

Abstract
N-Acyl O-amino phenol derivatives of CBI-TMI and CBI-indole2 are reported as prototypical members of a new class of reductively activated prodrugs of the duocarmycin and CC-1065 class of antitumor agents. The expectation being that hypoxic tumor environments, with their higher reducing capacity, carry an intrinsic higher concentration of “reducing” nucleophiles (e.g., thiols) capable of activating such derivatives (tunable N–O bond cleavage) increasing their sensitivity to the prodrug treatment. Preliminary studies indicate the prodrugs effectively release the free drug in functional cellular assays for cytotoxic activity approaching or matching the activity of the free drug, yet remain essentially stable and unreactive to in vitro DNA alkylation conditions (<0.1–0.01% free drug release), pH 7.0 phosphate buffer, and exhibit a robust half-life in human plasma (t½ = 3 h). Characterization of a representative O-(acylamino) prodrug in vivo indicate that they approach the potency and exceed the efficacy of the free drug itself (CBI-indole2) indicating that not only is the free drug effectively released from the inactive prodrug, but that they offer additional advantages related to a controlled or targeted release in vivo.
Introduction
CC-1065, the duocarmycins, and yatakemycin constitute exceptionally potent naturally occurring antitumor agents that derive their biological properties through a characteristic sequence-selective DNA alkylation reaction (Figure 1).1–10 The examination of the natural products, their synthetic unnatural enantiomers, their derivatives, and synthetic analogues have defined fundamental features that control the alkylation selectivity, impact the alkylation efficiency, and are responsible for DNA alkylation catalysis providing a detailed understanding of the relationships between structure, reactivity, and biological activity.6,11–13
Figure 1.
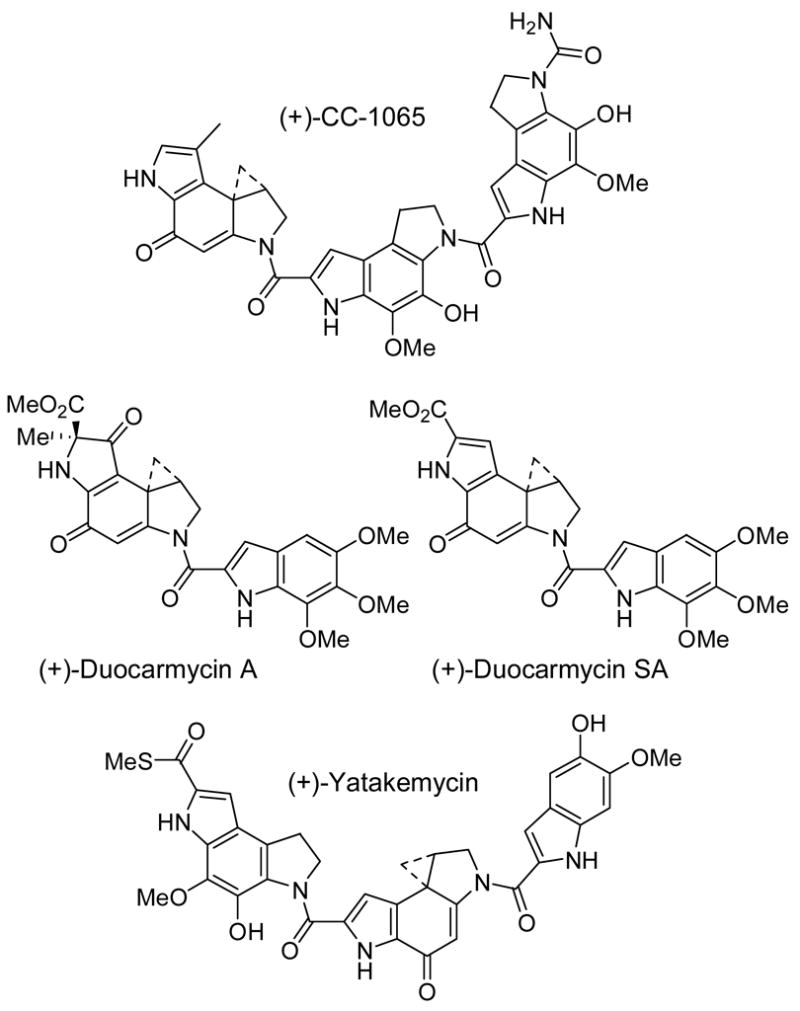
Natural products
One of the most important and widely explored class of analogues is CBI14 (1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one), being synthetically14,15 more accessible than the natural products, yet indistinguishable in its DNA alkylation selectivity (Figure 2).16 Moreover, the CBI derivatives proved to be four times more stable and, correspondingly, four times more potent than derivatives bearing the CC-1065 alkylation subunit (7-MeCPI) approaching the stability and potency of duocarmycin SA and yatakemycin derivatives, and they exhibit efficacious in vivo antitumor activity in animal models at doses that reflect this potency.17 Consequently, CBI and its derivatives have been the focus of much development as well as the prototype analogues on which new design concepts have been explored, developed, or introduced.14–32
Figure 2.
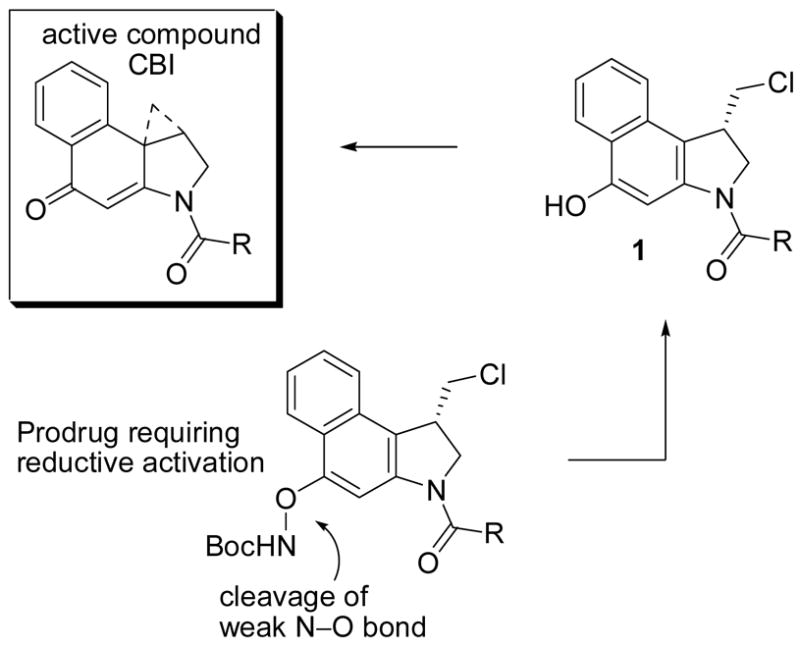
Prodrug design
A unique feature of this class of molecules including the natural products themselves is the observation that synthetic phenol precursors (e.g., 1) to the final products, entailing a Winstein Ar-3′ spirocyclization with displacement of an appropriate leaving group, exhibit biological properties typically indistinguishable from the cyclopropane-containing final products (DNA alkylation rate or efficiency, in vitro cytotoxic activity, and in vivo antitumor activity). This dependable behavior of the precursor phenols has provided the basis on which the development of useful, stable, or safe prodrugs has been conducted.33–35
One feature limiting the attractiveness of this class of cytotoxic agents is their remarkable potencies (IC50 5–20 pM) creating special requirements for their preparation and handling. In many instances, this has been addressed by the introduction of chemically stable phenol protecting groups that are readily cleaved at the final stage of their preparation or upon in vivo administration. Such protected phenol precursors are intrinsically much less potent, yet readily release an active precursor to the drug upon deprotection. Extensions of this protection and release strategy have been pursued in which the free phenol release in vivo is coupled to features that might facilitate tumor selective delivery or cleavage.36 Such inactive prodrugs serve the dual role of providing safer handling intermediates or final products as well as potentially enhancing the therapeutic index of the drug. As attractive and amenable as this approach is for this class of drugs, a surprisingly small series of such studies have been disclosed.25–32
Herein, we disclose a novel set of reductively activated phenol prodrugs for the CC-1065 and duocarmycin class of compounds that do not require enzymatic release and should prove general for other phenolic drugs that may benefit from such a designed activation. Alternative and prior efforts at incorporating a reductive activation into the CC-1065 and duocarmycin class includes the Denny disclosures of nitro precursors to aryl amine variants of the phenol precursors,30 Lee’s use of an ester subject to cleavage upon a tethered quinone reduction,31 and our own report of mitomycin-like quinone precursors to a reductively activated o-spirocyclization (versus p-spirocyclization) analogous to those observed with the duocarmycins or its analogues.32 Although the approaches have provided some increase in aerobic selectivity that results from the reductive activation, none effectively or clearly utilize an intrinsic enzyme activity that differentiated normal versus tumor cells and in many instances it may be the ease of reoxidation in normal cells that protects them from the effects of the drug, which occurs less readily in hypoxic tumors. The features contributing to and limiting reductively activated hypoxia-selective cytotoxic agents have been reviewed and such agents include mitomycin C and other aziridoquinones, nitro aromatics, N-oxides, various metal complexes, azides, and di- and trisulfides.37 Most related to the design disclosed herein are FR900482 and its related congeners (FR66979 and FK-317)38 as well as a dehydromonocrotaline progenitor39 bearing hydroxylamine hemiacetals which are irreversibly activated by reductive cleavage of a N–O bond.
The approach detailed herein was not designed for reversible or enzymatic reductive activation, but rather for irreversible activation by cleavage of a weak N–O bond by reducing nucleophiles (Figure 2). The expectation being that hypoxic tumor cells, with their higher reducing capacity, may carry an intrinsic higher concentration of “reducing” nucleophiles (i.e., thiols) capable of activating such derivatives making them more sensitive to the prodrug treatment.36 Moreover, as detailed below, the design lends itself to a rational tuning of the ease of reduction of the derivative allowing empirical experience with the series to guide future design.
Chemistry
Synthesis
A range of methods for direct conversion of a precursor phenol to the corresponding O-amino phenol were examined (O-amidation) and several routes to the final compounds were explored. It was anticipated that this might best be conducted on a seco-N-Boc-CBI derivative lacking the capabilities of spirocyclization (e.g., 11). However, the lability of the resulting N-acyl O-amino phenol derivatives to subsequent chemical transformations proved significant and this approach proved less viable than a surprisingly effective direct O-amidation reaction of seco-CBI-TMI or seco-CBI-indole2 (Scheme 1). Thus, low temperature phenol deprotonation of 2 (3 equiv of LiHMDS, 0 °C, ether–dioxane) followed by treatment with the amidating reagents TsONHBoc40 or TsONPhth41 provided 4 and 8 directly in good conversions. Competitive spirocyclization of 2 to CBI-TMI itself was observed if the deprotonation was carried out at higher reaction temperatures or in more polar solvents. It diminished as the solvent polarity was reduced (glyme > THF > dioxane–ether > ether, insoluble) and was less prominent with LiHMDS versus NaHMDS. In most instances, recovered starting phenol was present in the crude reaction product and was chromatographically close enough to the N-acyl O-amino phenols that special precautions were taken to ensure its removal. This entailed exposure of the product mixture to conditions that promote deliberate spirocyclization of the seco phenol derivatives (saturated aqueous NaHCO3–THF (1:1), 23 °C, 2 h)15 and subsequent chromatographic separation of the much more polar CBI-TMI or CBI-indole2. N-Acetylation of 4 (Ac2O, cat. DMAP, CH2Cl2, 23 °C, 12 h, 81%) provided 6 and subsequent Boc deprotection (TFA–CH2Cl2 (1:1), 23 °C, 3 h, 88%) afforded 5. In an analogous fashion, seco-CBI-indole2 (3) was directly converted to 8 (45%) upon LiHMDS deprotonation (3 equiv of LiHMDS, ether–dioxane, 0 °C, 30 min) and subsequent O-amidation with TsONHBoc.40
Scheme 1.
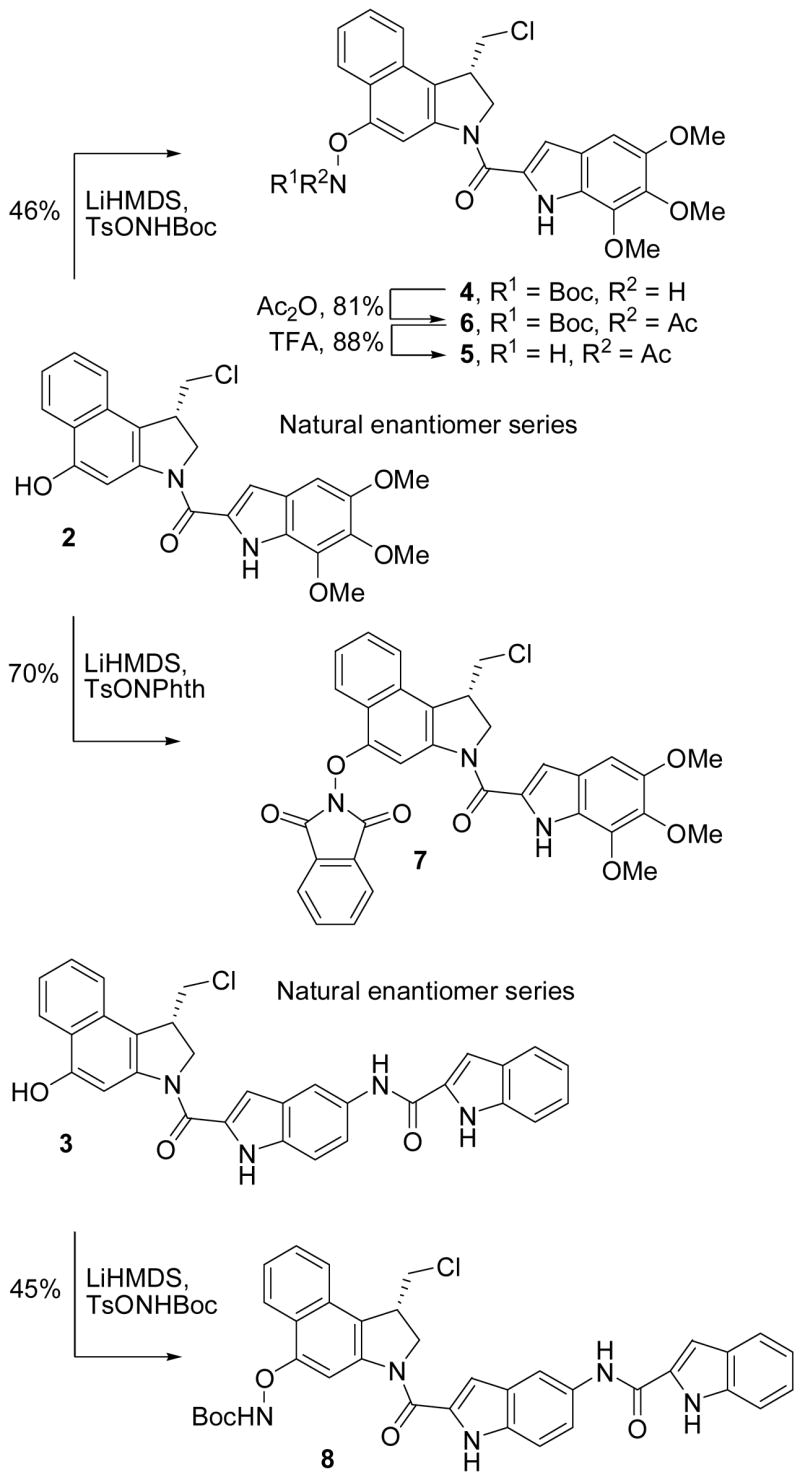
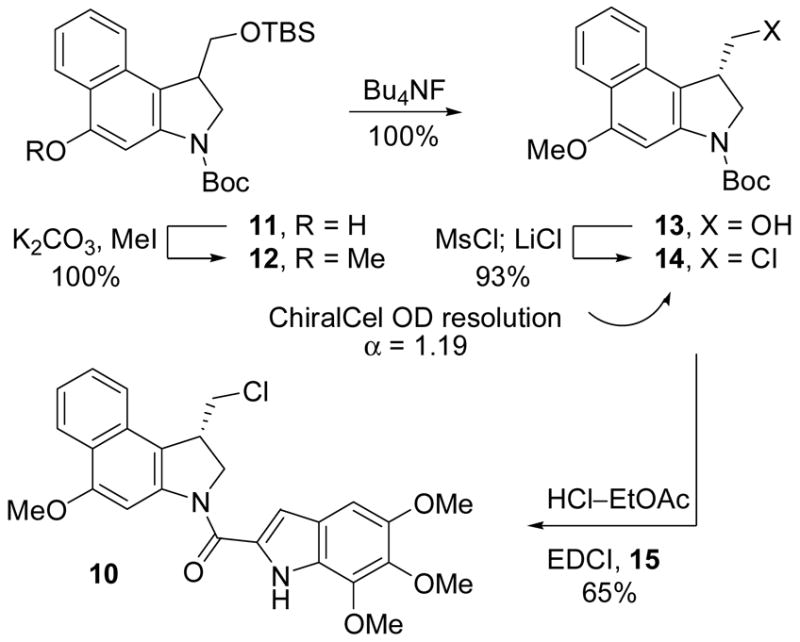
For comparison purposes, two analogues of seco-CBI-TMI were prepared that are incapable of spirocyclization to CBI-TMI itself. The first incorporates the C4 phenol protected as its methyl ether (10) and second contains no C4 substituent (9). The former was prepared from 1115i by phenol O-methylation, primary alcohol OTBS deprotection and subsequent conversion to the primary chloride 14, followed by N-Boc deprotection and coupling with 5,6,7-trimethoxyindole-2-carboxylic acid (15) to provide 10, Scheme 2. Throughout this sequence and as a result of the multiple purifications, the chances of residual, contaminant phenol (2) being present in the final product 10 are remote. Nonetheless, since even trace quantities of 2 can be misleadingly detected in the subsequent biological evaluations (e.g., 0.01%), the analogue 9 was also prepared for comparison and by an approach that precludes the presence of such a contaminate phenol. Thus, following a route analogous to that used for CBI itself,15 20 was prepared from 16 and converted to 21 enlisting a key 5-exo-trig aryl radical–alkene cyclization,15h Scheme 3. The product 21, like 14 (α = 1.19), was chromatographically resolved on a semipreparative ChiralCel OD column (α = 1.42) providing each enantiomer which were coupled with 5,6,7-trimethoxyindole-2-carboxylic acid (15) upon N-Boc deprotection to provide 9.
Scheme 2.
Scheme 3.
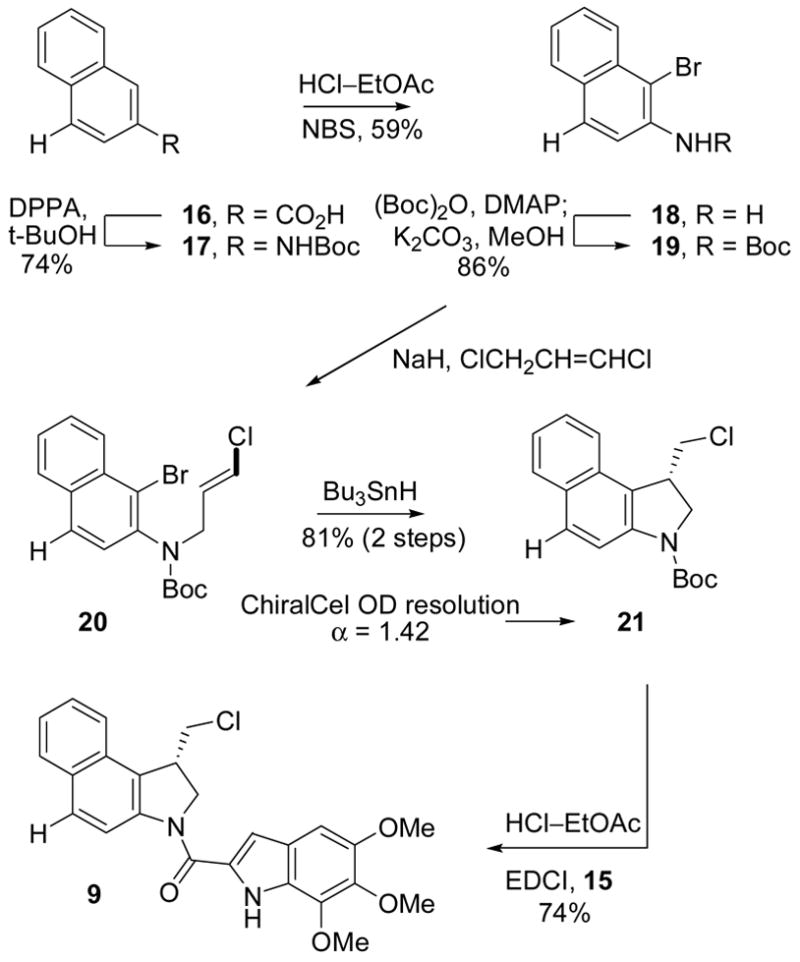
Stability and Reactivity of the N-Acyl O-Amino Phenol Derivatives
Clear from efforts directed at their preparation, the N-acyl amino phenol prodrugs displayed a useful range of stability, yet were susceptible to cleavage of the critical N–O bond. As might be anticipated, their relative stability followed the order of 4 > 5 > 6 > 7 with 4 and 5 withstanding even long term storage effectively, but with 7 noticeably deteriorating over time. Derivatives 4 and 6, as well as 7, proved surprisingly robust to acidic conditions (TFA–CH2Cl2, 4 N HCl–EtOAc), and stable to mild base treatment in nonpolar, aprotic solvents (Et3N or DMAP, CH2Cl2), but exhibited a diminished stability as the solvent polarity increases: stable to NaHCO3 in THF or THF–H2O, but cleaved in NaHCO3/DMF–H2O or H2O and DBU/CH2CN. Similarly, 4 proved stable in MeOH, but 2 was released slowly upon treatment with NaHCO3 or Na2CO3 in MeOH (2 h, 23 °C). Most pertinent to the potential source of cleavage under physiological conditions, 4 was stable to treatment with BnSH in THF (2–72 h, 23 °C) or MeOH (2–72 h, 23 °C), and stable to treatment with BnSH in THF even in the presence of insoluble NaHCO3 (2 h, 23 °C), but is cleaved to release 2 upon treatment with BnSH in MeOH in the presence of NaHCO3 (2 h, 23 °C). Significantly, the stability of 4 was assessed in pH 7.0 phosphate buffer and within the limits of detection (HPLC, UV), no significant cleavage of the prodrug was observed over the time monitored (72 h). Finally, the stability of 4 was monitored in human plasma (50 μg/100 μL, 10% DMSO) in which it displayed a half-life of 3 h with release of the free drug 2.
Biological Properties
Cytotoxic Activity
The O-amino phenol derivatives bearing the N–O prodrug linkages and the various N-acyl substituents were assayed for cytotoxic activity alongside the parents drugs CBI-TMI (2)18 and CBI-indole2 (3)17 as well as the two control standards 9 and 10 incapable of free phenol release. Three cell lines were examined including a standard L1210 cell line (mouse leukemia) as well as the mitomycin-sensitive (H460, expresses high levels of DT-Diaphorase) and resistant (H596, lacks DT-Diaphorase) non small cell lung cancer (NSCLC) cell lines.32, 17 Several important trends emerged from these studies. First, the natural enantiomer control standards 9 and 10, incapable of free phenol release, were inactive against all three cell lines (IC50 >100 nM) being ≥10,000-fold less active than the free drug 2 (seco-CBI-TMI), Figure 3. In sharp contrast, the natural enantiomers of the O-amino phenol prodrugs exhibited potent cytotoxic activity approaching that of the free drug itself (1–0.1 times the activity of 2) indicating its successful release under the assay conditions. Even more significantly, the relative potency of the prodrugs, when distinguishable, mirror the expected ease of N–O bond cleavage (e.g. L1210:7 > 6 > 5 > 4) suggesting fundamental chemical principles may be used to “tune” the reductive free drug release. Provocatively, the potency differences between the free drug 2 and the prodrugs may vary with the character of the cell line; 4 is 10-fold less potent than 2 against L1210, but 2 and 4 are essentially equipotent against H460/H596. More significantly and unlike mitomycin C, this reductive activation is not linked to the expression levels of DT-Diaphorase since 2 and 4–7 remain equipotent in the H460 or H596 cell lines, although H596 is 10-fold less sensitive than H460 to seco-CBI-TMI itself. This illustrates that DT-Diaphorase is not mediating the reductive release of the drug from the O-amino phenol prodrugs indicating that their utility is orthogonal to that of mitomycin. While this does not rule out enzymatic activation, the behavior is consistent with our suggestion that the activation is nonenzymatic and likely is mediated in situ by appropriate nucleophiles. Analogous trends are also observed with the CBI-TMI unnatural enantiomers albeit at potencies that are approximately 100 to 1000-fold higher than that of the natural enantiomers (Figure 3).
Figure 3.
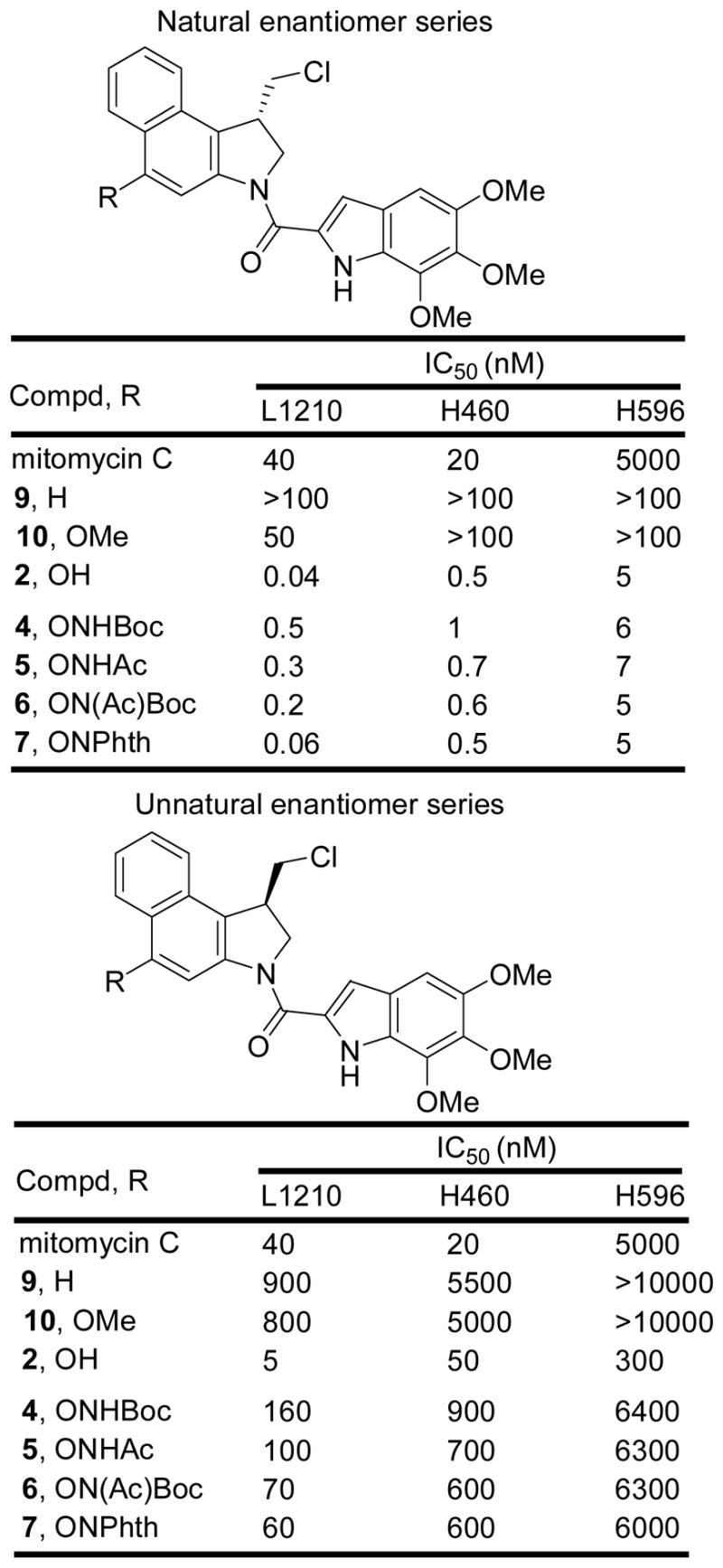
In vitro cytotoxic activity
Especially interesting was the behavior of the CBI-indole2 prodrug. For this CBI analogue, only the NHBoc derivative was examined since it was the most stable of the N-acyl O-amino phenol prodrugs examined (Figure 4). In each cell line examined, the prodrug 8 was essentially equipotent with CBI-indole2 itself indicating effective release of the free drug under the conditions of the assay. In addition it proved to be exceptionally potent being 100–1000 times more active than mitomycin C (IC50 = 30–200 pM vs 20–40 nM) and it remained remarkably active against the mitomycin-resistant H596 cell line (IC50 = 4 nM vs 5 μM). Even the unnatural enantiomer of 8, which was found to be 10–100 fold less active than the natural enantiomer, proved to be more active than mitomycin C. Given the efficacy of (+)-CBI-indole2 in animal tumor models,17 it was especially interesting to compare 8 with 3 in vivo.
Figure 4.
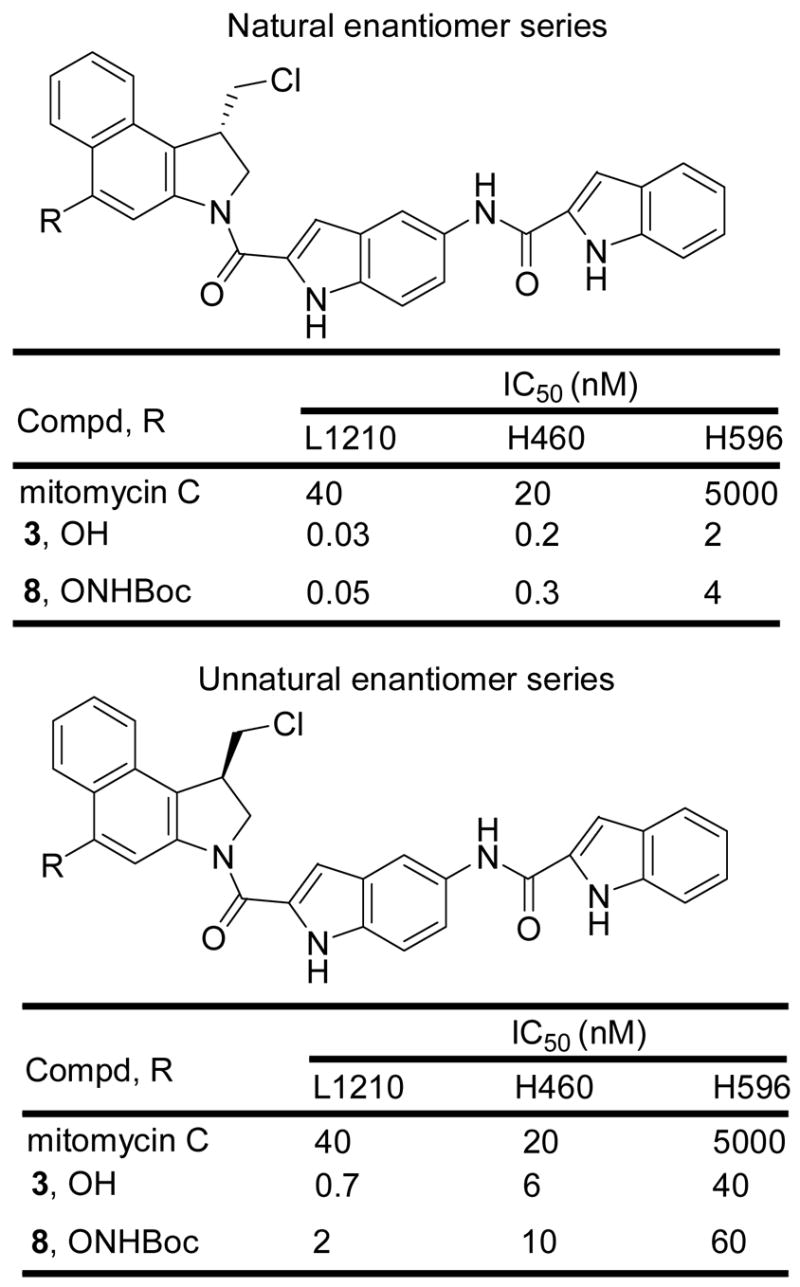
In vitro cytotoxic activity
DNA Alkylation Selectivity and Efficiency
The DNA alkylation properties of 4 were examined alongside the parent drug CBI-TMI (2), and the two control standards 9 and 10 (incapable of spirocyclization) within w794 duplex DNA42 for which results for an extensive series of duocarmycin analogues have been reported. The sites of DNA alkylation and its efficiency were directly assessed by thermally-induced singly 5′ end-labeled duplex DNA strand cleavage following incubation with the agents (Figure 5, natural enantiomers examined).
Figure 5.
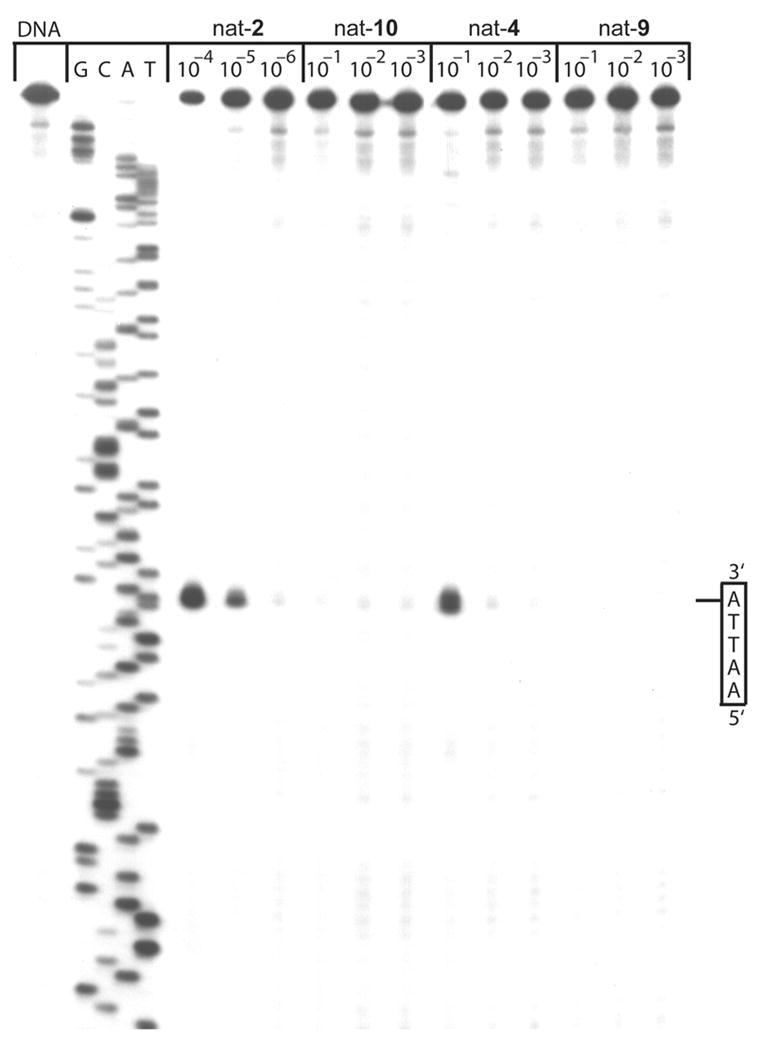
Thermally-induced strand cleavage of w794 DNA; DNA–agent incubation at 4 ºC for 18 h, removal of unbound agent by EtOH precipitation, and 30 min of thermolysis (100 ºC) followed by 8% denaturing PAGE and autoradiography. Lane 1, control DNA; lanes 2–5, Sanger G, C, A, and T sequencing reactions; lanes 6–8, 2 (1 × 10−4 to 1 × 10−6); lanes 9–11, 10 (1 × 10−1 to 1 × 10−3); lanes 12–14, 4 (1 × 10−1 to 1 × 10−3), lanes 15–17, 9 (1 × 10−1 to 1 × 10−3). All compounds possess the natural 1S-configuration.
The reductively activated agent 4 was found to alkylate w794 DNA with an identical sequence selectivity as the parent agent CBI-TMI (2), albeit with a substantially reduced efficiency (1,000–10,000 fold). Similarly, the O-methyl ether 10 as well as 9 lacking a C4 substituent failed to exhibit significant observable DNA alkylation. In fact, 9 showed no appreciable DNA alkylation even under forcing conditions (37 ºC, 18 h, data not shown), whereas the potentially more reactive O-methyl ether 10 (via assisted phenonium ion formation) displayed perhaps a trace amount of DNA alkylation (<0.01% that of 2) that could be attributed to either its direct, but much less facile, DNA alkylation or contaminant free phenol present in the synthetic sample of 10. With detection of DNA alkylation by the prodrug 4 at the level observed (0.1–0.01% of 2), we cannot distinguish whether this is due to direct alkylation by 4 itself, trace release of 2 from 4 under the DNA incubation conditions (in situ N–O cleavage), or attributable to trace contaminate 2 in the synthetic samples of 4. What the results do indicate is that 4 is incapable of significant DNA alkylation in its own right (requires N–O bond cleavage), and that 4 is essentially stable to the DNA alkylation conditions examined requiring deliberate N–O bond cleavage to initiate effective DNA alkylation. These observations are consistent with the stability of 4 observed in pH 7.0 phosphate buffer. Significantly, the results then suggest that the in vitro cytotoxic activity of 4, and by analogy that of the related O-amino phenol prodrugs which all approach that of the parent drug CBI-TMI (2), is derived from in situ intracellular cleavage of the N–O bond and productive release of the active drug under the cell culture conditions.
In Vivo Antitumor Activity
The prodrug 8 was examined for in vivo efficacy alongside the parent drug 3 in a standard antitumor model enlisting L1210 murine leukemia implanted i.p. into DBA/2J mice. This model has been reported to respond well to the parent drugs of related compounds43 and is a system that collaborators through the years have used to assess an extensive series of (+)-CBI-indole2 analogues. Although not published, these latter studies provided the foundation on which we based our examination of 8. With use the dose range (10–100 μg/kg) and the dosing schedule (administered three times i.p. on days 1, 5, and 9) found suitable for related parent drugs including (+)-CBI-indole2 (3),17 the prodrug 8 was examined (Figure 6). The dose at which a maximal response was observed for 8 corresponded closely to that of (+)-CBI-indole2 (3) while its efficacy was significantly improved. This indicates that the prodrug 8 (1) efficiently and effectively releases the free drug 3 in the in vivo model (reductive activation), and (2) that either the rate of release or the site of release enhances the efficacy of the drug. Moreover, the efficacy of 8 is extraordinary providing 5/6 long term survivors at 52 weeks (365 days, T/C >1550) at the optimal dosing examined (100 μg/kg). Notably, little distinction between 3 and 8 was observed at days 30–100 except that the prodrug treated animals appeared healthier displaying little or no weight loss which was evident with 3 at the highest dosing. With the prolonged management of the treated animals herein that exceeded the time frame typically allotted for such an in vivo antitumor assessment, we observed that the surviving mice at day 90 treated with the free drug 3, but not the prodrug 8, eventually expired due to drug administration related complications.44 While these would likely be capable of being managed with an optimized dosing schedule, this distinction between 3 and 8 in the long term cures (>90 days) suggests the prodrug 8 offers significant advantages over the free drug administration. Finally, it is worth noting that these compounds are extraordinarily potent requiring less than 1 mg of sample to conduct the entire in vivo antitumor testing suggesting that clinical supplies of such agents could easily be supplied by chemical synthesis.
Figure 6.
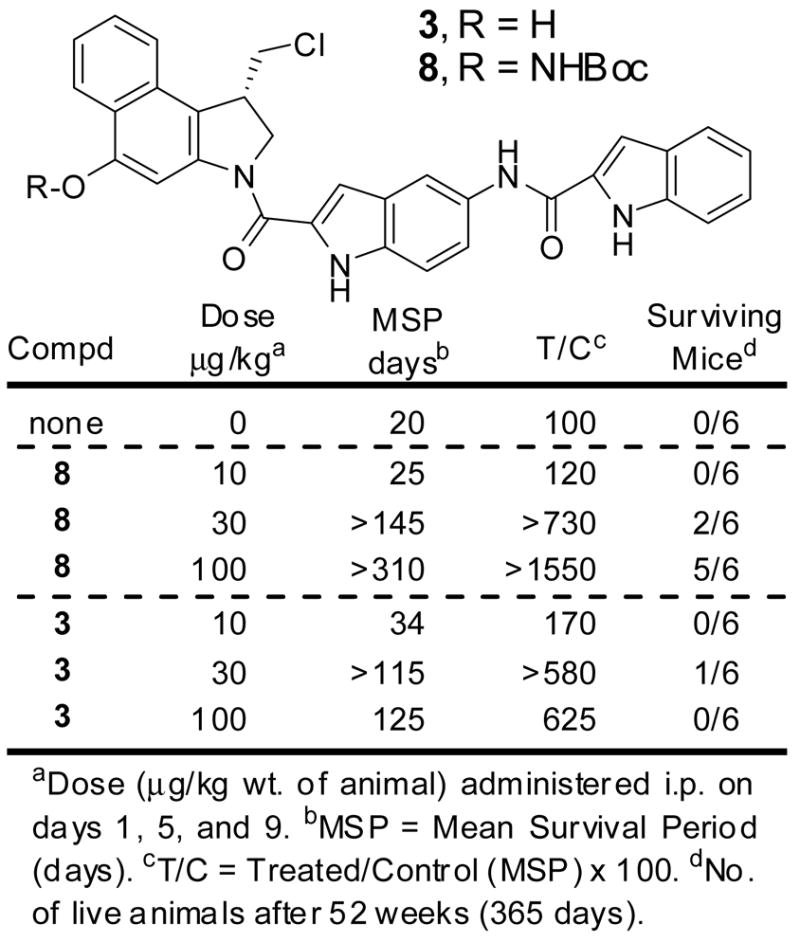
In vivo antitumor activity (L1210, i.p.)
Confirming these observations, an analogous antitumor assessment was carried out independently at a second site utilizing a slightly different and harsher protocol for drug administration (neat DMSO vs 30% DMSO in 0.1% glucose). Although this assessment was terminated after 120 days, it similarly indicates that administration of the prodrug 8 is significantly less toxic than free drug 3, and that it is comparable or superior in terms of reducing deaths due to the disease, and tumor burden (Figure 7). Again, 7/10 long term survivors were observed with 8 at day 120 at the optimal dosing (60 μg/kg).
Figure 7.
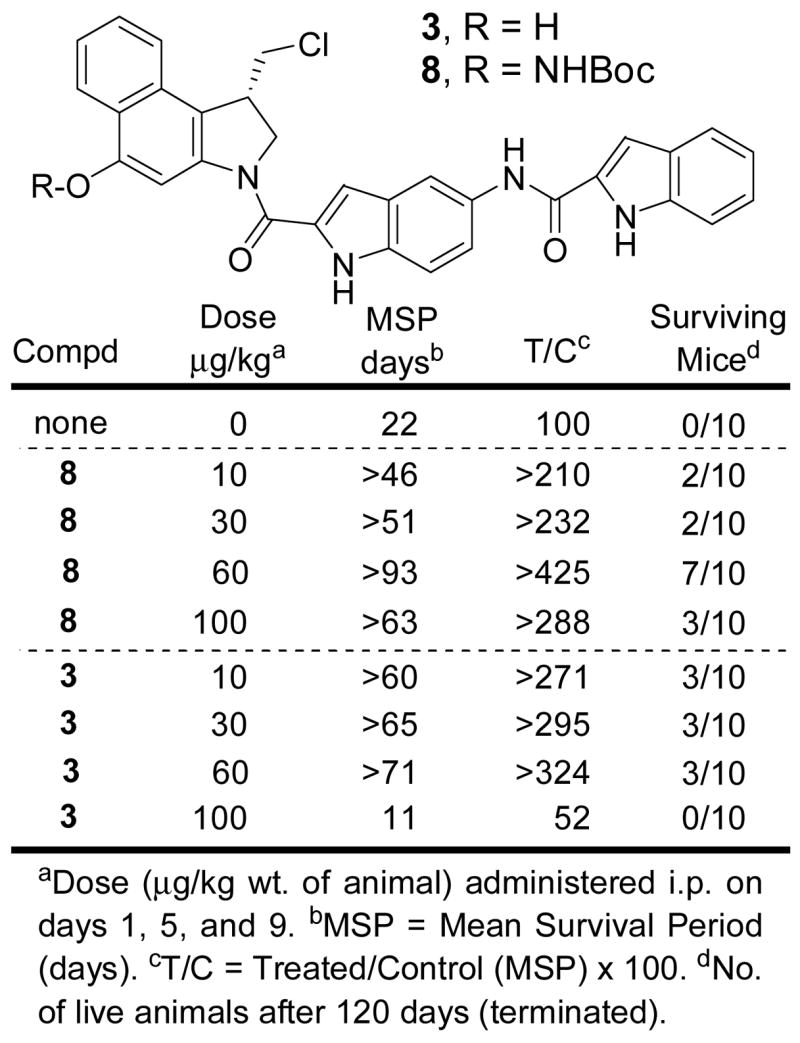
In vivo antitumor activity (L1210, i.p.)
Conclusions
A unique class of N-acyl O-amino phenol prodrugs of CBI-TMI and CBI-indole2 was explored as representative members of the duocarmycin and CC-1065 class of antitumor agents. The prodrugs, subject to reductive activation by nucleophilic cleavage of a weak N–O bond, effectively release the free drug in functional cellular assays for cytotoxic activity approaching or matching the activity of the free drug, yet remain essentially stable to ex vivo DNA alkylation conditions (< 0.1–0.01% free drug release), pH 7.0 phosphate buffer, and exhibiting a robust half-life in human plasma (t½ = 3 h for 4). Most impressively, assessment of the in vivo antitumor activity of a representative O-(acylamino) prodrug, 8, indicate that they approach the potency and exceed the efficacy of the free drug itself (CBI-indole2) indicating that the inactive prodrugs not only effectively release the free drug in vivo, but that they offer additional advantages related to a controlled or targeted release in vivo. With cleavage release of the free drug being easily tunable using fundamental chemical principles, the potential of developing derivatives selectively or most effectively released in a reducing hypoxic environment characteristic of solid tumors may further improve on these impressive observations. An additional unique feature of the O-amino phenol prodrugs is their capability to serve as the linkage site for conjugation to targeting molecules (i.e., antibodies). Thus, reductive cleavage of the drug may be capable of a tunable release 14 from a tethered, covalent delivery vehicle offering a unique alternative to linkers (e.g., disulfide or hydrazone) typically enlisted in studies to date.
Supplementary Material
Full experimental details and compound characterizations are provided.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA41986) and the Skaggs Institute for Chemical Biology. We thank the American Society for Engineering Education (NDSEG) for a predoctoral fellowship (J.D.T). J.D.T. is a Skaggs Fellow.
References
- 1.Chidester CG, Krueger WC, Mizsak SA, Duchamp DJ, Martin DG. J Am Chem Soc. 1981;103:7629. [Google Scholar]
- 2.Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. J Antibiot. 1988;41:1915. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 3.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. J Antibiot. 1990;43:1037. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 4.Yasuzawa T, Muroi K, Ichimura M, Takahashi I, Ogawa T, Takahashi K, Sano H, Saito Y. Chem Pharm Bull. 1995;43:378. doi: 10.1248/cpb.43.378. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai TJ. Antibiot. 2003;56:107. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]; Structure revision: Tichenor MS, Kastrinsky DB, Boger DL. J Am Chem Soc. 2004;126:8396. doi: 10.1021/ja0472735.
- 6.CC-1065: Warpehoski MA, Hurley LH. Chem Res Toxicol. 1988;1:315. doi: 10.1021/tx00006a001.Hurley LH, Needham-VanDevanter DR. Acc Chem Res. 1986;19:230.
- 7.CC-1065: Boger DL, Johnson DS, Yun W, Tarby CM. Bioorg Med Chem. 1994;2:115. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. J Am Chem Soc. 1990;112:4623.
- 8.Duocarmycin A: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1990;112:8961.Boger DL, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. Bioorg Med Chem Lett. 1992;2:759.
- 9.Duocarmycin SA: Boger DL, Johnson DS, Yun W. J Am Chem Soc. 1994;116:1635.
- 10.Yatakemycin: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. J Am Chem Soc. 2003;125:10971. doi: 10.1021/ja035984h.Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J Am Chem Soc. 2006;128:15683. doi: 10.1021/ja064228j.Trzupek JD, Gottesfeld JM, Boger DL. Nature Chem Biol. 2006;2:79. doi: 10.1038/nchembio761.
- 11.(b) Reviews: Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043.Boger DL, Garbaccio RM. Bioorg Med Chem. 1997;5:263. doi: 10.1016/s0968-0896(96)00238-6.Boger DL, Johnson DS. Angew Chem, Int Ed Engl. 1996;35:1438.Boger DL. Acc Chem Res. 1995;28:20.Boger DL, Johnson DS. Proc Natl Acad Sci, USA. 1995;92:3642. doi: 10.1073/pnas.92.9.3642.Boger DL. Chemtracts: Org Chem. 1991;4:329.
- 12.Review of synthetic studies: Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem Rev. 1997;97:787. doi: 10.1021/cr960095g.
- 13.Tse WC, Boger DL. Chem Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 14.(a) Boger DL, Ishizaki T, Wysocki RJ, Jr, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1989;111:6461. [Google Scholar]; (b) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823. [Google Scholar]
- 15.(a) Boger DL, Yun W, Teegarden BR. J Org Chem. 1992;57:2873. [Google Scholar]; (b) Boger DL, McKie JA. J Org Chem. 1995;60:1271. [Google Scholar]; (c) Drost KJ, Cava MP. J Org Chem. 1991;56:2240. [Google Scholar]; (d) Aristoff PA, Johnson PD. J Org Chem. 1992;57:6234. [Google Scholar]; (e) Mohamadi F, Spees MM, Staten GS, Marder P, Kipka JK, Johnson DA, Boger DL, Zarrinmayeh H. J Med Chem. 1994;37:232. doi: 10.1021/jm00028a005. [DOI] [PubMed] [Google Scholar]; (f) Ling L, Xie Y, Lown JW. Heterocyclic Commun. 1997;3:405. [Google Scholar]; (g) Boger DL, McKie JA, Boyce CW. Synlett. 1997:515. [Google Scholar]; (h) Boger DL, Boyce CW, Garbaccio RM, Searcey M. Tetrahedron Lett. 1998;39:2227. [Google Scholar]; (i) Kastrinsky DB, Boger DL. J Org Chem. 2004;69:2284. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]
- 16.Boger DL, Munk SA. J Am Chem Soc. 1992;114:5487. [Google Scholar]
- 17.CBI-indole2: Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Bioorg Med Chem Lett. 1991;1:115.Boger DL, Yun W, Han N. Bioorg Med Chem. 1995;3:1429. doi: 10.1016/0968-0896(95)00130-9.
- 18.CBI-TMI: Boger DL, Yun W. J Am Chem Soc. 1994;116:7996.
- 19.(a) Boger DL, Ishizaki T. Tetrahedron Lett. 1990;31:793. [Google Scholar]; (b) Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. BioorgMed Chem Lett. 1991;1:55. [Google Scholar]; (c) Boger DL, Munk SA, Ishizaki T. J Am Chem Soc. 1991;113:2779. [Google Scholar]; (d) Boger DL, Yun W. J Am Chem Soc. 1994;116:5523. [Google Scholar]; (e) Boger DL, Yun W, Han N, Johnson DS. Bioorg Med Chem. 1995;3:611. doi: 10.1016/0968-0896(95)00048-l. [DOI] [PubMed] [Google Scholar]; (f) Boger DL, Yun W, Cai H, Han N. Bioorg Med Chem. 1995;3:761. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]; (g) Boger DL, Han N. Bioorg Med Chem. 1997;5:233. doi: 10.1016/s0968-0896(96)00237-4. [DOI] [PubMed] [Google Scholar]; (h) Boger DL, Garbaccio RM, Jin Q. J Org Chem. 1997;62:8875. [Google Scholar]; (i) Boger DL, Santillan A, Jr, Searcey M, Jin Q. J Am Chem Soc. 1998;120:11554. [Google Scholar]; (j) Boger DL, Garbaccio RM. J Org Chem. 1999;64:5666. doi: 10.1021/jo990762g. [DOI] [PubMed] [Google Scholar]; (k) Boger DL, Santillan A, Jr, Searcey M, Jin Q. J Org Chem. 1999;64:5241. doi: 10.1021/jo990452y. [DOI] [PubMed] [Google Scholar]; (l) Boger DL, Hughes TV, Hedrick MP. J Org Chem. 2001;66:2207. doi: 10.1021/jo001772g. [DOI] [PubMed] [Google Scholar]; (m) Boger DL, Brunette SR, Garbaccio RM. J Org Chem. 2001;66:5163. doi: 10.1021/jo010309g. [DOI] [PubMed] [Google Scholar]; (n) Boger DL, Schmitt H, Fink BE, Hedrick MP. J Org Chem. 2001;66:6654. doi: 10.1021/jo010454u. [DOI] [PubMed] [Google Scholar]; (o) Boger DL, Stauffer F, Hedrick MP. Bioorg Med Chem Lett. 2001;11:2021. doi: 10.1016/s0960-894x(01)00372-9. [DOI] [PubMed] [Google Scholar]; (p) Parrish JP, Kastrinsky DB, Stauffer F, Hedrick MP, Hwang I, Boger DL. Bioorg Med Chem. 2003;11:3815. doi: 10.1016/s0968-0896(03)00194-9. [DOI] [PubMed] [Google Scholar]; (q) Parrish JP, Trzupek JD, Hughes TV, Hwang I, Boger DL. Bioorg Med Chem. 2004;12:5845. doi: 10.1016/j.bmc.2004.08.032. [DOI] [PubMed] [Google Scholar]; (r) Parrish JP, Hughes TV, Hwang I, Boger DL. J Am Chem Soc. 2004;126:80. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]
- 20.MCBI: Boger DL, McKie JA, Cai H, Cacciari B, Baraldi PG. J Org Chem. 1996;61:1710. doi: 10.1021/jo952033g. CCBI: Boger DL, Han N, Tarby CM, Boyce CW, Cai H, Jin Q, Kitos PA. J Org Chem. 1996;61:48–94.; Boger DL, McKie JA, Han N, Tarby CM, Riggs HW, Kitos PA. Bioorg Med Chem Lett. 1996;6:659. [Google Scholar]; CNA: Boger DL, Turnbull P. J Org Chem. 1997;62:5844. Iso-CI/Iso-CBI: Boger DL, Garbaccio RM, Jin Q. J Org Chem. 1997;62:8875. CBIn: Boger DL, Turnbull P. J Org Chem. 1998;63:8004. CPyI: Boger DL, Boyce CW. J Org Chem. 2000;65:4088. doi: 10.1021/jo000177b. CBA: Parrish JP, Kastrinsky DB, Hwang I, Boger DL. J Org Chem. 2003;68:8984. doi: 10.1021/jo035119f.Parrish JP, Kastrinsky DB, Boger DL. Org Lett. 2003;5:2577. doi: 10.1021/ol035000t.
- 21.(a) Kumar R, Lown JW. Org Biomol Chem. 2003;1:2630. doi: 10.1039/b303650m. [DOI] [PubMed] [Google Scholar]; (b) Kumar R, Rai D, Ching S, Ko C, Lown JW. Heterocyclic Commun. 2002;8:521. [Google Scholar]; (c) Kumar R, Lown JW. Org Lett. 2002;4:1851. doi: 10.1021/ol020047k. [DOI] [PubMed] [Google Scholar]; (d) Jia G, Lown JW. Bioorg Med Chem. 2000;8:1607. doi: 10.1016/s0968-0896(00)00088-2. [DOI] [PubMed] [Google Scholar]; (e) Jia G, Iida H, Lown JW. Synlett. 2000;5:603. [Google Scholar]; (f) Jia G, Iida H, Lown JW. Heterocyclic Commun. 1999;5:497. [Google Scholar]; (g) Jia G, Iida H, Lown JW. Heterocyclic Commun. 1998;4:557. [Google Scholar]; (h) Kumar R, Rai D, Marcus SL, Ko SCC, Lown JW. Lett Org Chem. 2004;1:154. [Google Scholar]
- 22.(a) Philips BJ, Chang AY, Dervan PB, Beerman TA. Mol Pharmacol. 2005;67:877. doi: 10.1124/mol.104.006254. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Dziegielewski J, Chang AY, Dervan PB, Beerman TA. J Biol Chem. 2002;277:42431. doi: 10.1074/jbc.M207179200. [DOI] [PubMed] [Google Scholar]; (c) Chang AY, Dervan PB. J Am Chem Soc. 2000;122:4856. [Google Scholar]
- 23.(a) Minoshima M, Bando T, Sasaki S, Shinohara K, Shimizu T, Fijimoto J, Sugiyama H. J Am Chem Soc. 2007;129:5384. doi: 10.1021/ja065235a. [DOI] [PubMed] [Google Scholar]; (b) Shinohara K, Bando T, Sasaki S, Sakakihara Y, Minoshima M, Sugiyama H. Cancer Sci. 2006;27:219. doi: 10.1111/j.1349-7006.2006.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bando T, Sasaki S, Minoshima M, Dohno C, Shinohara K, Narita A, Sugiyama H. Bioconjugate Chem. 2006;17:715. doi: 10.1021/bc060022w. [DOI] [PubMed] [Google Scholar]; (d) Bando T, Narita A, Sasaki S, Sugiyama H. J Am Chem Soc. 2005;127:13890. doi: 10.1021/ja052412j. [DOI] [PubMed] [Google Scholar]; (e) Bando T, Narita A, Asada K, Ayame H, Sugiyama H. J Am Chem Soc. 2004;126:8948. doi: 10.1021/ja049398f. [DOI] [PubMed] [Google Scholar]
- 24.(a) Wang Y, Yuan H, Wright SC, Wang H, Larrick JW. Bioorg Med Chem. 2003;11:1569. doi: 10.1016/s0968-0896(02)00603-x. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Li L, Ye W, Tian Z, Jiang W, Wang H, Wright SC, Larrick JW. J Med Chem. 2003;46:634. doi: 10.1021/jm0203433. [DOI] [PubMed] [Google Scholar]; (c) Wang Y, Yuan H, Ye W, Wright SC, Wang H, Larrick JW. J Med Chem. 2000;43:1541. doi: 10.1021/jm990514c. [DOI] [PubMed] [Google Scholar]; (d) Wang Y, Li L, Tian Z, Jiang W, Larrick JW. Bioorg Med Chem. 2006;14:7854. doi: 10.1016/j.bmc.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 25.Chari RVJ, Jackel KA, Bourret LA, Derr SJ, Tadayoni BM, Mattocks KM, Shah SA, Liu C, Blattler WA, Goldmacher VS. Cancer Res. 1995;55:4079. [PubMed] [Google Scholar]
- 26.Lillo AM, Sun C, Gao C, Ditzel H, Parrish J, Gauss C–M, Moss J, Felding–Habermann B, Wirshing P, Boger DL, Janda KD. Chem Biol. 2004;11:897. doi: 10.1016/j.chembiol.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 27.(a) Tietze LF, Herzig T, Feuerstein T, Schuberth I. Eur J Org Chem. 2002;10:1634. [Google Scholar]; (b) Tietze LF, Feuerstein T, Fecher A, Haunert F, Panknin O, Borchers U, Schuberth I, Alves F. Angew Chem Int Ed. 2002;41:759. doi: 10.1002/1521-3773(20020301)41:5<759::aid-anie759>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]; (c) Tietze LF, Herzig T, Fecher A, Haunert F, Schuberth I. ChemBioChem. 2001;2:758. doi: 10.1002/1439-7633(20011001)2:10<758::AID-CBIC758>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; (d) Tietze LF, Major F, Schuberth I. Angew Chem Int Ed. 2006;45:6574. doi: 10.1002/anie.200600936. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Yuan H, Wright SC, Wang H, Larrick JW. Bioorg Med Chem. 2003;11:1569. doi: 10.1016/s0968-0896(02)00603-x. [DOI] [PubMed] [Google Scholar]
- 28.Jeffrey SC, Torgov MY, Andreyka JB, Boddington L, Cerveny CG, Denny WA, Gordon KA, Gustin D, Haugen J, Kline T, Nguyen MT, Senter PD. J Med Chem. 2005;48:1344. doi: 10.1021/jm040137q. [DOI] [PubMed] [Google Scholar]
- 29.Kline T, Torgov MY, Mendelsohn BA, Cerveny CG, Senter PD. Mol Pharmaceut. 2004;1:9. doi: 10.1021/mp0340183. [DOI] [PubMed] [Google Scholar]
- 30.(a) Hay MP, Anderson RF, Ferry DM, Wilson WR, Denny WA. J Med Chem. 2003;46:5533. doi: 10.1021/jm030308b. [DOI] [PubMed] [Google Scholar]; (b) Tercel M, Stribbling SM, Sheppard H, Siim BG, Wu K, Pullen SM, Botting KJ, Wilson WR, Denny WA. J Med Chem. 2003;46:2132. doi: 10.1021/jm020526p. [DOI] [PubMed] [Google Scholar]; (c) Gieseg MA, Matejovic J, Denny WA. Anti-Cancer Drug Design. 1999;14:77. [PubMed] [Google Scholar]; (d) Hay MP, Sykes BM, Denny WA, Wilson WR. Bioorg Med Chem Lett. 1999;9:2237. doi: 10.1016/s0960-894x(99)00381-9. [DOI] [PubMed] [Google Scholar]; (e) Atwell GJ, Milbank JJB, Wilson WR, Hogg A, Denny WA. J Med Chem. 1999;42:3400. doi: 10.1021/jm990136b. [DOI] [PubMed] [Google Scholar]; (f) Atwell GJ, Tercel M, Boyd M, Wilson WR, Denny WA. J Org Chem. 1998;63:9414. [Google Scholar]; (g) Atwell GJ, Wilson WR, Denny WA. Bioorg Med Chem Lett. 1997;7:1493. [Google Scholar]
- 31.Townes H, Summerville K, Purnell B, Hooker M, Madsen E, Hudson S, Lee M. Med Chem Res. 2002;11:248. [Google Scholar]
- 32.Boger DL, Garbaccio RM. J Org Chem. 1999;69:8350. doi: 10.1021/jo991301y. [DOI] [PubMed] [Google Scholar]
- 33.Carzelesin: Aristoff PA. Adv Med Chem. 1993;2:67.
- 34.KW-2189: Kobayashi E, Okamoto A, Asada M, Okabe M, Nagamura S, Asai A, Saito H, Gomi K, Hirata T. Cancer Res. 1994;54:2404.Amishiro N, Nagamura S, Kobayashi E, Okamoto A, Gomi K, Okabe M, Saito H. Bioorg Med Chem. 2000;8:1637. doi: 10.1016/s0968-0896(00)00086-9.Amishiro N, Nagamura S, Kobayashi E, Gomi K, Saito H. J Med Chem. 1999;42:669. doi: 10.1021/jm980559y.Nagamura S, Asai A, Kanda Y, Kobayashi E, Gomi K, Saito H. Chem Pharm Bull. 1996;44:1723. doi: 10.1248/cpb.44.1723.Nagamura S, Kanda Y, Kobayashi E, Gomi K, Saito H. Chem Pharm Bull. 1995:43. doi: 10.1248/cpb.43.1530.
- 35.CBI: Boger DL, Boyce CW, Garbaccio RM, Searcey M, Jin Q. Synthesis. 1999:1505.
- 36.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q.Reviews on reductive activation: Papadopoulou MV, Bloomer WD. Drugs Future. 2004;29:807.Jaffar M, Stratford IJ. Exp Opin Ther Patents. 1999;9:1371.Patterson LH, Raleigh SM. Biomed Health Res. 1998;25:72.
- 37.Denny WA. Hypoxia-Selective Cytotoxins. In: Foye WO, editor. Cancer Chemotherapeutic Agents. ACS; Washington DC: 1995. pp. 483–500. [Google Scholar]
- 38.Paz MM, Hopkins PB. J Am Chem Soc. 1997;119:5999. [Google Scholar]; Williams RM, Rajski SR, Rollins SB. Chem Biol. 1997;4:127. doi: 10.1016/s1074-5521(97)90256-8. [DOI] [PubMed] [Google Scholar]
- 39.Tepe JJ, Kosogof C, Williams RM. Tetrahedron. 2002;58:3553. [Google Scholar]; Tepe JJ, Williams RM. Angew Chem, Int Ed Engl. 1999;38:3501. doi: 10.1002/(sici)1521-3773(19991203)38:23<3501::aid-anie3501>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 40.Greck C, Bischoff L, Girard A, Hajicek J, Genêt JP. Bull Soc Chim Fr. 1994;131:429. [Google Scholar]
- 41.Neumann U, Gütschow M. J Biol Chem. 1994;269:21561. [PubMed] [Google Scholar]
- 42.Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. Tetrahedron. 1991;47:2661. [PubMed] [Google Scholar]
- 43.Li LH, Kelly RC, Warpehoski MA, McGovern JP, Gebhard I, DeKoning TF. Invest New Drugs. 1991;9:137. doi: 10.1007/BF00175081. [DOI] [PubMed] [Google Scholar]
- 44.This appears to arise from damage to the intraperitioneal cavity or its organs that originate with the bolus drug administration.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and compound characterizations are provided.