionarily derived (13).
Fig. 1.

A) Functional evolution of corticosteroid receptors. Dose-response curves show transcription of a luciferase reporter gene by extant and resurrected ancestral receptors with varying doses (in log-M) of aldosterone (green), deoxycorticosterone (DOC, orange), and cortisol (purple). Black box indicates evolution of cortisol specificity. The number of sequence changes on each branch is shown (aa, replacement; Δ deletion). Bars, SEM of three replicates. Node dates from the fossil record (19, 20). For complete phylogeny and sequences, see Figs S10 and Table S5. B) Crystal structure of the AncCR ligand-binding domain (LBD) with bound aldosterone (green, with red oxygens). Helices are labeled; AF-H, activation-function helix. C) AncCR’s ligand-binding pocket. Side chains (<4.2 Å from bound ligand) are superimposed from crystal structures of AncCR with aldosterone (green), DOC (orange), and cortisol (purple). Oxygen and nitrogen atoms are red and blue; dashed lines, hydrogen bonds. Arrows show C11, C17, and C18 positions, which differ among the hormones.
To identify the structural mechanisms by which GR evolved this new function, we used x-ray crystallography to determine the structures of the resurrected AncCR ligand-binding domain in complex with aldosterone, DOC, and cortisol (16) at 1.9, 2.0, and 2.4 Å resolution, respectively (Table S1). All structures adopt the classic active conformation for nuclear receptors (17), with unambiguous electron density for each hormone (Fig. 1B, Figs. S1, S2). AncCR’s structure is extremely similar to the human MR (rmsd=0.9Å for all backbone atoms), and, to a lesser extent, to the human GR (rmsd=1.2Å). The network of hydrogen-bonds supporting activation in the human MR (18) is present in AncCR, indicating that MR’s structural mode of action has been conserved for >400 my (Fig. S3).
Because aldosterone evolved only in the tetrapods, tens of millions of years after AncCR, that receptor’s sensitivity to aldosterone was surprising (13). The AncCR-ligand structures indicate that the receptor’s ancient response to aldosterone was a structural by-product of its sensitivity to DOC, the likely ancestral ligand, which it binds almost identically (Fig. 1C). Key contacts for binding DOC involve conserved surfaces among the hormones, and no obligate contacts are made with moieties at C11, C17, and C18, the only variable positions among the three hormones. These inferences are robust to uncertainty in the sequence reconstruction: we modeled each plausible alternate reconstruction (posterior probability PP >0.20) into the AncCR crystal structures and found that none significantly affected the backbone conformation or ligand interactions. The receptor therefore had the structural potential to be fortuitously activated by aldosterone when that hormone evolved tens of millions of years later, providing the mechanism for evolution of the MR-aldosterone partnership by molecular exploitation, as described (13).
To determine how GR’s preference for cortisol evolved, we identified substitutions that occurred during the same period as the shift in GR function. We used maximum likelihood phylogenetics to determine the sequences of ancestral receptors along the GR lineage (16). The reconstructions had strong support, with mean PP >0.93 and the vast majority of sites with PP >0.90 (Tables S2, S3). We synthesized a cDNA for each reconstructed LBD, expressed it in cultured cells, and experimentally characterized its hormone sensitivity in a reporter gene transcription assay (16). GR from the common ancestor of all jawed vertebrates (AncGR1 in Fig. 1A) retained AncCR’s sensitivity to aldosterone, DOC, and cortisol. At the next node, however, GR from the common ancestor of bony vertebrates (AncGR2) had a phenotype like that of modern GRs, responding only to cortisol. This inference is robust to reconstruction uncertainty: we introduced plausible alternative states by mutagenesis, but none changed function (Fig. S4). GR’s specificity therefore evolved during the interval between these two speciation events, ∼420 to 440 mya (19, 20).
During this interval, there were 36 substitutions and one single-codon deletion (Figs. S5, S6). Four substitutions and the deletion are conserved in one state in all GRs that descend from AncGR2 and in another state in all receptors with the ancestral function. Two of these - S106P and L111Q - were previously identified as increasing cortisol specificity when introduced into AncCR (13). We introduced these substitutions into AncGR1 and found that they recapitulate a large portion of the functional shift from AncGR1 to AncGR2, radically reducing aldosterone and DOC response while maintaining moderate sensitivity to cortisol (Fig. 2A); the concentrations required for half-maximal activation (EC50) by aldosterone and DOC increased by 169- and 57-fold, respectively, while that for cortisol increased only 2-fold. A strong epistatic interaction between substitutions was apparent: L111Q alone had little effect on sensitivity to any hormone, but S106P dramatically reduced activation by all ligands. Only the combination switched receptor preference from aldosterone/DOC to cortisol. Introducing these historical substitutions into the human MR yielded a completely non-functional receptor, as did reversing them in the human GR (Fig. S7). These results emphasize the importance of having the ancestral sequence to reveal the functional impacts of historical substitutions.
Fig. 2.
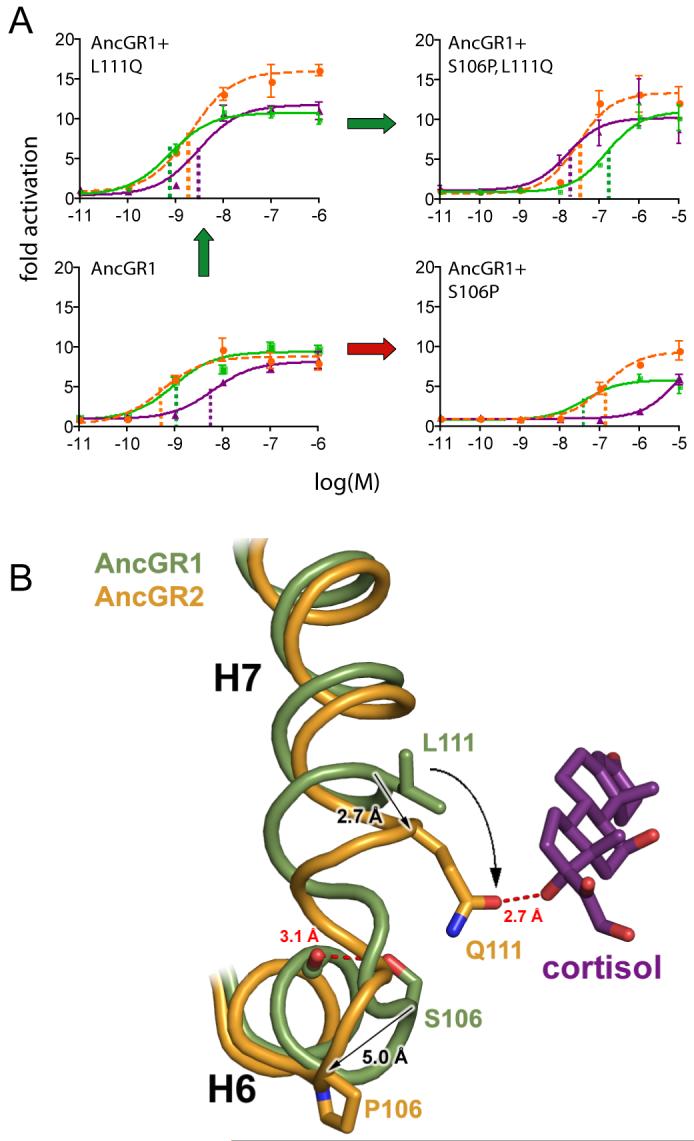
Mechanism for switching AncGR1’s ligand preference from aldosterone to cortisol. A) Effect of substitutions S106P and L111Q on the resurrected AncGR1’s response to hormones. Dashed lines indicate sensitivity to aldosterone (green), cortisol (purple), and DOC (orange) as the EC50 (concentration at which half-maximal reporter activation is achieved). Green arrow shows probable pathway through a functional intermediate; red arrow, intermediate with radically reduced sensitivity to all hormones. B) Structural change conferring new ligand specificity. Backbones of helices 6 and 7 from AncGR1 (green) and AncGR2 (yellow) in complex with cortisol are superimposed. Substitution S106P induces a kink in the interhelical loop of AncGR2, repositioning sites 106 and 111 (arrows). In this background, L111Q forms a new hydrogen bond with cortisol’s unique C17-hydroxyl (dotted red line).
To determine the mechanism by which these two substitutions shift function, we compared the structures of AncGR1 and AncGR2, which were generated by homology modeling and energy minimization based on the AncCR and human GR crystal structures, respectively (16). These structures are robust to uncertainty in the reconstruction: modeling plausible alternate states did not significantly alter backbone conformation, interactions with ligand, or intra-protein interactions. The major structural difference between AncGR1 and AncGR2 involves helix 7 and the loop preceding it, which contain S106P and L111Q and form part of the ligand pocket (Figs. 2B, S8). In AncGR1 and AncCR, the loop’s position is stabilized by a hydrogen bond between S106 and M103’s backbone carbonyl. Replacing S106 with proline in the derived GRs breaks this bond and introduces a sharp kink into the backbone, which pulls the loop downward, repositioning and partially unwinding helix 7. By destabilizing this crucial region of the receptor, S106P impairs activation by all ligands. The movement of helix 7, however, also dramatically repositions site 111, bringing it close to the ligand. In this conformational background, L111Q generates a hydrogen bond with cortisol’s C17-hydroxyl, stabilizing the receptor-hormone complex. Aldosterone and DOC lack this hydroxyl, so the new bond is cortisol-specific. The net effect of the S106P/L111Q combination is to destabilize the aldosterone/DOC-bound receptor and restore stability in a cortisol-specific fashion, switching AncGR2’s preference to that hormone. We call this mode of structural evolution conformational epistasis, because one substitution remodels the protein backbone and repositions a second site, changing the functional effect of substitution at the latter.
Although S106P and L111Q (“group X” for convenience) recapitulate the evolutionary switch in preference from aldosterone to cortisol, the receptor retains some sensitivity to MR’s ligands, unlike AncGR2 and extant GRs. We hypothesized that the other three strictly conserved changes that occurred between AncGR1 and AncGR2 (L29M, F98I, and deletion S212Δ) would complete the functional switch. Surprisingly, introducing these “group Y” changes into the AncGR1 and AncGR1+X backgrounds produced completely non-functional receptors that cannot activate transcription, even in the presence of high ligand concentrations (Fig. 3A). Additional epistatic substitutions must have modulated the effect of group Y, providing a permissive background for their evolution that was not yet present in AncGR1.
Fig. 3.

Permissive substitutions in the evolution of receptor specificity. A) Effects of various combinations of historical substitutions on AncGR1’s transcriptional activity and hormone-response in a reporter gene assay. Group Y (L29M, F98I, and S212Δ) abolishes receptor activity unless groups X (S106P, L111Q) and Z (N26T and Q105L) are present; the XYZ combination yields complete cortisol-specificity. Table shows hormone sensitivity as the EC50 (in nM). Parentheses, 95% confidence interval. Dash, no activation. B) Structural prediction of permissive substitutions. Models of AncGR1 (green) and AncGR2 (yellow) are shown with cortisol. Group X and Y substitutions (circles and rectangles) yield new interactions with the C17-hydroxyl of cortisol (purple) but destabilize receptor regions required for activation. Group Z (underlined) impart additional stability to the destabilized regions. C) Restricted evolutionary paths through sequence space. Nodes of the cube represent states for residue sets X, Y, and Z. Edges represent pathways from the ancestral sequence (AncGR1) to the cortisol-specific combination (+XYZ). Filled circles at vertices show sensitivity to aldosterone (green), DOC (orange), and cortisol (purple); empty circles, no activation. Red octagons, paths through non-functional intermediates; arrows, paths through functional intermediates with no change (white) or switched ligand preference (green).
The AncCR crystal structure allowed us to identify these permissive mutations by analyzing the effects of group Y substitutions (Fig. 3B). In all steroid receptors, transcriptional activity depends on the stability of an activation-function helix (AF-H), which is repositioned upon ligand binding to create the interface for transcriptional coactivators. The stability of this orientation is determined by a network of interactions among the loop preceding AF-H, the ligand, and helix 3 (17). Group Y substitutions compromise activation because they disrupt this network. S212Δ eliminates a hydrogen bond that directly stabilizes the AF-H loop, and L29M on helix 3 creates a steric clash and unfavorable interactions with the D-ring of the hormone. F98I opens up space between helix 3, helix 7, and the ligand; the resulting instability is transmitted indirectly to AF-H, impairing activation by all ligands (Fig. 3B). If the protein could tolerate group Y, however, the structures suggest these mutations would enhance cortisol specificity: L29M forms a hydrogen bond with cortisol’s unique C17-hydroxyl, and the additional space created by F98I relieves a steric clash between the repositioned loop and M108, stabilizing the key interaction between Q111 and the C17-hydroxyl (Fig. 3B).
We hypothesized that historical substitutions which add stability to the regions destabilized by group Y might have permitted the evolving protein to tolerate group Y mutations and complete the GR phenotype. Structural analysis suggested two candidates (group Z): N26T generates a new hydrogen bond between helix 3 and the AF-H loop, and Q105L allows helix 7 to pack more tightly against helix 3, stabilizing the latter and, indirectly, AF-H (Fig. 3B). As predicted, introducing group Z into the non-functional AncGR1+X+Y receptor restored transcriptional activity, indicating that Z is permissive for Y (Fig. 3A). Further, AncGR1+X+Y+Z displays a fully GR-like phenotype that is unresponsive to aldosterone and DOC and maintains moderate cortisol sensitivity. Both N26T and Q105L are required for this effect (Table S4). Strong epistasis is again apparent: adding group Z substitutions in the absence of Y has little or no effect on ligand-activated transcription, presumably because the receptor has not yet been destabilized (Fig. 3A). Evolutionary trajectories that pass through functional intermediates are more likely than those involving non-functional steps (21), so the only historically likely pathways to AncGR2 are those in which the permissive substitutions of group Z and the large-effect mutations of group X occurred before group Y was complete (Fig. 3C).
Our discovery of permissive substitutions in the AncGR1-AncGR2 interval suggested that other permissive mutations might have evolved even earlier. We used the structures to predict whether any of the 25 substitutions between AncCR and AncGR1 (Fig. S5) might be required for the receptor to tolerate the substitutions that later yielded GR function. Only one was predicted to be important: Y27R, which is conserved in all GRs, stabilizes helix 3 and the ligand pocket by forming a cation-π interaction with Y17 (Fig. 4A). When we reversed Y27R in the GR-like AncGR1+X+Y+Z, activation by all ligands was abolished (Fig. 4B). In contrast, introducing Y27R into AncCR (Fig. 4B) or AncGR1 (Fig. S9) had negligible effect on the receptor’s response to any hormone. By conferring increased stability on a crucial part of the receptor, Y27R created a permissive sequence environment for substitutions that, millions of years later, remodeled the protein and yielded a new function.
Fig. 4.
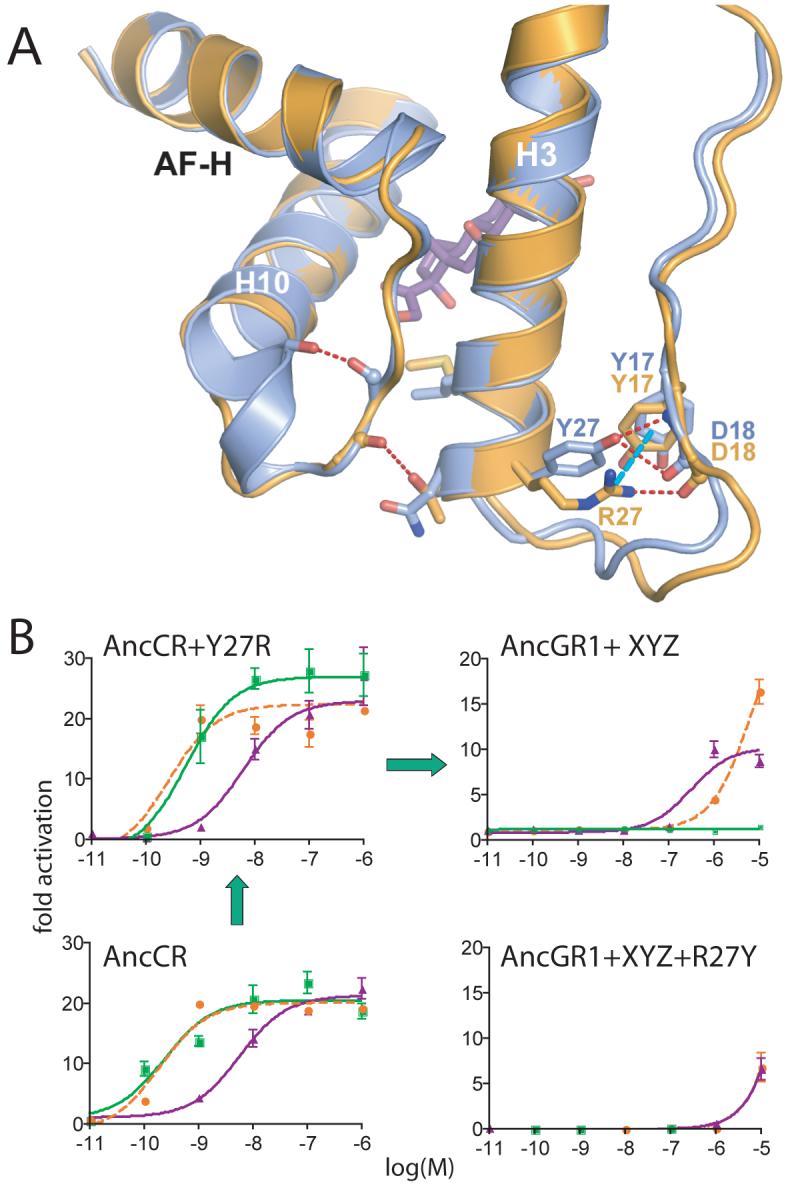
Structural identification of an ancient permissive substitution. A) Comparison of the structures of AncCR (blue) and AncGR2 (yellow). Y27R generates a novel cation-π interaction in AncGR2 (dotted cyan line), replacing the weaker ancestral hydrogen bond (dotted red) and imparting additional stability to helix 3. B) Y27R is permissive for the substitutions that confer GR function. Reporter gene activation by AncGR1+XYZ (upper right) is abolished when Y27R is reversed (lower right). Left, Y27R has negligible effect in the AncCR background (or in AncGR1,Fig. S9). Green, orange, and purple lines show aldosterone, DOC, and cortisol response. Green arrows, likely pathway through functional intermediates.
These results shed light on long-standing issues in evolutionary genetics. One classic question is whether adaptation proceeds by mutations of large or small effect (22). Our findings are consistent with a model of adaptation in which large-effect mutations move a protein from one sequence optimum to the region of a different function, which smaller-effect substitutions then fine-tune (23, 24); permissive substitutions of small immediate effect, however, precede this process. The intrinsic difficulty of identifying mutations of small effect creates an ascertainment bias in favor of large-effect mutations; the ancestral structures allowed us isolate key combinations of small-effect substitutions from a large set of historical possibilities.
A second contentious issue is whether epistasis makes evolutionary histories contingent on chance events (25, 26). We found several examples of strong epistasis, where substitutions that have very weak effects in isolation are required for the protein to tolerate subsequent mutations that yield a new function. Such permissive mutations create “ridges” connecting functional sequence combinations and narrow the range of selectively accessible pathways, making evolution more predictable (27). Whether a ridge is followed, however, may not be a deterministic outcome. If there are few potentially permissive substitutions and these are nearly neutral, then whether they will occur is largely a matter of chance. If the historical “tape of life” could be played again (28), the required permissive changes might not happen, and a ridge leading to a new function could become an evolutionary road not taken.
Our results provide insights into the structural mechanisms of epistasis and the historical evolution of new functions. GR’s functional specificity evolved by substitutions that destabilized the receptor structure with all hormones but compensated with novel interactions specific to the new ligand. Compensatory mutations have been thought to occur when a second substitution restores a lost molecular interaction (29). Our findings support this notion, but in a reversed order: permissive mutations stabilized specific structural elements, allowing them to tolerate later destabilizing mutations which then conferred a new function (9, 10, 30). We also observed a more striking mechanism: conformational epistasis, by which one substitution repositions another residue in three-dimensional space and changes the effects of mutations at that site. It is well known that mutations may have non-additive effects on protein stability (31), and fitness (9,32), but we are aware of few cases (11,33) specifically documenting new functions or epistasis via conformational remodeling. This may be due to the lack of ancestral structures, which allow evolutionary shifts in the position of specific residues to be determined. Conformational epistasis may be an important theme in structural evolution, playing a role in many cases where new gene functions evolve via novel molecular interactions.
Supplementary Material
Acknowledgments
We thank D. Ornoff and J. Bischof for technical assistance, and the Thornton, Redinbo, Phillips and Cresko labs for comments. Supported by NIH-R01-GM081592, NSF-IOB-0546906, and a Sloan fellowship (JWT), NIH-R01-DK622229 (MRR), and NIH-F32-GM074398 (JTB). AncCR crystal structure has PDB IDs 2Q1H, 2Q1V, and 2Q3Y.
Footnotes
The structure of a 450 million year-old protein, “resurrected” using computational and biochemical methods, was determined using x-ray crystallography, revealing the mechanisms by which modern-day hormone receptors evolved their specific functions.
References
- 1.Golding GB, Dean AM. Mol Biol Evol. 1998;15:355. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- 2.Stern DL. Evolution Int J Org Evolution. 2000;54:1079. doi: 10.1111/j.0014-3820.2000.tb00544.x. [DOI] [PubMed] [Google Scholar]
- 3.Perutz MF. Mol Biol Evol. 1983;1:1. doi: 10.1093/oxfordjournals.molbev.a040299. [DOI] [PubMed] [Google Scholar]
- 4.Glasner ME, Gerlt JA, Babbitt PC. Curr Opin Chem Biol. 2006;10:492. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Kinch LN, Grishin NV. Curr Opin Struct Biol. 2002;12:400. doi: 10.1016/s0959-440x(02)00338-x. [DOI] [PubMed] [Google Scholar]
- 6.Vatzaki EH, et al. Eur J Biochem. 1999;260:176. doi: 10.1046/j.1432-1327.1999.00142.x. [DOI] [PubMed] [Google Scholar]
- 7.Khersonsky O, Roodveldt C, Tawfik DS. Curr Opin Chem Biol. 2006;10:498. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Campbell-Valois FX, Tarassov K, Michnick SW. J Mol Biol. 2006;362:151. doi: 10.1016/j.jmb.2006.06.061. [DOI] [PubMed] [Google Scholar]
- 9.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Nature. 2006;444:929. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 10.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Proc Natl Acad Sci U S A. 2006;103:5869. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner JM, Graziano J, Spraggon G, Schultz PG. Proc Natl Acad Sci USA. 2006;103:6483. doi: 10.1073/pnas.0601756103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thornton JW. Nat Rev Genet. 2004;5:366. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- 13.Bridgham JT, Carroll SM, Thornton JW. Science. 2006;312:97. [Google Scholar]
- 14.Bentley PJ. Comparative Vertebrate Endocrinology. Cambridge University Press; 1998. [Google Scholar]
- 15.Sturm A, et al. Endocrinology. 2005;146:47. doi: 10.1210/en.2004-0128. [DOI] [PubMed] [Google Scholar]
- 16.Methods are described in supplemental online material.
- 17.Wagner RL, et al. Nature. 1995;378:690. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 18.Bledsoe RK, et al. J Biol Chem. 2005;280:31283. doi: 10.1074/jbc.M504098200. [DOI] [PubMed] [Google Scholar]
- 19.Benton MJ. Vertebrate Palaeontology. Blackwell Science; 2005. [Google Scholar]
- 20.Janvier P. Early Vertebrates. Clarendon Press; Oxford: 1996. [Google Scholar]
- 21.Smith JM. Nature. 1970;225:563. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 22.Orr HA. Nat Rev Genet. 2005;6:119. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- 23.Charlseworth B. In: Evolutionary innovations. Nitecki M, editor. University of Chicago Press; Chicago: 1990. pp. 47–70. [Google Scholar]
- 24.Orr HA. Evolution Int J Org Evolution. 2002;56:1317. doi: 10.1111/j.0014-3820.2002.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 25.Provine WB. Origins of Theoretical Population Genetics. University of Chicago; 1971. [Google Scholar]
- 26.Whitlock MC, Phillips PC, Moore FBG, Tonsor SJ. Annu Rev Ecol Syst. 1995;26:601. [Google Scholar]
- 27.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Science. 2006;312:111. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 28.Gould SJ. Wonderful Life: The Burgess Shale and the Nature of History. Norton; 1989. [Google Scholar]
- 29.Kern AD, Kondrashov FA. Nat Genet. 2004;36:1207. doi: 10.1038/ng1451. [DOI] [PubMed] [Google Scholar]
- 30.Haag ES, Molla MN. Evolution Int J Org Evolution. 2005;59:1620. [PubMed] [Google Scholar]
- 31.Reichmann D, et al. Proc Natl Acad Sci U S A. 2005;102:57. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lunzer M, Miller SP, Felsheim R, Dean AM. Science. 2005;310:499. doi: 10.1126/science.1115649. [DOI] [PubMed] [Google Scholar]
- 33.Hedstrom L. Biol Chem. 1996;377:465. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.