

Abstract
Exchange proteins directly activated by cAMP (Epac) is a family of cAMP-binding domain containing proteins that play important roles in mediating the effects of cAMP through the activation of down-stream small GTPases, Ras-proximate proteins. To delineate the mechanism of Epac activation, we probed the conformation and structural dynamics of Epac using amide hydrogen/deuterium (H/D) exchange coupled with Fourier transform infrared spectroscopy (FT-IR) and structural modeling. Our studies show that unlike that of cAMP-dependent protein kinase (PKA), the classic intracellular cAMP receptor, binding of cAMP to Epac does not induce significant changes in overall secondary structure and structural dynamics, as measured by FT-IR and the rate of H/D exchange, respectively. These results suggest that Epac activation does not involve significant changes in the amount of exposed surface areas as in the case of PKA activation and conformational changes induced by cAMP in Epac are most likely confined to small local regions. Homology modeling and comparative structural analyses of the CBDs of Epac and PKA lead us to propose a model of Epac activation. Based on our model, Epac activation by cAMP employs the same underlying structural principal utilized by PKA although the detailed structural and conformational changes associated with Epac and PKA activation are significantly different. In addition, we predict that during Epac activation the first β strand of the switchboard switches conformation to a α-helix, which folds back to the β barrel core of the CBD and interacts directly with cAMP to form the base of the cAMP-binding pocket.
Keywords: cyclic adenosine 3′,5′-monophosphate (cAMP); exchange protein directly activated by cAMP (Epac); Fourier transform infrared spectroscopy (FT-IR); cAMP-dependent protein kinase (PKA); hydrogen/deuterium exchange
Exchange proteins directly activated by cAMP (Epac1) and cAMP-dependent protein kinase (PKA) are two cAMP receptors that mediate the intracellular actions of the second messenger cyclic AMP in eukaryotic cells. While PKA phosphorylates multiple down-stream substrates, Epac proteins act as guanine nucleotide exchange factors (GEFs) for the small GTP-binding proteins Rap1 and Rap2. The discovery of Epac proteins in 1998 as the second family of intracellular cAMP receptors (1;2) has opened up a new chapter for the studies of cAMP-mediated signaling as Epac proteins have been implicated in a myriad of cAMP-related cellular functions, such as exocytosis (3–5), cell adhesion (6;7), cell-cell junction (8;9), and differentiation (10). Furthermore, Epac and PKA have been shown to exert synergistic or antagonistic effects on down-stream signaling targets depending upon the specific cellular contexts (11;12). The existence of two highly coordinated cAMP effectors therefore provides a mechanism for a more precise and integrated control of the cAMP signaling pathways in a spatial and temporal manner (13).
Both Epac and PKA are regulated by a cAMP binding domain (CBD) that is evolutionally conserved to the CBD of the bacterial transcriptional factor, cAMP receptor protein. CBD, the only common structural module between the PKA and Epac, acts as a molecular switch for sensing the intracellular second messenger cAMP levels. In the case of PKA, the regulatory (R) and catalytic (C) components of PKA are encoded as two separated polypeptides. Binding of cAMP to the tandem CBDs of the R subunit induces a conformational change that leads to dissociation of the holoenzyme (R2C2) and subsequent activation of PKA (14). X-ray crystal structures of PKA holoenzyme complex and individual subunits reveal a molecular mechanism for cAMP-mediated activation of PKA (15–17). The R and C subunits form a large interface in the PKA holoenzyme complex with several key residues (Tyr247 and Trp196) of the C subunit binding directly to the phosphate binding cassette (PBC) of the CBD-A in the R subunit (17). cAMP not only competes directly with the C subunit for these interactions but also induces major conformational changes in the R subunit, particularly the helical subdomain of CBD, the inhibitor sequence and the linker region (16;17). These conformational changes lead to the reorientation and displacement of the inhibitor sequence from the active site cleft of C, consequently the activation of PKA. On the other hand, the CBD in Epac is covalently connected to the catalytic GEF domain as a single polypeptide chain and the intramolecular interaction between the CBD and GEF domains sterically blocks the access of downstream effector, Rap. Recently, the crystal structural of Epac2 is solved in the absence of cAMP (18). In this auto-inhibited Epac2 structure, the second CBD of Epac2, which is common in Epac1 and Epac2, is loosely connected to the CDC-25 homology guanine nucleotide exchange (GEF) domain by the central “switchboard” and an ionic “latch” (18). Unlike the extensive interface between the R and C subunits of PKA holoenzyme, the intramolecular interaction between the regulatory and catalytic regions in Epac2 is surprisingly brief, suggesting that the detailed mechanisms of PKA and Epac activation by cAMP will most likely be different at the structural level.
In an earlier study, we probed the conformation states of PKA holoenzyme complexes using amide hydrogen-deuterium (H/D) exchange Fourier transform infrared spectroscopy (FT-IR) and chemical protein footprinting (19). Consistent with the crystal structural study of PKA holoenzyme complex, significant conformational changes were observed during PKA activation. In addition, binding of cAMP to PKA holoenzymes leads to a downshift in the IR wavenumber for both the alpha-helix and beta-strand bands, suggesting that R and C subunits become overall more dynamic in the holoenzyme complexes (19). Since the structure of the Epac in its active state is not currently available, the molecular mechanism of Epac activation is not known. In the current study, we investigated the conformational and dynamic states of Epac1 in the presence and absence of cAMP using H/D exchange FT-IR and structural modeling to delineate the mechanism of Epac activation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Δ(1–148)Epac1 was expressed as a GST-fusion protein (pGEX4T2) in E. coli and purified as described previously (20). Briefly, E. coli BL21-DE3 cells carrying Δ(1–148)Epac1 expression vector were grown in LB medium containing 100 μg/ml ampicillin at 37 °C. Protein expression was induced with 100 μM IPTG at room temperature for 9 h after A600 reached between 0.8 and 1.2. Cells were harvested, resuspended in PBS buffer with 10 % (v/v) glycerol, 1 mM EDTA, 5 mM DTT and 1 mM PMSF and lysed using a sonicator. After centrifugation the supernatant was loaded onto a pre-equilibrated GSH column (Pharmacia). The column was washed sequentially with 1) 360 mM NaCl in PBS; 2) 50 mM Tris-HCl (pH 7.6), 100 mM KCl, 10 mM MgCl2, 10 % glycerol, 5 mM DTT, 2 mM ATP; and 3) 50 mM Tris-HCl (pH 7.6), 50 mM NaCl, 10 % glycerol, 5 mM DTT. GST-Δ(1–148)Epac1 eluted from the column was cleaved with 150 units of immobilized thrombin for three hours at 4°C to remove the GST tag. After removing immobilized thrombin beads by centrifugation, Δ(1–148)Epac1 was further purified by gel filtration on Superdex 200 in buffer A containing 50 mM Tris-HCl (pH 7.6), 50 mM NaCl, 5 % glycerol, and 5 mM DTT. Purified proteins were judged at least 95% pure by SDS-polyacrylamide gel electrophoresis.
FT-IR Spectroscopy
FT-IR spectra were measured with a Bomem MB series Fourier Transform Infrared Spectrometer (Quebec, Canada) equipped with a dTGS detector and purged constantly with dry air. Protein samples (~15 mg/ml) were warmed up to room temperature and loaded in a CaF2 cell with a 7.5 μm spacer. For each spectrum, a 256-scan interferogram was collected in single-beam mode with a 4 cm−1 resolution at a rate of 3 scans per second. Reference spectra were recorded under identical conditions with only the corresponding buffer in the cell. Protein spectra were obtained using a previously established protocol (21). A straight base line between 2000 and 1750 cm−1 was used as the standard to judge the success of water subtraction. Second-derivative spectra were obtained with a seven-point Savitsky-Golay derivative function, baseline-corrected and area-normalized as described (21). Secondary structure content of Δ(1–148)Epac1 was calculated by curve-fitting analysis of the inverted second-derivative spectrum in the amide I band range from 1600 to 1700 cm−1 (22). This band is ascribed to >C=O stretching vibration of the peptide bond (23). It was assumed that the fraction of residues composing each secondary structural element is proportional to the relative percent area of the associated vibrational band (24;25).
Hydrogen-Deuterium Exchange Measurement
100 μl of protein in buffer A was lyophilized at room temperature to dryness. There was no significant difference in the catalytic activity and the contents of α-helices and β-sheets between the lyophilized (after rehydration) and the unlyophilized proteins measured by FT-IR. Samples for H/D exchange experiments were prepared by dissolving the lyophilized 100 μl of Δ(1–148)Epac1 or buffer solution with or without 300 μM of cAMP in same volume of D2O. The reconstituted sample was injected immediately into an IR cell with a pathlength of 50 μm. One minute after the addition of D2O, single-beam spectra were recorded using kinetic scanning mode. FT-IR spectra were recorded at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 15, 20, 30, 40, 50, 60, 90, 120, 150, and 180 min in D2O. Eight scans were collected for each time interval between 1 to 10 min while 64 and 128 scans for each time interval between 11 to 90 min and longer, respectively. In order to compare the FT-IR spectra in H2O and D2O, we normalized the amide I band in H2O to the amide I band in D2O at 1 min. The spectrum collected after 24 hours exchange was used as the fully deuterated spectrum.
Calculation of Amide Proton Exchange Rate
We monitored the H/D exchange of Epac1 by following apparent intensity changes of the amide II band, located around 1548 cm−1 that is attributed to a combination of N-H in-plane bending and C-N stretching vibrations in peptide bond, because this band dose not adversely interfere with absorption bands of H2O, HOD, or D2O (24). As N-H in protein is exchanged into N-D in D2O, the absorption peak of N-D bending vibration at about 1450 cm−1 is strengthened while the N-H absorption peak is decreased. The fraction of unexchanged amide proton, F, was calculated at various time intervals using Equ. 1
| (1) |
where AI and AII are the absorbance maximum of the amide I and II bands, respectively. AII∞ is the amide II absorbance maximum of fully deuterated protein; and ω is the ratio of AIIo/AIo, with AIIo and AIo being the respective absorbance maximum for the amide II and amide I bands of PKA in H2O (26).
The exchange kinetic parameters were fitted from equation below:
| (2) |
where F is amide proton fraction at given time t; k1 and k2 are the intermediate and slow exchange rate, respectively; A1, A2 and C are constants. F0 is the remaining amide proton fraction at 1 min.
Structural Modeling of Epac1
Epac1 structural model is initially generated using homology modeling software MODELLER ((27) http://www.salilab.org/modeller/). The recently solved Epac2 three-dimensional coordinates (2BYV) (18) was used as the template. The initial Epac1 model was further refined using the molecular dynamics simulation package GROMACS (http://www.gromacs.org/) to obtain the final Epac1 model.
RESULTS
FT-IR Spectra of Δ(1–148)Epac1 in H2O Solution
We used Δ(1–148)Epac1 in this study as published literatures so far indicate that the function of the N-terminal 148 residues is mainly associated with cellular targeting of Epac1 in vivo (28;29), which is further supported by extensive biophysical and biochemical characterization by Wittinghofer and colleagues showing that Δ(1–148)Epac1 retains all measurable biochemical properties such as cAMP binding and Rap activation (30). Removal of the first 148 residues significantly improves Epac1 solubility and increases expression levels in E. coli while the full-length Epac1 protein cannot be expressed and purified in sufficient quantity ((31) and unpublished data). The FT-IR absorption spectra of Δ(1–148)Epac1 in the presence and absence of 300 μM of cAMP in buffer A were shown in Figure 1A. The second-derivative amide I spectra of Δ(1–148)Epac1 (Figure 1B) all exhibited basic band components that can be assigned to secondary structure components. Quantitative analysis of the secondary structure of Δ(1–148)Epac1 by curve-fitting revealed that Δ(1–148)Epac1 contained 65% α helices, 14% β strands, and 21% β turns (Figure 2A, Table I). Binding of cAMP to Δ(1–148)Epac1 did not lead to significant changes in FT-IR second-derivative spectra and overall secondary structure (Figure 2B, Table 1).
Figure 1. FT-IR spectra of Epac1 in the absence and presence of cAMP.

(A) IR absorption spectra of Δ(1–148)Epac1 in the absence (solid line) and presence of 300 μM cAMP (doted line). (B) Secondary-derivative amide I spectra of Δ(1–148)Epac1 in the absence (solid line) and presence of 300 μM cAMP (doted line).
Figure 2. Calculation of secondary structural components of PKA holoenzymes.

Deconvolution of inverted secondary-derivative amide I spectra of Δ(1–148)Epac1 in the absence (A) and presence of 300 μM cAMP (B). Secondary structure contents were obtained from curve fitting of the second derivative FT-IR spectra using a 7-point Savitsky-Golay derivative function.
Table 1.
Secondary Structure Contents of Δ(1–148)Epac1 Determined by FT-IRa.
| Δ(1–148)Epac1 | Δ(1–148)Epac1 with 300 μM cAMP | ||||
|---|---|---|---|---|---|
| Frequency (cm−1) | Percent | Assignment | Frequency (cm−1) | Percent | Assignment |
| 1690.1–1684.3 | 11.9± 0.4 | β Turn | 1689.1–1683.7 | 12.1± 1.2 | β Turn |
| 1671.4±0.2 | 8.8± 2.0 | β Turn | 1671.2±0.4 | 6.9± 0.1 | β Turn |
| 1654.2±0.1 | 65.4± 0.3 | α helix | 1654.2±0.1 | 66.8± 1.1 | α helix |
| 1634.5±0.3 | 13.9± 2.5 | β Strand | 1634.9±0.4 | 14.2± 2.9 | β Strand |
Secondary structure contents were obtained from curve fitting of the second derivative FT-IR spectra using a 7-point Savitsky-Golay derivative function as shown in Figure 2.
H/D Exchanges of Δ(1–148)Epac1 Monitored by FT-IR
To explore the intrinsic protein dynamics and conformational flexibility of Epac1, H/D exchange of Δ(1–148)Epac1 were monitored by FT-IR. Figure 3A showed an overlay of the representative absorption spectra of Δ(1–148)Epac1 recorded at 1, 3, 5, 10, 30, 60, 120, and 180 min in D2O with spectra of the proteins in H2O and fully deuterated in D2O plotted as references. While the Δ(1–148)Epac1 spectra in H2O exhibited characteristic amide I and II band maxima at 1652 and 1548 cm−1, respectively, H/D exchange in D2O led to a time-dependent isotopic shift of the amide II band from 1548 to 1455 cm−1. This effect is indicative of NH to ND exchange of peptide backbone groups that causes a downshift of approximately 100 cm−1 in the vibrational frequency of the amide II band (32).
Figure 3. H/D exchange of Epac1 as monitored by FT-IR.

Infrared spectra of Δ(1–148)Epac1 in the absence (A) and presence of 300 μM cAMP (B) at various time intervals after being dissolved in D2O at room temperature. Secondary-derivative amide I spectra of Δ(1–148)Epac1 in the absence (C) and presence of 300 μM cAMP (D) at various time intervals after being dissolved in D2O at room temperature. The spectra of proteins in H2O and in D2O after 24 hours of exchange drawn in solid and dashed lines, respectively, were included for comparison.
To dissect the overlapping band components of the amide I region, second-derivative analyses were performed. Figure 3B showed an overlay of second-derivative spectra of Δ(1–148)Epac1 as a function of H/D exchange time, respectively. Almost immediately after dissolving the proteins in D2O, the 1655 cm−1 band in the α helix spectral region of Δ(1–148)Epac1 underwent an initial large drop in intensity and 2 cm−1 downshift to 1653 cm−1 at the 1 minute time measurement, followed by a gradual intensity decrease and downshift to 1651 cm−1 accompanied by a shoulder around 1646 cm−1. The downshift and decrease in intensity of the 1655 cm−1 band were completed in about 60 min. On the other hand, the effects of deuterium exchange on the β–sheet spectral region were different. The 1634.5 cm−1 β–sheet band increased in intensity initially at the 1 minute time measurement, followed by a gradual downshifted to form a shoulder around 1629 cm−1 in D2O (Figure 3B).
H/D Exchanges of Δ(1–148)Epac1 in the Presence of camp
The overall H/D exchange absorption profiles of Δ(1–148)Epac1 in the presence of cAMP were very similar to those in the absence of cAMP (Figure 3C & D). The secondary derivative spectra of Δ(1–148)Epac1 H/D exchange in the presence of cAMP were also similar but with noticeable differences (Figure 3B & D). Unlike the spectral shifts in the absence of cAMP, the downshift and decrease in intensity of the 1655 cm−1 band were slower and didn’t complete after 180 min. In addition, the 1655 cm−1 band downshifted 5 cm−1 in the presence of cAMP instead of 4 cm−1 as observed in the absence of cAMP and the shoulder around 1646 cm−1 observed in the absence of cAMP became much more prominent in the presence of cAMP to form a noticeable peak at 1643 cm−1. Similar to that of Δ(1–148)Epac1 in the absence of cAMP, the 1634.5 cm−1 β–sheet band also increased in intensity initially at the 1 minute time measurement followed by a gradual downshifted as in the absence of cAMP. However, instead of a shoulder around 1629 cm−1, a distinct peak at 1627 cm−1 was observed for the fully deuterated Δ(1–148)Epac1 in the presence of cAMP.
Differences in Overall H/D Exchange between Δ(1–148)Epac1 and Δ(1–148)Epac1-cAMP
The overall H/D exchange rates of Δ(1–148)Epac1 in the absence and presence of cAMP were estimated by plotting the fraction of unexchanged amide protons, calculated from amide II band data using Equation 1, as a function of time. In general, all amide protons in proteins can be divided into three classes (33–35): 1) fast exchange protons, which are most likely located on the surface of protein or in regions that are easily solvent accessible; 2) amide protons with intermediate rates located in flexible buried regions; 3) the slow exchange fraction located in the core region of the protein. The fraction of the unexchanged amide protons at the first exchange time point (1 min) for Δ(1–148)Epac1 in the absence and presence of cAMP were around 20% as shown in Figure 4, suggesting that the majority of the amide protons exchanged so rapidly that their exchange was completed within the time interval of the acquisition of the first time point. Therefore, only the intermediate and slow exchange protons can be practically monitored semi-quantitatively over the time range employed in this study. A two-exponential decay model (Equation 2) was used to describe the exchange reaction of the remaining amide protons within the experimental time frame and the resolved parameters were summarized in Table 2. Because of the complexity of the overall H/D exchange reaction in protein, no attempt was made to quantitatively associate these parameters with any actual physical properties; instead, they were only used qualitatively to assess the overall dynamics of Δ(1–148)Epac1 in the absence and presence of cAMP. Overall, there are no significant differences in H/D exchange rates between active and inactive Epac1 states (Table 2 and Figure 4).
Figure 4. Amide proton exchange rates of Epac1.

Fraction of unexchanged amide protons as a function of exposure time in D2O for the Δ(1–148)Epac1 in the absence (triangles) and presence (inverted triangles) of 300 μM cAMP. The lines represent best fits to the data points using a two-exponential function as described in Equation 2.
Table 2.
Fitted Exchange Parameters for Δ(1–148)Epac1a.
| Parametersb | Δ(1–148)Epac1 | Δ(1–148)Epac1+cAMP |
|---|---|---|
| A1 | 8.0 ± 0.3 | 7.9 ± 0.3 |
| A2 | 9.6 ± 0.3 | 9.9 ± 0.3 |
| k1(min−1) | 0.21 ± 0.02 | 0.26 ± 0.02 |
| k2(min−1) | 0.02 ± 0.01 | 0.02 ± 0.01 |
| c | 0.8 ± 0.2 | 1.3 ± 0.1 |
| F0 (%) | 17.02 | 17.17 |
Parameters were derived from fitting the exchange data in Figure 4 to a two-exponential model as described by Equation 2.
k1 and k2 are the intermediate and slow exchange rates, respectively; A1, A2 and C are the constants while F0 is the remaining amide proton fraction after 1 min of D2O exposure.
Structural Modeling of Epac1
Recently, the three-dimensional structural of Epac2 in its cAMP-free, inactive state have been solved (18). Since Epac1 is more than 50% identical to Epac2 in amino acid sequence, we constructed an Epac1 three-dimensional model based on the crystal structure of Epac2 using homology modeling (Figure 5A). The final model of Epac1 has a Ramachandran Z-score of −0.572, which is similar to that of the Epac2 template structure (−0.411). Because of the extensive sequence homology between Epac1 and 2, the three-dimensional model of Epac1 should be reliable for further structural analysis. It is not surprising that the overall structural architecture of Epac1 is almost identical to the crystal structure of Epac2. Important contact points between the catalytic core and regulatory regions revealed by the Epac2 structure are also preserved in the Epac1 model. For example, the three-stand β-sheet formed by the C-terminus of CBD and the N-terminus of the REM domain interacts with the loop of the helical hairpin of the catalytic core through an extensive hydrogen bonding network to form the “switchboard” (Figure 5B) while the second helix of the hairpin is anchored by Phe 797 and Ile 785, which forms a hydrophobic core with the two helices of the REM domain (Figure 5C). Like Epac2, there is only one brief contact point between the CBD and the catalytic core of Epac1. However, the detailed molecular interactions for this specific contact point are different between Epac1 and 2. While the hydrogen bonding between Asp 750 (Asp 863 in Epac2) of the catalytic core and Gln 168 (Gln 303 in Epac2) of the CBD is preserved, the Arg 886 residues that interacts with the Asp 307 and Glu 332 in Epac2 is replaced by a tryptophan in Epac1 (Figure 5D). Ionic interactions between Arg 886 and Asp 307 and between Arg 886 and Glu 332 in Epac2 are substituted by potential hydrogen bonding between Trp 753 and His 200 and Trp 753 and Gln 168, as well as ionic interactions between Arg 756 and Asp 196 in Epac1 (Figure 5D).
Figure 5. Structural Model of Epac1.

(A) Ribbon diagram of Epac1. The DEP, CBD, REM, RA, and CDC-25 homology domain are colored in pink, red, green, blue, and cyan, respectively. (B) The 31 hydrogen bonding network (dotted lines) of the switchboard, a five-strand, β-sheet like structure formed by the C-terminus of CBD (red), the N-terminus of the REM domain (green) and the loop of the helical hairpin of the catalytic core (cyan). (C) The hydrophobic core formed by the C-terminal helix of the helical hairpin of the catalytic core (cyan) and the REM domain (green). (D) Interactions between CBD and CDC-25 homology domain of Epac1. Hydrogen bonding and ionic interactions are shown with dotted and dash lines, respectively.
Mechanism of Epac Activation
Our amide H/D exchange and FT-IR studies suggest that Epac activation by cAMP likely involves local motion, such as hinge movements. Therefore, we subjected Epac1 model to further structural analysis using the HingeMaster server, which predicts hinge locations in single protein structures using algorithm combines FlexOracle, TLSMD, StoneHinge and NSHP hinge predictors for maximum accuracy (36) (The Yale Morph Server (http://molmovdb.org)). Our analysis revealed a major hinge in Epac1 between residues 310–345 (Figure 6A). This region has been identified as the “lid” region at the C-terminus of CBD that plays an important role in the communication between the regulatory and catalytic domains and is pivotal for the activation of Epac by cAMP (29;37;38). Based on extensive structural, biochemical and biophysical studies by Bos and Wittinghofer groups, it was proposed that regulation of Epac is dictated by the equilibrium between an inactive and an active state. In the absence of cAMP, Epac exists mostly in the inactive states, in which the lid region, particularly the conserved VLVLE motif, interacts with the catalytic domain and prevents the interaction between Epac and Rap. Binding of cAMP to Epac leads to a conformational change that allow the lid/VLVLE motif to move away from the catalytic core and closer to the cAMP binding pocket to interact directly with the cAMP, and consequently releases the inhibitory effect of the VLVLE and permits the binding of Rap (38). The crystal structure of the full-length Epac2 in its inactive, ligand-free state verified the existence of the lid region as part of the “switchboard”, a five-strand, β-sheet like structure formed by the C-terminus of CBD, the N-terminus of the REM domain and the loop of the helical hairpin of the catalytic core (18). However, the lid/VLVLE motif is not in the form of a helix as in the case of PKA, nor does the lid/VLVLE motif interact directly with catalytic core as original proposed (38). To further elucidate the mechanism of Epac activation, we carefully analyzed the structural and sequence homology between Epac and PKA at the lid region. As shown in Figure 6B, although the lid region is not conserved in sequence among all CBD proteins, there is significant sequence homology between Epac and the first cAMP binding domain of type I regularity subunit of PKA (PKA-RI) in this specific region. Interestingly, while the lid regions of Epac and PKA-RI are similar in sequences, the secondary structure components at this region are different between Epac and PKA-RI. The lid of Epac assumes a β-sheet structure. On the other hand, the lid of PKA-RI is helical both in the presence and absence of cAMP (16;39) (Figure 6B). In fact, the corresponding lid regions of all known CBD proteins except Epac exist as helical structures although they may differ in the 3-dimensional arrangement (40). Based on these comparative analyses, we proposed the following based on a previously suggested model by Rehemann et al (18;37;38): In the absence of cAMP, the regulatory and the catalytic regions of Epac are hold together by the switchboard to keep the CBD in close proximity of the catalytic core to prevent the binding of Rap. The lid/VLVLE motif is anchored and stabilized by the N-terminus of REM in a three-strand β-sheet structure as part of the switch board. Binding of cAMP drags the highly conserved PBC in the direction of the cAMP and reorients B-helix, tethered to the PBC by a hydrophobic hinge, toward the β barrel of the CBD. This hinge movement pulls the β strand 1 away from strands 2 and 3 of the three-strand β-sheet of the switchboard. We further predict that break away of the β strand 1 from the rest of β-sheet enables it to switch to an α-helix, which folds back to the β barrel and interacts directly with cAMP to form the base of the cAMP-binding pocket. The net conformational changes induced upon cAMP binding result in an open and active Epac conformation, in which the reorientation of the CBD/DEP relative to the rest of the Epac molecule relieves the catalytic core from the inhibitory contact imposed by the CBD. This transition between the close/inactive and open/active Epac conformation induced by cAMP has been experimentally demonstrated by an apparent cAMP-induced decrease in florescence resonance energy transfer between enhanced cyan fluorescent protein and citrine fused to the N- and C-terminus of Epac1, respectively (41).
Figure 6. Structure and sequence analyses of the lid region of Epac.
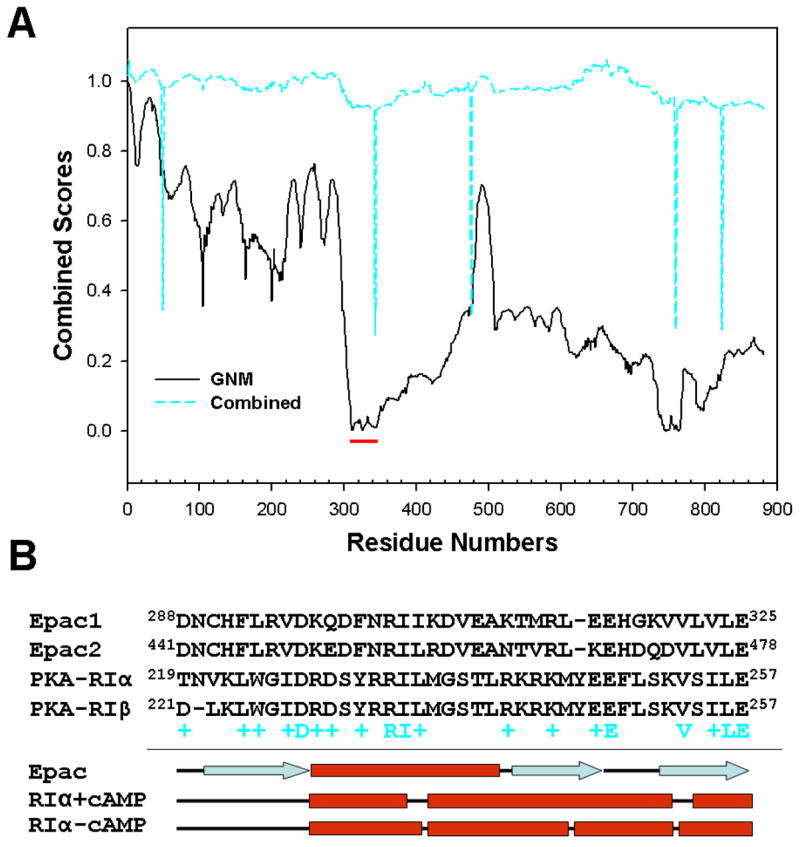
(A) Hinge prediction results of Epac1 structure derived from analysis using the HingeMaster server (http://molmovdb.org). Solid black line: The Gaussian Network Model (GNM) first normal mode displacement score). Minima correspond to hinges. Cyan dashed line: combined predictor score using algorithm combines FlexOracle, TLSMD, StoneHinge and NSHP hinge predictors. Minima correspond to hinges. Red bar highlights the lid region of Epac1. (B) Primary sequence and secondary structure alignments of the lid regions of human Epac and PKA-RI proteins. Secondary structures for Epac and PKA-RI are assigned using X-ray crystal structures 2BYV, 1RGS, and 1RL3.
DISCUSSION
The cAMP-binding domain, conserved from bacteria to human, is an ancient structural module that acts as a molecular switch for controlling various biological functions. While CBD is a highly conserved and compact structural entity, the functionalities that it regulates varies from DNA binding in the cAMP-receptor protein and protein phosphorylation in PKA to membrane potential in cyclic-nucleotide-regulated ion channels. The Epac family proteins represent the newest members of the CBD-containing proteins. Unlike PKA, the classic eukaryotic intracellular cAMP receptor, Epac proteins are encoded by single polypeptide chains with the CBD being covalently linked to catalytic domain, the guanine nucleotide exchange factor domain (1;2). On the other hand, CBD acts as an inhibitory unit for the catalytic component in both PKA and Epac in the ligand-free state and cAMP binding leads to the activation of both proteins. One important question that reminds to be answered is whether similar structural mechanisms are employed by the CBD during the activation of Epac and PKA.
Extensive biochemical and structural analyses of PKA, particularly the crystal structures of the cAMP-bound regulatory subunit and a partial PKA holoenzyme complex, have revealed a clear picture regarding the interactions between CBD and the catalytic subunit and detailed mechanism of PKA activation by cAMP (16;17). In the PKA holoenzyme complex of RIα(91–244):C, The CBD-A of RIα(91–244) makes extensive contact with the C-subunit. The B and C-helices of CBD-A snap into a single extended helix that is anchored and stabilized by the catalytic subunit, in particular, the activation loop. The PBC is stretched away from the β barrel to interact directly with the G helix of the C subunit in the complex (17). Activation of PKA holoenzyme, initiated by the binding of cAMP to the R subunit, is accompanied by widespread conformational changes of the R subunit, demonstrated extensively by X-ray crystallographic analyses (16;17) and biophysical studies in solution (19). Binding of the cAMP results in the retraction of the PBC in the direction of cAMP binding pocket and global reorientation of the sub-helical domain of CBD-A. The pivot motion around the hydrophobic hinge dislodges the single extended B/C helix, and subsequently the inhibitor sequence, from the docking site on the C-subunit. In the absence of stabilization/anchoring effects of the C subunit, the B/C helix bends in the middle to form two individual helices with the C-helix portion folds back onto the β barrel to form the “lid” of the cAMP-binding pocket. These extensive cAMP-induced conformation changes eventually cause the dissociation of the PKA holoenzyme.
Since the structure of cAMP-bound Epac in its active state is not currently available, we decided to apply H/D exchange analysis to investigate the conformational and dynamic changes associated with Epac1 activation. Amide H/D exchange has been used extensively to analyze structural dynamics and conformational changes in proteins as the rate at which an amide proton exchanges with the solvent is largely determined by the flexibility and motion around the exchanging proton. To our surprise, unlike PKA, no significant changes in overall secondary structure and structural dynamics, as measured by FT-IR and the rate of H/D exchange, respectively, were observed for Epac1 between cAMP-bound and ligand-free state. These results suggest that Epac1 activation does not involve significant changes in the amount of exposed surface areas as in the case of PKA activation and conformational changes induced by cAMP in Epac1 are most likely local such as hinge motion. These conclusions are consistent with previous biochemical studies as well as the recently solved ligand-free Epac2 structure (18;29;38). In its auto-inhibited state, there are only one direct contact point between the CBD and catalytic core of Epac, described as the “ionic latch”. Because of its proximity to the Rap1-binding surface, this ionic latch is believed to be responsible for the inhibitory effect exerted by the CBD on the catalytic activity. Interestingly, the key Arg residue involved in ionic interactions with the CBD in Epac2 is not conserved and replaced by a tryptophan in Epac1, which instead forms hydrogen bonds with the CBD so that the overall structure and function of the ionic latch is preserved in Epac1 (Figure 5D). The CBD is anchored to the catalytic core indirectly by the REM domain through the so called “switchboard”, which is a five-strand β-sheet like structure consisted of the C-terminus lid of the CBD, the N-terminus of the REM domain and hairpin of the catalytic core (18). The first part of the switchboard, corresponding to the C-terminus of CBD, described as the lid region (B and C helices) of the cAMP binding pocket in PKA, has been suggested to play a critical role in the activation of Epac. On the other hand, the second part of the switchboard, including the N-terminus of REM domain and the hairpin of the catalytic core, functions to anchor the CBD but most likely does not play important roles during Epac activation by cAMP as it has been demonstrated that isolated CBD halve, cleaved between the CBD and REM domains, can completely inhibit the catalytic activity of the GEF halve and this inhibition is relieved in response to cAMP binding just like in the intact Epac1 (29).
The cAMP-free Epac2 structure reveals an unexpected observation: despite the lid region of Epac2 and PKA share extensive sequence homology, the corresponding c helix of PKA-RI CBD-A in Epac actually forms the β-strand one of the switchboard. In light of this substantial divergence of the lid structure between Epac and PKA, we performed an in-depth comparative sequence and structure analyses of Epac and PKA, our study leads to a refined model of Epac activation, in which upon binding of cAMP the β strand 1 breaks away from strands 2 and 3 of the three-strand β-sheet of the switchboard, folds back as an α helix towards the β barrel and interacts directly with cAMP to form the base of the cAMP-binding pocket. This localized hinge motion coupled with a switch of the secondary structure of the lid reorients the GEF domain relative to CBD that leads to the ultimate exposure of the catalytic core of Epac for the access of Rap GTPase, therefore the activation of Epac. This model is based on the initial model proposed by Rehmann and colleagues (18;37) and supported by several lines of evidences. First, secondary structure prediction based on several most commonly used algorithms all predicted α helical structure, instead of a β strand for the Epac lid region. Second, the corresponding homologous region of all other known CBD proteins all exist as helical structures despite they may differ in the 3-dimensional arrangement (40). The unique β strand structure observed in Epac is most likely due to the stabilization effect provided by the strands 2 and 3 of the three-strand β-sheet of the switchboard. In addition, replacing the conserved VLVLE motif in strand 2 with high helical propensity amino acid side chains such as alanines results in the constitutive activation of Epac (38). It is conceivable that these high helical propensity amino acid substitutions may disrupt the three-strand β-sheet structure, destabilize the interactions between strands 1 and 2 and consequently allow the lid (strand 1) switch back to a helix because of its natural helical forming potential. Finally, our model also suggests that while the detailed structural configurations may differ, the same underline principal is employed by CBDs to control a diverse array of functionalities in different CBD proteins, as demonstrated in the cAMP receptor protein, PKA and cyclic-nucleotide-regulated ion channels systems (16;17;42;43).
Acknowledgments
We thank Dr. James C. Lee for the use of the FT-IR instrument and Dr. R. Bryan Sutton for advice in structural modeling.
Footnotes
This work is supported by a grant from the National Institute of Health GM060170.
The abbreviations used are: cAMP, cyclic adenosine 3′, 5′-monophosphate; CBD, cAMP binding domain; Epac, exchange protein directly activated by cAMP; DEP, Dishevelled, Egl-10, Pleckstrin domain; FT-IR, Fourier transform infrared spectroscopy; GEF, guanine nucleotide exchange factor; PBC, phosphate binding cassette; PBS, phosphate-buffered saline; PKA, cAMP-dependent protein kinase; C, catalytic subunit of PKA; R, regulatory subunit of PKA; RA, Ras-association domain; Rap, Ras-proximate; REM, Ras-exchange motif.
References
- 1.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 2.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 3.Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- 4.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 5.Maillet M, Robert SJ, Cacquevel M, Gastineau M, Vivien D, Bertoglio J, Zugaza JL, Fischmeister R, Lezoualc’h F. Crosstalk between Rap1 and Rac regulates secretion of sAPPalpha. Nat Cell Biol. 2003;5:633–639. doi: 10.1038/ncb1007. [DOI] [PubMed] [Google Scholar]
- 6.Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J Cell Biol. 2003;160:487–493. doi: 10.1083/jcb.200209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enserink JM, Price LS, Methi T, Mahic M, Sonnenberg A, Bos JL, Tasken K. The cAMP-Epac-Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the alpha3beta1 integrin but not the alpha6beta4 integrin. J Biol Chem. 2004;279:44889–44896. doi: 10.1074/jbc.M404599200. [DOI] [PubMed] [Google Scholar]
- 8.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- 9.Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 10.Kiermayer S, Biondi RM, Imig J, Plotz G, Haupenthal J, Zeuzem S, Piiper A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol Biol Cell. 2005;16:5639–5648. doi: 10.1091/mbc.E05-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem. 2002;277:11497–11504. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Dillon TJ, Pokala V, Mishra S, Labudda K, Hunter B, Stork PJ. Rap1-Mediated Activation of Extracellular Signal-Regulated Kinases by Cyclic AMP Is Dependent on the Mode of Rap1 Activation. Mol Cell Biol. 2006;26:2130–2145. doi: 10.1128/MCB.26.6.2130-2145.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 15.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 16.Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 17.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 18.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 19.Yu S, Mei FC, Lee JC, Cheng X. Probing cAMP-dependent protein kinase holoenzyme complexes I alpha and II beta by FT-IR and chemical protein footprinting. Biochemistry. 2004;43:1908–1920. doi: 10.1021/bi0354435. [DOI] [PubMed] [Google Scholar]
- 20.Mei FC, Cheng XD. Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Molecular Biosystems. 2005;1:325–331. doi: 10.1039/b511267b. [DOI] [PubMed] [Google Scholar]
- 21.Dong A, Caughey WS. Infrared methods for study of hemoglobin reactions and structures. Methods Enzymol. 1994;232:139–175. doi: 10.1016/0076-6879(94)32047-0. [DOI] [PubMed] [Google Scholar]
- 22.Dong A, Malecki JM, Lee L, Carpenter JF, Lee JC. Ligand-induced conformational and structural dynamics changes in Escherichia coli cyclic AMP receptor protein. Biochemistry. 2002;41:6660–6667. doi: 10.1021/bi020036z. [DOI] [PubMed] [Google Scholar]
- 23.Jung C. Insight into protein structure and protein-ligand recognition by Fourier transform infrared spectroscopy. J Mol Recognit. 2000;13:325–351. doi: 10.1002/1099-1352(200011/12)13:6<325::AID-JMR507>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 24.Susi H, Byler DM. Resolution-enhanced Fourier transform infrared spectroscopy of enzymes. Methods Enzymol. 1986;130:290–311. doi: 10.1016/0076-6879(86)30015-6. [DOI] [PubMed] [Google Scholar]
- 25.Dong A, Huang P, Caughey WS. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 1990;29:3303–3308. doi: 10.1021/bi00465a022. [DOI] [PubMed] [Google Scholar]
- 26.Barksdale AD, Rosenberg A. Acquisition and interpretation of hydrogen exchange data from peptides, polymers, and proteins. Methods Biochem Anal. 1982;28:1–113. doi: 10.1002/9780470110485.ch1. [DOI] [PubMed] [Google Scholar]
- 27.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 28.Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. Cell cycle dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem. 2002;277:26581–26586. doi: 10.1074/jbc.M203571200. [DOI] [PubMed] [Google Scholar]
- 29.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 30.Kraemer A, Rehmann HR, Cool RH, Theiss C, de Rooij J, Bos JL, Wittinghofer A. Dynamic interaction of cAMP with the Rap guanine-nucleotide exchange factor Epac1. J Mol Biol. 2001;306:1167–1177. doi: 10.1006/jmbi.2001.4444. [DOI] [PubMed] [Google Scholar]
- 31.Kraemer A, Rehmann HR, Cool RH, Theiss C, de Rooij J, Bos JL, Wittinghofer A. Dynamic interaction of cAMP with the Rap guanine-nucleotide exchange factor Epac1. J Mol Biol. 2001;306:1167–1177. doi: 10.1006/jmbi.2001.4444. [DOI] [PubMed] [Google Scholar]
- 32.Blout ER, de Lozé C, Asadourian A. Journal of American Chemical Society. 1961;83:1895–1900. [Google Scholar]
- 33.Kim KS, Fuchs JA, Woodward CK. Hydrogen exchange identifies native-state motional domains important in protein folding. Biochemistry. 1993;32:9600–9608. doi: 10.1021/bi00088a012. [DOI] [PubMed] [Google Scholar]
- 34.de Jongh HH, Goormaghtigh E, Ruysschaert JM. Tertiary stability of native and methionine-80 modified cytochrome c detected by proton-deuterium exchange using on-line Fourier transform infrared spectroscopy. Biochemistry. 1995;34:172–179. doi: 10.1021/bi00001a021. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Cheng X, Lee JC. Structure and dynamics of the modular halves of Escherichia coli cyclic AMP receptor protein. Biochemistry. 2002;41:14771–14778. doi: 10.1021/bi026383q. [DOI] [PubMed] [Google Scholar]
- 36.Flores S, Echols N, Milburn D, Hespenheide B, Keating K, Lu J, Wells S, Yu EZ, Thorpe M, Gerstein M. The Database of Macromolecular Motions: new features added at the decade mark. Nucleic Acids Res. 2006;34:D296–D301. doi: 10.1093/nar/gkj046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- 38.Rehmann H, Rueppel A, Bos JL, Wittinghofer A. Communication between the regulatory and the catalytic region of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:23508–23514. doi: 10.1074/jbc.M301680200. [DOI] [PubMed] [Google Scholar]
- 39.Wu J, Brown S, Xuong NH, Taylor SS. RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure. 2004;12:1057–1065. doi: 10.1016/j.str.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 40.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKay DB, Steitz TA. Structure of catabolite gene activator protein at 2.9 A resolution suggests binding to left-handed B-DNA. Nature. 1981;290:744–749. doi: 10.1038/290744a0. [DOI] [PubMed] [Google Scholar]
- 43.Clayton GM, Silverman WR, Heginbotham L, Morais-Cabral JH. Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell. 2004;119:615–627. doi: 10.1016/j.cell.2004.10.030. [DOI] [PubMed] [Google Scholar]