

Abstract
Kinetic and structural analyses of 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid (HOPDA) hydrolase from Burkholderia xenovorans LB400 (BphDLB400) provide insight into the catalytic mechanism of this unusual serine hydrolase. Single turnover stopped-flow analysis at 25 °C showed that the enzyme rapidly (1/τ1 ∼ 500 s−1) transforms HOPDA (λmax = 434 nm) to a species with electronic absorption maxima at 473 and 492 nm. The absorbance of this enzyme-bound species (E:S) decayed in a biphasic manner (1/τ2 = 54 s−1, 1/τ3 = 6 s−1 ∼ kcat) with simultaneous biphasic appearance (48 and 8 s−1) of an absorbance band at 270 nm characteristic of one of the products, 2-hydroxypenta-2,4-dienoic acid (HPD). Increasing solution viscosity with glycerol slowed 1/τ1 and 1/τ2, but affected neither 1/τ3 nor kcat, suggesting that 1/τ2 may reflect diffusive HPD dissociation, while 1/τ3 represents an intramolecular event. Product inhibition studies suggested that the other product, benzoate, is released after HPD. Contrary to studies in a related hydrolase, we found no evidence that ketonized HOPDA is partially released prior to hydrolysis, and therefore postulate that the biphasic kinetics reflect one of two mechanisms, pending assignment of E:S (λmax = 492 nm). Crystal structures of wild type, the S112C variant, and S112C incubated with HOPDA were each determined to 1.6 Å resolution. The latter reveals interactions between conserved active site residues and the dienoate moiety of the substrate. Most notably, the catalytic residue His265 is hydrogen-bonded to the 2-hydroxy/oxo substituent of HOPDA, consistent with a role in catalyzing ketonization. The data are more consistent with an acyl-enzyme mechanism than with the formation of a gem-diol intermediate.
The microbial degradation of aromatic compounds is crucial to maintaining the global carbon cycle (1). Aerobic degradation typically involves oxygenation of the aromatic ring to produce a catechol followed by a dioxygenase-catalyzed ring-opening reaction. In one type of degradation pathway, ring-opening yields a meta-cleavage product (MCP)1. A serine hydrolase then catalyzes carbon-carbon bond hydrolysis of the MCP, generating 2-hydroxypenta-2,4-dienoic acid (HPD). This strategy is utilized in the Bph pathway, responsible for the degradation of biphenyl (Figure 1). HPD is further transformed to pyruvic acid for the TCA cycle, thereby allowing microorganisms to grow on aromatic compounds (2). The MCP hydrolases are therefore important catabolic enzymes.
Figure 1.

The aerobic microbial degradation of biphenyl by the upper Bph pathway is typical of aromatic compound degradation via the meta-cleavage pathway. The meta-cleavage product, HOPDA, is hydrolyzed in an unusual reaction by the final enzyme, BphD.
MCP hydrolases are of interest to the fields of biodegradation and pathogenesis. First, like other degradation enzymes, they are able to transform the metabolites of some xenobiotic compounds. One important example is the degradation of the metabolites of polychlorinated biphenyls (PCBs) within the Bph pathway. In this case, overall degradation of PCBs may be limited in part by the poor turnover of certain chlorinated 2- hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acids (HOPDAs) by the HOPDA hydrolase, BphD (3). Thus, structure-function studies of BphD should advance protein engineering efforts to improve its activity towards chlorinated substrates, facilitating the biodegradation of PCBs (2, 4). With respect to pathogenesis, a close homolog of BphD was recently found to be critical for survival of Mycobacterium tuberculosis in the macrophage (5). The mycobacterial homolog catalyzes HOPDA hydrolysis (6), and appears to be involved in cholesterol degradation (Van der Geize et al., unpublished results). Greater understanding of BphD and its inhibitors could thus be useful to develop novel therapeutics for tuberculosis.
BphD from Burkholderia xenovorans LB400 (BphDLB400), a potent PCB-degrader, exemplifies MCP hydrolases, catalyzing the unusual cleavage of a carbon-carbon bond adjacent to a carbonyl (Figure 1). It is a member of the α/β-hydrolase superfamily (7-9) and contains conserved Ser-His-Asp residues that might be expected to constitute a catalytic triad enabling the acyl-enzyme mechanism common to many well characterized hydrolytic enzymes. In this mechanism, the hydrogen-bonding network between the triad of conserved residues serves to activate the serine hydroxyl for nucleophilic attack at the carbonyl carbon of the scissile bond. Expulsion of the leaving group results in acylated serine, which is then deacylated by histidine-mediated hydrolysis. However, it is unlikely that BphD and other MCP hydrolases follow this exact mechanism. For example, many hydrolases attack substrates endowed with electron sinks, such as alcohols (esterases and lipases), amines (proteases), and α-ketones (β-ketolases); protonation of these entities provides good leaving groups. By contrast, the substrates for MCP hydrolases apparently have no such electron sink available upon carbon-carbon bond cleavage, but rather an electron-rich dienoate. Therefore Bugg and colleagues proposed a two-phase mechanism that includes a preliminary enol-keto tautomerization to generate an electron sink prior to hydrolysis (Figure 2) (10). This requires additional catalytic residues to participate in acid-base chemistry, or an alternative use of the triad residues to generate the keto-intermediate. In addition, they proposed that the catalytic serine serves as a base to activate water as the nucleophile, obviating the acyl-serine intermediate. Elucidating the mechanism of BphD is therefore of considerable interest.
Figure 2.

The proposed enol-keto tautomerization and subsequent hydrolytic C-C cleavage catalyzed by the MCP hydrolases.
Early mechanistic investigations advanced the idea of enol-keto tautomerization as a key feature of catalysis in MhpC, the MCP hydrolase in the phenylpropionic acid degradation pathway. Enzyme-catalyzed deuterium exchange at H-5 of the substrate provided indirect support for the existence of the keto-intermediate, and also suggested its partial release during the catalytic cycle (11). Stopped-flow kinetic analysis at acidic pH demonstrated a biphasic decay of the substrate, consistent with a rapid ketonization followed by slower hydrolysis. The stopped-flow data supported a model that included partial release of the keto-intermediate, which is consistent with the deuterium exchange results (12).
Experimental evidence favoring a gem-diol intermediate, rather than an acyl-enzyme mechanism, has been obtained in MhpC. Specifically, radiochemical trapping provided low stoichiometry of a covalent intermediate, 18O was partially incorporated into both oxygen atoms of the carboxylic acid product, and 18O was exchanged into the carbonyl of a non-hydrolyzable substrate analog (13). In BphD, the processing of a reduced substrate required base-catalysis (14), and the negative ρ value obtained from Hammett analysis is opposite in sign to values obtained for hydrolases using a nucleophilic serine mechanism (14, 15). Overall, these results are more consistent with a mechanism involving the generation of a gem-diol intermediate formed via base-catalyzed attack of water (Figure 2) (16) than a classical acyl-enzyme hydrolytic mechanism.
Crystal structures of four MCP hydrolases have been reported: BphDRHA1 from Rhodococcus sp. strain RHA1 (17); CumDIP01 from Pseudomonas fluorescens IP01, involved in cumene degradation (18); CarCJ3 from Janthinobacterium sp. strain J3, involved in carbazole degradation (19); and MhpC (20). All enzymes have an α/β-hydrolase fold with the conserved Ser, His, and Asp located between the “core” and “lid” domains. The structure of MhpC in complex with an inhibitor lacking the dienoate moiety has provided some insight into possible catalytic residues (20). Although two orientations of the inhibitor were observed in the active site, the orientation likely mimicking dienoate binding indicated that the C-1 carboxylate interacts with conserved Arg190 and Asn51 residues (BphDLB400 numbering). The catalytic His was close to both the C-2 hydroxyl and C-5, implying that it may be involved in proton transfer between these positions during tautomerization. On the basis of the structural data and stopped-flow kinetic studies of MhpC mutants, the authors proposed that His265 catalyzes both tautomerization and deprotonation of water for C-C cleavage, while Ser112 may stabilize the gem-diolate tetrahedral intermediate via hydrogen bonding (21). Different crystal forms of CumD show a shift in position of the His residue, lending support to this hypothesis (18). Although the MhpC inhibitor complex advances our understanding of the catalytic mechanism of the MCP hydrolases, a complex with the unmodified dienoate moiety would more clearly delineate the catalytic roles of active site residues.
Herein we investigate the catalytic mechanism of BphDLB400 using steady state and transient kinetic analyses together with structural analyses of the wild type enzyme, a S112C variant, and a complex prepared by incubation of the latter with HOPDA. This study provides the first full spectra of catalytic intermediates for an MCP hydrolase. Further, it reveals key enzyme:substrate interactions, allowing us to predict the catalytic roles of active site residues.
MATERIALS AND METHODS
Chemicals
HOPDA was enzymatically generated from 2,3-dihydroxybiphenyl (DHB) using dihydroxybiphenyl dioxygenase (DHBD). Briefly, ∼50 mg DHB in 500 mL buffer (potassium phosphate, I = 100 mM, pH 7.5) was quantitatively converted to HOPDA by DHBD. The HOPDA solution was acidified to pH 2−3 using 2 N HCl and extracted 3 times with 0.5 volumes of ethyl acetate. The organic extract was dried over anhydrous MgSO4 and rotary evaporated to dryness. If necessary, the HOPDA was purified by HPLC from a methanol:water (0.5% H3PO4) mixture (80:20) using a Waters 2695 separation module fitted with a Prodigy 10-μm ODS-Prep column (21.2 × 250 mm) (Phenomenex, Torrance, CA). HOPDA was eluted in the same methanol/water (0.5% H3PO4) mixture at a flow rate of 7 ml/min. The HOPDA-containing fractions were pooled, extracted into ethyl acetate, dried, and evaporated as described above, yielding the purified HOPDA as a powder. 2,3-Dihydroxybiphenyl was prepared as described previously (22). Deuterium oxide was purchased from Cambridge Isotope Laboratories (Andover, MA, USA), and protein sequencing grade trypsin and 3-methylcatechol from Aldrich (Mississauga, ON, Canada). All other chemicals were of analytical grade. Catechol 2,3-dioxygenase was used to prepare 2-hydroxy-6-oxo-2,4-heptadienoic acid from 3-methylcatechol as previously described (23).
Mutagenesis, protein expression and purification
BphDLB400 and TodF (an MCP hydrolase from the toluene degradation pathway of Pseudomonas putida F1) were expressed and purified as previously described (24). The S112C variant of BphDLB400 was expressed and purified as described for the wild type enzyme. Selenomethionyl BphD was expressed in E. coli DL41 that was grown in minimal medium supplemented with selenomethionine (25). The active site serine of BphD was mutated to cysteine (S112C) using the Transformer site-directed mutagenesis method (Clontech Laboratories, Palo Alto, CA, USA). Briefly, the S112C mutagenic primer (primer S112C: 5’-GCGCCCCCCATGCAGTTGCCGACCAG-3’) and a second mutagenic selection primer to remove an EcoRI site (primer ODM: 5’-AGCTCGAATTGGTAATCATGG-3’) were mixed with the pEMBL18 plasmid containing the BphDLB400 gene (pSS184) (23). After second strand synthesis and ligation using T4 DNA polymerase and T4 DNA ligase (Pharmacia, Uppsala, Sweden), respectively, the DNA was digested with EcoRI to linearize unmutated plasmid, and transformed into E. coli BMH 71−18 mutS. Plasmid isolated from this first transformation was subjected to a second round of EcoRI digestion and transformation, from which S112C mutants were isolated. The nucleotide sequence of the mutated gene was confirmed at the University of British Columbia DNA sequencing facility using an ABI 373 Stretch DNA sequencer (Applied Biosystems, Foster City, CA) and a BigDye v3.1 Terminator kit. Concentration of purified enzyme was determined using the molar absorptivity of BphDLB400 (ε280 = 55.4 mM−1 cm−1) calculated from the absorbance at 280 nm of a sample whose concentration had been determined by amino acid analysis at the Advanced Protein Technology Centre (Hospital for Sick Children, Toronto, ON, Canada).
Steady state kinetics
Steady state measurements were performed by monitoring absorbance on a Varian Cary 1E spectrophotometer equipped with a thermostatted cuvette holder (Varian Canada, Mississauga, ON, Canada) maintained at 5 ± 3 °C, or 25 ± 1 °C controlled by Cary WinUV software version 2.00. The cuvette holder was modified to deliver streams of nitrogen gas over both sides of the cuvette, thus minimizing condensation on the cuvette surface at low temperature. For HPD inhibition experiments, HPD was generated in situ from TodF-catalyzed hydrolysis of freshly prepared 2-hydroxy-6-oxo-2,4-heptadienoate; the concentration of the latter was calculated from the previously determined molar absorptivity (23). Sufficient TodF enzyme was added to ensure complete formation of HPD in approximately 2 min, after which time HOPDA and BphDLB400 were immediately added, and the BphD-catalyzed hydrolysis of HOPDA was spectrophotometrically monitored at 434 nm. The decay of HPD was not greatly accelerated by TodF: ∼10% loss of HPD occurred over the course of the 5 min measurement. BphD-catalyzed HOPDA hydrolysis was not affected by the presence of the TodF enzyme or by the presence of acetate at 100 μM. Steady state rate equations were fit to data using the program LEONORA (26).
Transient kinetic experiments
Experiments were conducted using an SX.18MV stopped-flow reaction analyzer (Applied Photophysics Ltd., Leatherhead, UK) equipped with a photodiode array detector. The temperature of the drive syringe chamber and optical cell was controlled by circulating water. For wavelengths below 350 nm, a deuterium lamp was employed to improve the signal-to-noise ratio, while a Xe lamp was used for wavelengths above 350 nm. Concentrations of reactants and buffer conditions were varied as described in Results. For each experiment, the data are averages of three replicates, and each replicate is generated by averaging at least five shots of the stopped-flow drive syringe. Single or double exponential equations were fit to the averaged time courses for single wavelength data using the SX18MV software running on the system's Acorn A5000 computer and the RISC OS 3.7 operating system. These fits provided the reciprocal relaxation times (1/τ) and amplitudes; good fits are characterized by random variation in the fit residuals. Multiple wavelength data from the time courses of single turnover experiments were exported to Excel (Microsoft, Redmond, WA, USA) and then averaged. Selected single wavelengths of the averaged data were analyzed using the SX18MV software.
HPLC assay for coupling of substrate consumption to product formation
Separations were performed on a Waters 2695 system (Waters, Mississauga, ON, Canada) equipped with a Hewlett Packard ODS Hypersil C18 column (5 μm, 125 × 4 mm) column, operating at a flow rate of 1 ml/min. Sample injections of 95 μl were eluted with 55% methanol and 45% water containing 0.5% phosphoric acid. Benzoic acid was quantified by comparing peak areas at 230 nm to those of standards containing known concentrations (5, 20, 100, and 250 μM) of benzoic acid. To 4 ml of potassium phosphate buffer (I = 0.1 M), pH 7.5 containing 94.6 μM HOPDA (calculated using ε = 25.7 mM−1 cm−1 (3)) was added sufficient BphDLB400 to permit complete conversion in 35 min. The reaction was monitored spectrophotometrically at 434 nm, and a 200 μl aliquot was removed at each increment of 10% conversion (judged by A434), and immediately quenched into 244 μl methanol (containing 0.41% phosphoric acid). The quenched reaction solutions were analyzed by HPLC as described above (retention times: HPD, 1.6 min; benzoic acid, 2.0 min; HOPDA, 2.9 min).
Deuterium incorporation
Deuterated buffer was prepared by evaporating 700 μl of potassium phosphate buffer (I = 5 mM), pH 7.5 and resuspending in the same volume of deuterium oxide. To 700 μl of deuterated or non-deuterated buffer was added a small volume of concentrated HOPDA dissolved in ethanol, such that the final HOPDA concentration was 1 mM, and the solution contained 2% ethanol. BphDLB400 (8 μl) was added and the reaction was followed spectrophotometrically at 434 nm until completion (< 4 min). Immediately upon reaction completion, 7 μl of 2 N HCl was added, and the reaction was extracted 3 times with 0.5 volumes of ethyl acetate and then evaporated under a stream of nitrogen gas. The dried sample was redissolved in acetone-d6 and transferred to an NMR tube for analysis using a Varian 500 MHz NMR spectrometer (Varian Inc., Mississauga, ON, Canada). A reaction was also quenched approximately 1 min after completion to allow observation of ketonized HPD ((E)-2-oxo-3-pentenoic acid). Found for HPD: 1H NMR (acetone-d6, 500 MHz) δ 5.18 (1H, dd, J4,5E = 10.3 Hz, J5Z,5E = 1.9 Hz, H-5E), 5.39 (1H, dd, J4,5Z = 17.2 Hz, J5Z,5E = 1.9 Hz, H-5Z), 6.21 (1H, d, J3,4 = 11.0 Hz, H-3), 6.77 (1H, dt, J4,5Z = 17.0 Hz, J3,4 = 10.7 Hz, J4,5E = 10.7 Hz, H-4); for (E)-2-oxo-3-pentenoic acid: 1H NMR (acetone-d6, 500 MHz) δ 6.59 (1H, d, J3,4 = 15.6 Hz, H-3), 7.15 (1H, dq, J3,4 = 15.8 Hz, J4,5 = 6.7 Hz, H-4). The acetone signal obscured the signal from H-5 of the latter compound (expected at 2.15 ppm). Nevertheless, the H-3 and H-4 chemical shifts and coupling constants agreed with a previous report (27).
Mass spectrometric analysis of BphDLB400 S112C
To check for Cys112 oxidation in the BphDLB400 S112C crystals, the protein was digested with trypsin, and the proteolytic fragment containing Cys112 was analyzed by mass spectrometry. About 70 μg of protein (∼2 mg/ml) in buffer (130 mM Tris-Cl, pH 8.0, 5.8 M urea) was heated at 95 °C for 15−20 min. To the denatured enzyme (36 μl) was added 182 μl of buffer (50 mM Tris-Cl, pH 8.0, 1 mM CaCl2) to dilute the urea to <1 M. 20 μg of this diluted denatured protein solution was then mixed with acetonitrile (10% v/v), and trypsin (0.3 μg). The trypsin digestion occurred overnight at 37 °C, and complete digestion was confirmed by SDS-PAGE. Masses of HPLC-resolved peptides were determined using an Applied Biosystems API 300 triple quadrupole mass spectrometer with an electrospray ionization source at the Laboratory of Molecular Biophysics, University of British Columbia.
Crystallization, preparation of complex, and data collection
Crystallization experiments were performed at 20 °C using the hanging drop vapor diffusion method (28). Suitable crystals for diffraction experiments were obtained using reservoir solutions containing 1.8−2.4 M ammonium sulfate and 6−10% ethanol in 100 mM Tris buffer, pH 8.2. Selenomethionyl BphDLB400, wild type BphDLB400, and BphDLB400 S112C were all crystallized using similar conditions. Crystals could diffract to ∼2.0 Å with Cu-Kα X-rays from a 5-kW rotating anode X-ray generator (Rigaku/MSC). To prepare crystals for flash freezing, they were loop-transferred from the growth drop into 2−5 μl of the reservoir solution augmented with 20−25% glycerol and immediately immersed in liquid nitrogen. To introduce substrate, S112C crystals were first loop-transferred from the growth drop into 200−300 μl of the reservoir solution augmented with ∼10 mM HOPDA for about one hour. Thereafter the crystals were loop-transferred into 200−300 μl of the reservoir solution plus 20−25% glycerol for several minutes before immersion in liquid nitrogen.
All final diffraction data were collected at cryogenic temperatures (ca. 100K) using flash-frozen crystals and various beamlines at the Advanced Photon Source (APS), Argonne National Laboratory (Argonne, IL). For wild type BphDLB400, high-resolution data (1.6 Å) were collected at BioCARS beamline 14-BMD using a Quantum-1 CCD detector (Area Detector Systems Corporation, Poway, CA). Multiwavelength anomalous diffraction (MAD) data were collected from a single crystal of selenomethione-substituted enzyme, also by the use of a Quantum-1 CCD detector at beamline 14-BMD. Both S112C and S112C+HOPDA complex data to 1.6 Å were collected at the Structural Biology Center beamline 19-ID. Programs from the HKL and HKL2000 software packages were used for diffraction data analysis (29). In addition, a data set was obtained at beamline 14-BMD from an S112C crystal crystallized and frozen under anaerobic conditions. This experiment was a successful attempt to prevent oxidation of active site cysteine, which was consistently observed for S112C crystals grown in air and exposed to X-rays. For this experiment, the cryopreservative solution was augmented with 10 mM ascorbate, which was added to provide a free radical scavenger (30). A short exposure time was used to reduce total X-ray exposure. HKL2000 was used for data processing (29).
Structure determination and refinement
The structure of selenomethionine-modified BphDLB400 was determined using the multiple wavelength anomalous diffraction (MAD) method. BphDLB400 crystals have space group P64 with two monomers per asymmetric unit. The program SOLVE (31) was used to locate 19 crystallographically unique Se atoms by analysis of data from the three wavelength MAD experiment. The Se positions were refined with MLphare from the CCP4 suite (32). The initial phases were improved through solvent-flattening and histogram-matching by use of the program DM, also from the CCP4 suite. The secondary structure was easily recognized in the electron density maps, and the initial model with side chains was constructed with the O program (33), which was used for all subsequent electron density interpretation and model fitting. All atomic models were refined using the CNS program (34). After one round of refinement at 2.0 Å resolution, the phases were extended to 1.6 Å.
RESULTS
Single turnover stopped-flow kinetic analysis
BphDLB400 catalyzed the hydrolysis of HOPDA to benzoate and HPD with a kcat of 6.5 ± 0.5 s−1 at 25 °C. This value is slightly higher than previously reported (3, 23, 35), which may result from a more accurate determination of protein concentration (see Materials and Methods). We performed stopped-flow experiments using a diode array detector to resolve and spectrophotometrically characterize intermediates during a single catalytic cycle. Figure 3 illustrates data from an experiment performed under ‘slow’ conditions (T = 3.2 °C, 20% glycerol). Using a 2:1 ratio of enzyme (8 μM) to substrate (4 μM), the HOPDA enolate (λmax = 434 nm) was rapidly transformed to a species with absorbance maxima at 473 and 492 nm (Figure 3A). Indeed, at 25 °C the transformation was approximately 50% complete within the dead time of the instrument (∼1.2 ms), indicating a process occurring with reciprocal relaxation time, 1/τ1 ∼ 500 s−1 (36).2 The red-shifted spectrum of the E:S species may arise from a deprotonated form of HOPDA (enolate) or a ketonized HOPDA, as discussed below. The E:S intermediate decayed in a biphasic manner (at 25 °C, 1/τ2 = 54 ± 4 s−1, 1/τ3 = 5.8 ± 0.9 s−1), with similar phase amplitudes (Figure 3B, Table 1). Concurrent with E:S decay was a biphasic increase at 270 nm, the absorbance maximum of HPD (at 25 °C, 1/τ2 = 48 ± 12 s−1, 1/τ3 = 8.3 ± 0.7 s−1), followed by a slower monotonic decay (Figure 3C). The decay of HPD is due to ketonization to the more stable α/β-unsaturated ketone, a process which occurs non-enzymatically on a timescale of minutes (37). The faster rate observed in this experiment is due to enzyme-catalyzed ketonization, as observed with MhpC (37). In contrast to the relatively equal distribution of phase amplitudes for 1/τ2 and 1/τ3 at 492 nm, the amplitude of 1/τ3 at 270 nm was only ∼10% of the total absorbance change (Table 1). We attribute this to the enzyme-catalyzed decay of HPD, which reduces the amplitude associated with phase 3, and thereby makes 1/τ2 and 1/τ3 difficult to resolve at 270 nm. Performing the experiment at 3.2 °C better resolved the two phases at 270 nm. Under these conditions, BphDLB400 catalyzed HOPDA hydrolysis with a kcat = 0.98 ± 0.06 s−1 and Km = 0.30 ± 0.08 μM. Indeed, this experiment generated values of 1/τ2 (∼10 s−1) and 1/τ3 (∼1.5 s−1) with corresponding amplitudes at 270 nm that matched those at 492 nm (Table 1).
Figure 3.

A representative stopped-flow experiment illustrating a single turnover of HOPDA (4 μM) by BphDLB400 (8 μM) at 3.2 °C in potassium phosphate buffer supplemented with 20% glycerol (I = 100 mM), pH 7.5. A) Time-resolved spectral changes show free HOPDA (λmax = 434 nm) transformed to an enzyme-bound form, E:S (λmax = 492 nm). B) Absorbance at 492 nm versus time shows the decay of the E:S intermediate. The solid line denotes the double exponential fit. The inset shows the single exponential fit for the formation of the E:S intermediate. C) Absorbance at 270 nm versus time for the formation/decay of the HPD product. Data points are shown together with the fit of a single exponential (solid line) describing HPD decay. Inset shows the formation of HPD and its fit to a double exponential.
Table 1.
Reciprocal relaxation times and amplitudes for single turnover reactions.a
| T (° C) | Glycerol (% v/v) | Wavelength (nm) | Phase 1b | Phase 2c (% total amplitude) | Phase 3c (% total amplitude) | Phase 4d |
|---|---|---|---|---|---|---|
| 25 | 0 | 270 (HPD) | - | 48 ± 12 s−1 (89) | 8.3 ± 0.7 s−1 (11) | 0.59 ± 0.04 s−1 |
| 492 (E:Sk) | ∼500 s−1 | 54 ± 4 s−1 (68) | 5.8 ± 0.9 s−1 (32) | - | ||
| 20 | 270 (HPD) | - | 34 ± 2 s−1 (51) | 5.3 ± 0.3 s−1 (49) | 0.71 ± 0.01 s−1 | |
| 492 (E:Sk) | ∼200 s−1 | 33 ± 2 s−1 (66) | 5.5 ± 0.4 s−1 (34) | - | ||
| 3.2 | 0 | 270 (HPD) | - | 12 ± 2 s−1 (51) | 2.3 ± 0.6 s−1 (49) | 0.22 ± 0.01 s−1 |
| 492 (E:Sk) | ∼200 s−1 | 8.7 ± 0.3 s−1 (49) | 1.3 ± 0.2 s−1 (51) | - | ||
| 20 | 270 (HPD) | - | 3.8 ± 0.4 s−1 (68) | 1.0 ± 0.1 s−1 (32) | 0.25 ± 0.03 s−1 | |
| 492 (E:Sk) | 66 ± 6 s−1 | 4.5 ± 0.8 s−1 (49) | 1.2 ± 0.1 s−1 (51) | - | ||
| 20e | 270 (HPD) | - | 4.0 | - | 0.41 s−1 | |
| 492 (E:Sk) | ∼130 | 4.4 s−1 (45) | 1.1 s−1 (55) | - |
Errors are reported as the standard deviation of 3 measurements.
Phase 1 represents the rapid formation of E:S.
The amplitudes are reported as a percentage of the total amplitude of phase 2 and phase 3 combined.
Represents the enzyme-catalyzed ketonization of HPD.
Single measurement performed with 6:1 ratio of enzyme (20 μM) to substrate (4 μM).
Kinetic experiments were performed in the presence of 20% glycerol to help assign relaxations to bimolecular events. Specifically, increasing solvent viscosity should slow diffusive events that reflect association/dissociation from the enzyme. The presence of glycerol did not affect the steady state kinetic parameters. In contrast, glycerol slowed two of the observed relaxations at 3.2 °C: 1/τ1, the rate of formation of E:S was reduced from ∼200 s−1 to 66 ± 6 s−1, and 1/τ2 was reduced by ∼50% at both 270 and 492 nm (Table 1). Even at the slowed rate, the formation of E:S occurred with an apparent isosbestic point at 460 nm, implying a single step transformation from HOPDA (Figure 3A). The reduction in 1/τ2 is consistent with this step representing HPD dissociation. The inverse relationship of solvent viscosity and rate of diffusive product release from an enzyme is well documented. For example, viscosity effects on kcat have been used to demonstrate the rate-determining dissociation of the ADP product from protein kinases (38, 39). Finally, 1/τ3 was not affected by glycerol, suggesting that this step involves an intramolecular event rather than diffusive product release.
In the event that the high-wavelength spectrum represents enzyme-bound ketonized HOPDA (E:Sk), it is possible that the glycerol-induced decrease of 1/τ2 reflected dissociation of ketonized HOPDA (Sk) from the enzyme, as proposed for MhpC (Scheme 1) (12). In such a scenario, Sk dissociation and hydrolysis would have similar rate constants (k2 ∼ k3 ∼ 25 s−1 at 25 °C), such that 50% of substrate is initially turned over to product while 50% is released as Sk. Thus, re-enolization of the released Sk (k4 ∼ 6 s−1) would limit the rate of hydrolysis of 50% of the substrate such that 1/τ3 ∼ 6 s−1 would comprise 50% of the amplitude of E:Sk decay. However, 1/τ2 exhibits the same behavior at both 492 nm (E:Sk decay) and 270 nm (HPD formation), suggesting that E:Sk processing is directly coupled to HPD formation. Hence a kinetic model involving release of Sk does not satisfactorily account for the biphasic relaxation. Release of Sk was further ruled out as described below.
Scheme 1.
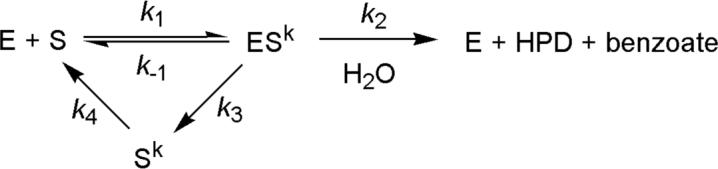
To investigate whether the biphasic kinetics reflected multiple turnovers, an experiment was conducted at 3.2 °C with 20% glycerol using a 6:1 ratio of enzyme (20 μM) to substrate (4 μM). In this experiment, the values and amplitudes of 1/τ2 and 1/τ3 were essentially identical to those observed using a 2:1 ratio (Table 1), indicating that the relaxations do not reflect multiple turnovers.
We also investigated whether the biphasic kinetics resulted from (a) a product complex absorbing at 492 nm, but with a molar absorptivity of ∼50% of E:S, or (b) the use of phosphate buffer. In a stopped-flow experiment performed by mixing enzyme (8 μM) with excess HPD (∼500 μM) and benzoate (1.0 mM), no absorbance at 492 nm was detected (data not shown). Moreover, in a single turnover experiment (E = 8 μM; S = 4 μM) performed using Na-HEPES buffer (I = 100 mM, pH 7.5), the amplitudes and rates of the observed relaxations were essentially identical to those observed using phosphate buffer. Thus, neither phosphate nor the absorbance of a product complex influenced the kinetic behavior of BphD.
Coupling of substrate consumption to product formation
A branched pathway (Scheme 1) involving partial release of the keto-intermediate (Sk) has been proposed for MhpC based on deuterium exchange and stopped-flow studies (11, 12). Subsequent crystallographic observation of apparent half-sites reactivity suggested that release occurs from an inactive subunit (20). Such release followed by rate-limiting re-enolization of Sk in solution may account for the biphasic kinetics observed in the single-turnover stopped-flow experiments, even though it was not supported by the apparent coupling of E:Sk decay (492 nm) and HPD formation (270 nm) (see above). Significantly, this model predicts a considerable steady state population of Sk. In an attempt to detect Sk accumulation during turnover of BphDLB400, we simultaneously monitored HOPDA consumption and benzoate production in a reaction performed at 25 °C (I = 100 mM potassium phosphate buffer, pH 7.5). More specifically, HOPDA consumption was monitored by absorbance at 434 nm. As the reaction proceeded, aliquots were removed at intervals corresponding to increments of 10% completion, quenched in acidic solution, and benzoate was quantified by HPLC analysis. At all intervals, the amount of benzoate detected corresponded to the amount of HOPDA consumed within the margin of error (±5%; data not shown). These results indicate that significant amounts of Sk did not accumulate in solution during turnover. Failure to detect significant quantities of Sk in solution during turnover suggests two possibilities: (1) Sk is not released from the enzyme, or (2) the re-enolization of Sk in solution is faster than release, in which case it could not be responsible for the biphasic kinetics.
Deuterium incorporation into HPD
To further investigate the possibility of Sk release during catalytic turnover and to confirm the stereochemical course of the reaction with native enzyme, we analyzed the incorporation of deuterium from D2O into HPD during the BphDLB400-catalyzed hydrolysis of HOPDA. Studies using His6-tagged BphDLB400 (40) have demonstrated that the enzyme stereospecifically incorporates deuterium into the H-5E position of HPD. To minimize non-specific exchange at H-5Z of HOPDA that might occur via phosphate-catalyzed ketonization similar to that reported for 2-hydroxymuconate (41), we lowered the phosphate buffer concentration to I = 5 mM. Under these conditions, deuterium exchange at H-5 of HOPDA occurred with a half-life of ∼10 min, which was about twice as fast as exchange into H-3. These rates were approximately one-third as fast as at I = 100 mM phosphate. Moreover, sufficient amounts of BphDLB400 were added to ensure HOPDA hydrolysis was complete in less than 4 min.
Figure 4 shows the 1H NMR spectrum of HPD from a reaction performed in D2O. Peak integration revealed that ∼10% signal loss occurred at H-5Z during the course of an enzyme-catalyzed reaction. This small amount of deuterium incorporation corresponded to what was observed over a similar time period in the absence of BphDLB400. By contrast, almost complete deuterium incorporation occurred at H-5E, as predicted.3 Hence the enzyme stereospecifically incorporates a proton at H-5E, and the non-specific incorporation at H-5Z can be explained by non-enzymatic exchange prior to enzymatic hydrolysis. If the reaction was allowed to continue after the complete conversion of HOPDA, ketonized HPD ((E)-2-oxo-3-pentenoate) appeared in the NMR spectrum (data not shown). Indeed, addition of BphDLB400 to a solution of HPD confirmed that the enzyme catalyzes ketonization of HPD, as reported for MhpC (37).
Figure 4.

Top: The 1H NMR spectrum of HPD generated from BphDLB400-catalyzed hydrolysis of HOPDA in 97% D2O (potassium phosphate, pD = 7.5, I = 5 mM). Peak integration demonstrates ∼10% deuterium incorporation into H-5Z and almost complete incorporation into H-5E. Bottom: The ∼10% non-specific deuterium incorporation into H-5Z is consistent with non-enzymatic exchange prior to enzymatic catalysis.
In summary, three independent experiments indicate that Sk is not released from the enzyme in significant amounts: (1) single turnover stopped-flow showed matching biphasic behavior for both HPD formation and E:Sk decay; (2) the amount of benzoate produced in steady state assays corresponded to the amount of HOPDA hydrolyzed; and (3) deuterium was not incorporated into HPD at H-5Z above background levels. These results suggest that the BphDLB400-catalyzed hydrolysis of HOPDA proceeds via a linear model as opposed to the branched model involving Sk release proposed for MhpC (12).
Product inhibition
To facilitate the interpretation of transient state kinetic data, product inhibition studies were performed (potassium phosphate buffer (I = 100 mM), pH 7.5) under two conditions representative of those used for the single turnover experiments: 1) 25 ± 1 °C, and 2) 5 ± 3 °C, 20% glycerol. In these experiments, inhibition by each product (benzoate and HPD) was examined by steady state kinetics so as to probe the order of product release. At 25 °C, benzoate was found to competitively inhibit BphDLB400-catalyzed hydrolysis of HOPDA with Kic = 165 ± 20 μM (data not shown). Under these conditions, however, HPD was not sufficiently stable to reliably characterize inhibition. At 5 °C in the presence of 20% glycerol, benzoate also competitively inhibited the BphDLB400-catalyzed hydrolysis of HOPDA with Kic = 220 ± 30 μM (Figure 5A). This result indicates that benzoate competes for the active site of free enzyme, and therefore may be the last product released from the enzyme. Under these same conditions, HPD inhibited the BphDLB400–catalyzed hydrolysis of HOPDA in a mixed fashion with Kic = 84 ± 41 μM, Kiu = 120 ± 30 μM (Figure 5B). This indicates that HPD is released in a step that generates an enzyme species other than that which binds free substrate (42). The competitive component of the mixed inhibition suggests that HPD may also bind to the free enzyme (E:HPD). Consistent with this hypothesis, BphDLB400 catalyzed the ketonization of HPD (see single turnover stopped-flow results). The results should be regarded with some caution as there will inevitably be some ketonized HPD present in the reaction even at low temperature. However, the product inhibition experiments are consistent with a mechanism in which HPD is released prior to benzoate, but the former may re-associate with the free enzyme to undergo ketonization.
Figure 5.

Cornish-Bowden plots illustrating the type of inhibition of the BphDLB400-catalyzed hydrolysis of HOPDA by reaction products. (A) Inhibition by benzoate described by competitive inhibition (Kic = 220 ± 30 μM; Km = 0.21 ± 0.02 μM; V = 5.9 ± 0.2 U/mg). Reaction rates determined using 0.24 (■), 0.57 (O), 1.1 (◆), 2.2 (△), and 4.6 μM (●) HOPDA. (B) Inhibition by HPD described by mixed inhibition (Kic = 84 ± 41 μM; Kiu = 120 ± 30 μM; Km = 0.45 ± 0.08 μM; V = 4.0 ± 0.2 U/mg). Reaction rates were measured using 0.23 (●), 0.56 (□), 0.82 (◆), and 5.9 μM (△) HOPDA. Equations describing competitive, uncompetitive, and mixed inhibition were each fit to the data using the least squares, dynamic weighting options of the LEONORA program, and the type of inhibition determined by comparing the quality of fit based on non-random trends in the residuals. The solid lines represent the best fit parameters of the global fit at each HOPDA concentration. Conditions: potassium phosphate buffer (I = 100 mM), 20% glycerol, pH 7.5, 5 ± 3 °C.
Kinetic behavior of Ser112Cys mutant
To help obtain a crystal structure approximating the E:S complex, the active site Ser112 of BphDLB400 was substituted with cysteine. This substitution was expected to lower the rate of HOPDA hydrolysis, perhaps to the point where the complex could be formed in the crystal and trapped by rapid freezing. Attempts to measure steady state kinetic parameters for the S112C variant were confounded by the non-linearity of the progress curves, indicative of strong product inhibition or enzyme inactivation. The inclusion of 1 mM DTT did not affect the non-linearity, suggesting that the latter was not due to oxidation of Cys112. Nevertheless, estimates of initial velocities at 25 °C generate kcat = 0.27 ± 0.02 s−1, and Km = 2.8 ± 0.6 μM, and a specificity constant (kcat/Km = 9.5 ± 2.2 × 104 M−1 s−1) approximately 240-fold lower than wild type enzyme. In a single turnover stopped-flow experiment at 25 °C, the high wavelength intermediate observed in wild type failed to accumulate. However, the HOPDA signal at 434 nm decayed in a biphasic manner (1/τ1 = 2.6 s−1, amplitude = 0.063; 1/τ2 = 0.25 s−1, amplitude = 0.019 where 1/τ1 and 1/τ2 apparently correspond to 1/τ2 and 1/τ3, respectively, in wild type). As in wild type, HPD (at 270 nm) also displayed biphasic formation (1/τ1 = 4.7 s−1, amplitude = 0.012; 1/τ2 = 1.0, amplitude = 0.014). The S112C substitution apparently slowed the initial reaction, since the red-shifted spectrum of E:S did not accumulate. Nevertheless, the biphasic disappearance of the HOPDA signal seems approximately coupled to the formation of HPD – a pattern not unlike that observed in wild type.
Crystal structures
Table 2 summarizes the crystallographic data used for initial phase determination and model refinement. The final phasing powers were 4.59 and 2.47 for acentric and centric reflections measured at the wavelength corresponding to the peak of the absorption spectrum and 4.26 (acentric) and 2.20 (centric) for data measured at the wavelength of the inflection point of the absorption edge. The mean figure of merit was 0.75 for all data after MAD phasing and 0.85 after density modification..
Table 2.
X-ray diffraction data
| Crystal Type | Wild-type | SeMet BPHD: MAD Phasing | S112C | S112C+HOPDA | S112Canaerobic | ||
|---|---|---|---|---|---|---|---|
| Inflection | Peak | Remote | |||||
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9793 | 0.9537 | 0.9783 | 0.9783 | 0.9000 |
| Resolution (Å) | 50−1.6 | 50.0−2.0 | 50.0−2.0 | 50.0−2.0 | 50−1.6 | 50−1.6 | 50−1.9 |
| Space Group | P64 | P64 | P64 | P64 | P64 | P64 | P64 |
| a (Å) | 135.00 | 136.50 | 136.50 | 136.48 | 135.08 | 135.51 | 135.06 |
| c (Å) | 66.73 | 66.03 | 66.03 | 66.03 | 66.28 | 66.62 | 66.30 |
| Observations | 750379 | 1610683 | 1637855 | 1684790 | 2008833 | 1663889 | 1387384 |
| Unique reflections | 88102 | 47880 | 48007 | 47694 | 90566 | 91070 | 52538 |
| Rsym (%)a (Last shell) | 6.0 (35.9) | 4.9 (30.9) | 5.1 (34.1) | 4.4 (28.6) | 8.9 (41.8) | 4.5 (39.7) | 8.2 (62.5) |
| Average I/σ(I) (Last shell) | 22.6 (2.7) | 27.6 (2.9) | 26.2 (2.7) | 29.9 (3.4) | 20.1 (2.1) | 38.8 (3.4) | 14.2 (1.8) |
| Completeness(%) (Last shell) | 96.6 (73.9) | 92.1 (59.2) | 92.5 (59.3) | 95.4 (72.9) | 99.8 (98.9) | 99.7 (99.4) | 96.3 (90.9) |
Rsym =
The refined models of wild type, S112C, S112C (anaerobic), and S112C+HOPDA are of high reliability as demonstrated by the data in Table 3. The two crystallographically independent molecules of BphDLB400 are labeled A and B. The atomic model of A does not include the N-terminal residue, and that of B does not include the three N-terminal residues.
Table 3.
Refinement statistics and characteristics of the refined atomic models
| Crystal Type | Wild-type | S112C | S112C+HOPDA | S112Canaerobic |
|---|---|---|---|---|
| Model Content | ||||
| Protein residues | 568 | 568 | 568 | 568 |
| Substrate (HOPDA) | 0 | 0 | 1 | 0 |
| Glycerol molecules | 1 | 3 | 3 | 1 |
| Ethanol molecules | 3 | 9 | 6 | 1 |
| Sulfate molecules | 4 | 2 | 1 | 3 |
| Water molecules | 469 | 404 | 338 | 336 |
| Refinement statistics | ||||
| Resolution range (Å) | 50−1.6 | 50−1.6 | 50−1.6 | 50−1.97 |
| R, Rfree (%) | 20.8, 22.5 | 20.3, 21.4 | 20.8, 22.5 | 19.7, 22.5 |
| Last Shell R, Rfree (%) | 28.8, 34.7 | 26.4, 29.4 | 27.9, 29.1 | 25.9, 25.5 |
| Average B-factors (Å2) | ||||
| Protein atomsa | A 16.0 | A 17.4 | A 21.2 | A 26.1 |
| B 37.1 | B 34.8 | B 42.9 | B 46.2 | |
| Water atoms | 32.2 | 32.4 | 35.3 | 40.4 |
| All atoms | 27.1 | 26.5 | 32.3 | 36.5 |
| rmsdb from restraints | ||||
| Bond lengths (Å) | 0.0048 | 0.0049 | 0.0048 | 0.0054 |
| Bond angles (°) | 1.16 | 1.17 | 1.16 | 1.15 |
The labels A and B identify atoms from crystallographically independent monomers.
Root-mean-square deviation.
Quaternary structure
As observed in solution (24), BphDLB400 is a tetrameric oligomer in the crystal (Figure 6). The tetramer is formed by the back-to-back interaction of two A-B dimers related by crystallographic two-fold symmetry. Two non-crystallographic two-fold axes relate A to B or B'. These axes are perpendicular to each other and to the crystallographic two-fold axis, thereby generating a tetramer with approximate 222 (D2) point group symmetry. Of the three interfaces in the tetramer, the AB interface has the largest buried surface area: 2039 Å2 as calculated using GRASP (43). Considerably smaller surface areas of 1135 and 964 Å2 were calculated for the AB' interface and the AA' interface, respectively. The A-B dimer interaction involves the β8 strands from both subunits forming an antiparallel β pair, and corresponds to the 1−5 interaction of the BphDRHA1 octamer (17). Similar interactions were also found in CumDIP01 (18), CarCJ3 (19), and MhpC (20), suggesting the A-B dimer is a basic structural unit from which some members of this family, such as BphDLB400 and BphDRHA1, may construct higher oligomers.
Figure 6.

Left: A schematic diagram of BphDLB400 shown as a tetramer with 222-point group symmetry. The view is down a crystallographic 2-fold axis parallel to the c axis of the crystal. A and B are crystallographically independent: crystallographic 2-fold symmetry transforms A to A' and B to B'. By contrast, A is related to B or B' by non-crystallographic 2-fold symmetry. Right: A ribbon diagram of the BphDLB400 tetramer viewed in the same direction. The four subunits are colored green, yellow, cyan, and blue. Arrows highlight the close contacts between lid domains of A and B' (and A' and B).
The crystallographically independent monomers (A and B) have highly similar structures (RMSD of 0.53 Å for the superposed Cα atoms). Subunit A has better electron density and lower B factors (Table 3). In addition, α9 of the lid domain is closer to the core domain in subunit A. These differences can be explained by crystal packing: the A subunits pack against each other and are further surrounded by B subunits, whereas each B subunit is open to one of the large solvent channels located about the crystal's 64 screw axes.
Monomer structure
BphDLB400 comprises two domains: a core domain (residues 2−145 and 213−286) containing the canonical α/β hydrolase fold (7-9); and a lid domain (residues 146−212), (Figure 7). The core domain primarily consists of a highly twisted eight-stranded β sheet with five helices on each side of the sheet. The lid domain consists of five helices, including one 310 helix, and occurs as an insertion in the core domain's canonical fold.
Figure 7.

A ribbon diagram of BphDLB400 subunit A. The lid domain (residues 146−212) is shown at the top, and the key catalytic residues (Asp237, His265 and Ser112) are shown in ball-and-stick representation.
The fold of BphDLB400 is similar to other MCP hydrolases. Overlay of Cα atoms of BphDLB400, BphDRHA1, MhpC, CarCJ3 and CumDIP01 showed that the β sheet and some α helices superpose well; as expected, some loop regions do not. The RMSDs for Cα atoms between BphDLB400 and MhpC (PDB entry 1U2E), CumDIP01 (PDB entry 1IUP), BphDRHA1 (PDB entry 1C4X), and CarCJ3 (PDB entry 1J1I) are 1.01, 1.44, 1.54 and 1.48 Å, respectively. The core domains superpose with high fidelity such that only a few short segments are not superposable. These include the N-terminal eight residues and loop regions between β1 and β2 (residues 16−22), between β2 and β3 (residues 32−34), and between β4 and α2 (residues 79−86; α and β numbered according to reference (17)). Greater structural divergence is observed among the lid domains, which is consistent with low sequence identity in this region. The observed conformational variability of the lid domain may also partially result from differential crystal packing among the five structures.
As expected, the overall subunit structures of S112C in both aerobic and anaerobic conditions, and S112C+HOPDA are almost identical to the wild-type structure, with the RMSD of Cα atoms between them less than 0.24 Å.
Active site
The active site is located at the cleft between the lid and core domains and includes conserved Ser112, His265, and Asp237, reminiscent of the catalytic triad of serine proteases. The active site can be divided into two sub-sites, P (polar) and NP (non-polar), located on each side of the catalytic serine. The P and NP sub-sites bind the dienoate moiety and phenyl ring of the substrate, respectively, and correspond to the proximal and distal parts identified in a study of BphDRHA1 (17). The P sub-site is lined with polar groups from the side chains of residues that are conserved in MCP hydrolases: Asn51, Asn111, Arg190, and Trp266. The NP sub-site is lined with non-polar side chains Ile153, Leu156, Phe175, Leu213, Trp216, and Phe239.
The hydrogen bonding among the catalytic residues Asp237, His265 and Ser112 is of significant mechanistic interest. In the wild-type BphDLB400 structure, the Oδ2 atom of Asp237 forms a hydrogen bond with the Nδ1 atom of His265. The Nε2 atom of His265 forms a hydrogen bond with Oδ1 of Asn111 instead of the Oγ atom of Ser112 in subunit A and does not form any hydrogen bond in subunit B. Consistent with these data, the catalytic His and Ser are not hydrogen bonded in the BphDRHA1 and CarCJ3 structures (17, 19). The His and Ser are hydrogen bonded in the MhpC structure, but the bond is longer (3.3 Å) than typically observed in serine hydrolases (20). Although the observed states of these molecules may differ significantly from the catalytic states, the lack of a prominent His-Ser interaction is consistent with an alternative use of the catalytic triad.
In the structure of wild-type BphDLB400, a water molecule is hydrogen bonded to Oγ Ser112, a phenomenon which was also observed in the structure of native MhpC (20). By contrast, unexplained electron density was observed adjacent to the Oγ atom of the active site serine in BphDRHA1 (17) and certain structures of MhpC (20), suggesting covalent modification. In the structure of the S112C variant of BphDLB400 crystallized aerobically, the electron density suggests that Cys112 is oxidized to the sulfonic acid in subunit A and to the sulfenic acid in subunit B (data not shown). The oxidation apparently occurred during X-ray exposure because mass spectrometric analysis of crystallized S112C before X-ray exposure indicated that Cys112 was not oxidized. Active site thiol oxidation by X-rays has been previously noted for cysteinyl hydrolases (44), including dienelactone hydrolase (45) and papain (46). Since dioxygen may be involved in the cysteine oxidation, we anaerobically crystallized S112C and collected diffraction data from the liquid nitrogen-frozen crystals maintained at 100K under a gaseous nitrogen stream. It is clear that Cys112 is not oxidized in the anaerobic S112C structure, suggesting the oxidation requires both X-rays and dioxygen. Interestingly, in the active site of the anaerobic S112C structure, a water molecule is hydrogen bonded to the Sγ atom of Cys112 as in the wild-type structure.
Enzyme-substrate interactions
In the crystal structure of S112C exposed to HOPDA, clear electron density is observed for the dienoate moiety of HOPDA in the active site of subunit A while the electron density for the phenyl moiety is partial and fragmented. The dienoate moiety is well defined as a keto tautomer since C6 clearly lies out of the C1-C2-C3-C4-C5 plane, implying saturation at C5. Similar electron density was observed in subunit B, but was less clearly defined. Although the density permitted modeling of a complete substrate molecule into the active site of subunit A and subsequent refinement, it only marginally supports inclusion of the phenyl moiety in the model. More significantly, the density is continuous between C112 Sγ and C6 of the ligand, and includes a feature that could represent the C6 hydroxyl substituent bound in the vicinity of the oxyanion hole. The density affords two possible interpretations. In one, the structure would be described as a covalent, tetrahedral intermediate with C6 of the substrate bonded to Cys112 (Figure 8). However, the discrete existence of such a transient catalytic intermediate on the timescale of the HOPDA soak-in (∼1 h) is unlikely. A more probable interpretation is suggested by the absence of clear density for the phenyl ring and the X-ray-induced oxidation of Cys112 observed in the absence of substrate. In this case, the density is explained by the combination of density for the product (HPD) in some but not all active sites and density for oxidized C112, perhaps in different conformations with and without product present. Hence, we refer to this structure as S112C+HOPDA to indicate how the crystal was prepared and to emphasize that the existence of a catalytically relevant tetrahedral intermediate at C6 in this structure is unlikely. In any case, the most important feature of this structure is the clear density for the dienoate moiety, which suggests important enzyme-substrate interactions in this region of the active site.
Figure 8.

Stereo view of HOPDA bound to the active site of BphDLB400 S112C mutant. Top: The refined structure is shown along with (2Fo - Fc) exp(iαc) electron density (1.0 σ). Bottom: Ball-and-stick stereo model of enzyme-substrate interactions. O, N and S atoms are colored in red, blue, and green, respectively. C atoms are colored in brown for the catalytic residues Asp237-His265-Cys112, gray for the substrate, and yellow for the rest of the residues. Potential hydrogen bonds are shown as dotted lines.
The S112C+HOPDA structure reveals a series of hydrogen bonds between conserved residues in the P sub-site (Gly43, Asn51, Arg190, His265, Trp266, and Asn111) and the dienoate moiety of the substrate (Figure 8, Table 4). The Nη1 and Nη2 atoms of Arg190, the N atom of Gly43, the Nδ2 atom of Asn51, and the Nε1 atom of Trp266 interact with the C1 carboxylate of the substrate, while the Nδ2 atom of Asn111, the Nε2 atom of His265, and the Nε1 atom of Trp266 interact with the C2 hydroxyl of the substrate. Significantly, a sulfate ion occupies the same position as the carboxylate and C2 hydroxyl of the substrate in both the wild-type and S112C mutant structures, indicating the important role of this enzyme pocket in stabilizing negative charge, and thereby positioning the substrate in the correct orientation.
Table 4.
Enzyme-substrate hydrogen bonds in the S112C+HOPDA complex.
| Substrate | Active site residues | Distance (Å) |
|---|---|---|
| C1-carboxylate O1 | Arg190 Nη2 | 3.0 |
| C1-carboxylate O1 | Gly43 N | 3.0 |
| C1-carboxylate O2 | Arg190 Nη1 | 3.0 |
| C1-carboxylate O2 | Trp266 Nε1 | 3.2 |
| C1-carboxylate O2 | Asn51 Nδ2 | 3.0 |
| C2-carbonyl O3 | His265 Nε2 | 3.3 |
| C2-carbonyl O3 | Asn111 Nδ2 | 2.9 |
| C2-carbonyl O3 | Trp266 Nε1 | 3.0 |
| C6-carbonyl O4 | Gly42 N | 2.9 |
| C6-carbonyl O4 | Met113 N | 2.8 |
The crystal structure also shows that the main chain amides from residues Met113 and Gly42 are appropriately positioned to form the oxyanion hole that is typical for mechanistically related hydrolases and thus would stabilize an oxyanion at C6 . Met113 is located at the N terminus of helix α3, suggesting that the partial positive charge at this end of α3 also may help stabilize the negative charge of the substrate oxyanion. Gly42 is located in the loop between β3 and α2. In this loop, the highly conserved His40 side chain is hydrogen bonded to the Gly41 carbonyl, thereby directing the Gly42 amide towards the substrate oxyanion.
Although the density for the substrate's phenyl ring is poor, the model places this part of the substrate so that it is entirely surrounded by hydrophobic residues (Val240, Trp216, Leu213, Ile153, Phe157, Leu156, and Phe239) in the NP sub-site of the active site (Figure 8). Comparison of the wild-type and S112C structures with the S112C+HOPDA structure suggests residues Phe175, Phe239, His265, Trp266 and Leu213 may undergo conformational changes upon substrate binding.
Examination of each of the BphDLB400 structures indicates that the conformation of the active site is adaptable. For example, electron density maps for each of the three structures clearly indicate that important active site residues occur in multiple conformations within subunit A or within subunit B of single crystals: Trp266 in subunit A, and Met135, Trp266 in subunit B of wild-type BphDLB400; Met135, Phe175, Phe239, and His265 in subunit A of BphDLB400 S112C; Met135, Phe175, Val240, Trp266 in subunit B of the BphDLB400 S112C+HOPDA complex. Comparisons of active sites between subunits in the same crystal or between the same subunit in different crystals also reveal differences in conformation for several residues (Asn111, Phe175, Leu213, Phe239, Val240, His265, Trp266). Thus, differences occur between A and B of wild-type BphDLB400, between subunits A of wild-type and S112C mutant, or between subunits A of wild-type and S112C+HOPDA. Taken together, this variability suggests conformational changes involving these residues may be important for substrate binding or catalysis. It is interesting to note that His252 and Trp253 in CumDIP01, corresponding to His265 and Trp266 in BphDLB400, have different conformations in various CumD structures (18).
The potential dual role of His265 in deprotonating HOPDA at C-2 and protonating it at C-5, as suggested in MhpC (20, 21), raises the issue of this residue's conformational flexibility. As described above, a clear interaction is observed between His265 and the C-2 hydroxyl/oxo of HOPDA, consistent with the role of His265 as a base. To examine the possible role of His265 in protonating C-5, the distance between C-5 and His265 Nε2 was measured in both subunits of the S112C+HOPDA structure. Moreover, the structures of S112C and wild type were least square fitted to the S112C+HOPDA structure using O, and the distance between His265 and C-5 was calculated for all structures. The data (Table 5) reveal the distance varied from 2.9 Å (deduced from subunit B of S112C) to 4.5 Å (observed in subunit A of S112C+HOPDA).
Table 5.
The conformational flexibility of His265.a
| Structure |
Monomer |
Distance, H265 Nε2 to HOPDA atoms (Å) |
|
|---|---|---|---|
| |
|
HOPDA C-2 |
HOPDA C-5 |
| S112C+HOPDA |
A | 3.3 | 4.5 |
| B |
3.5 |
3.8 |
|
| S112C |
A | 3.9 | 3.5 |
| B |
3.7 |
2.9 |
|
| Wild type | A | 3.4 | 4.5 |
| B | 3.8 | 3.1 | |
HOPDA coordinates from the S112C+HOPDA structure were fit into the S112C and wild type structures as described in the text.
DISCUSSION
The current study provides important new insight into the mechanism of MCP hydrolases. Prior studies have provided indirect evidence for a mechanism involving enzyme-catalyzed tautomerization of substrate to Sk, partial Sk release (11, 12), and hydrolytic C-C cleavage via a gem-diol intermediate (Figure 2, Scheme 1) (13). Our single turnover stopped-flow analysis of BphDLB400 shows rapid formation of E:S, an intermediate having an electronic absorption maximum that is strongly red-shifted (λmax = 492 nm) compared to the HOPDA enolate in aqueous solution (λmax = 434 nm). We observed biphasic decay of this intermediate coupled to formation of the HPD product, with the second phase similar to kcat. However, in contrast to previous work on MhpC (11, 12), we were unable to detect release of Sk, thereby precluding uncoupling as the source of the biphasic behavior. Product inhibition is consistent with HPD release prior to benzoate, and solvent viscosity did not affect kcat, suggesting that benzoate may be released in a rate-determining, intramolecular step. Key active site interactions with the dienoate moiety of the substrate are observed crystallographically, suggesting that His265 may play an important role in substrate ketonization.
Nature of the red-shifted intermediate
Based on the current evidence, we cannot unequivocally identify the rapidly formed E:S species possessing the red-shifted electronic absorption maximum (492 nm). One possibility is that E:S represents an enzyme-bound HOPDA enolate. The conjugated π electron system of the enolate extends from the C-1 to C-6 carbonyls of HOPDA, and in aqueous solution absorbs maximally in the visible region at 434 nm. The red-shift observed upon binding to BphD may arise from active site interactions, or a twisted conformation of the substrate. A second possibility is that E:S represents ketonized HOPDA. Ketonization disrupts the conjugated π electron system of HOPDA and therefore would be predicted to blue-shift the latter's spectrum. However, the enzyme active site environment may perturb the energy of this transition by polarizing the π electrons of the α,β-unsaturated keto tautomer. For example, a 90 nm red-shift produced by an α,β-unsaturated thiol ester inhibitor when bound to the active site of crotonase was attributed to π electron polarization (47). Analogous polarization of the α,β-unsaturated moiety of ketonized HOPDA may occur in the active site of BphD. However, we expect that this red shift would have to be greater than 90 nm to compensate for the blue shift expected upon ketonization. Alternatively, the BphD-bound keto tautomer may be in equilibrium with another form of the bound substrate. Importantly, no evidence for a red-shifted species was observed when excess HPD was mixed with the enzyme, suggesting that the C5-C6 bond is intact in the absorbing species. Recent spectroscopic analyses of HOPDA bound to mutant enzymes indicate that the E:S species with the 492 nm feature possesses a ketonized HOPDA (Horsman & Eltis, unpublished results).
Enzyme-substrate interactions
The S112C+HOPDA structure clearly illustrates important enzyme-substrate interactions. Previously, a hypothetical model for the BphDRHA1:HOPDA complex (17), and the crystal structure of inhibitor bound to MhpC (20) predicted the importance of Arg190 (BphDLB400 numbering) in anchoring the C-1 carboxylate, and the latter study also included the contribution to this function of Asn51. Our structure with the natural substrate reveals interactions of the dienoate moiety with three additional conserved residues: Asn111, His265, and Trp266. The conservation of these five residues among all known MCP hydrolases highlights their catalytic significance. Specifically, the hydrogen bonding to the C-2 oxygen demonstrates the capacity of some of these residues (Asn111, Trp266) to stabilize developing negative charge at this position during ketonization.
The ketonization reaction carried out by the MCP hydrolases is not well understood, but recent crystallographic and kinetic analysis of MhpC mutants suggested that His265 is the most likely candidate for direct transfer of a proton from the C-2 hydroxyl to C-5 (20, 21). Our crystallographic observation of a hydrogen bond between His265 and the C-2 hydroxyl of the dienoate moiety significantly advances the proposed role of His265 as a general base for substrate ketonization. Although the His265 to C-5 distance is rather far in the most clearly defined density of subunit A of the S112C+HOPDA structure (4.5 Å), multiple His265 positions were observed in the various structures, including one which predicts a His265 to C-5 distance of 2.9 Å (Table 5). The flexibility in this histidine, which was also noted in structural studies of CumD (18), appears to be sufficient to accommodate its additional role as a general acid in protonation of C-5. Furthermore, the native BphDLB400-catalyzed deuterium incorporation into the H-5E position of HPD, also seen in MhpC (10) and His6-BphDLB400 (40), suggests the transfer of a proton to the proS position of C-5 and fragmentation onto the re face – a stereochemical course consistent with His265-mediated proton transfer (Figure 9), rather than, for example, a general acid role for Ser112 in protonating HOPDA C-5.
Figure 9.

A possible mechanism of BphDLB400. The free enzyme (E) binds and ketonizes HOPDA via His265-catalyzed transfer of a proton (H*) to the proS position at C-5, generating E:Sk. Nucleophilic attack on E:Sk generates the first tetrahedral intermediate (Tet1), the negative charge of which is stabilized by the main chain amide protons of the ‘oxyanion hole’ residues, Met113 and Gly42. Collapse of Tet1 involves C-C fragmentation onto the re face of the double bond, generating the acyl-enzyme intermediate (E:B), and releasing HPD with the inserted proton at the H-5E position. His265 activates water to attack at the acyl-enzyme carbonyl, generating the second tetrahedral intermediate (Tet2), collapse of which releases benzoate and regenerates free enzyme. For clarity, the conserved residues Asp237 (of the catalytic triad) and Asn51 (interacts with the HOPDA carboxylate) are omitted.
The origin of the biphasic kinetics
The biphasic behavior of E:S decay (492 nm) and HPD formation (270 nm) could not be satisfactorily explained by either Sk release or the absorbance of a product complex. Thus, in contrast to what has been observed for another MCP hydrolase, MhpC (11, 12), three independent approaches indicated that BphDLB00 does not detectably release Sk: (1) HPD formation was apparently coupled to E:S depletion in single turnover stopped-flow experiments; (2) HPLC analysis revealed no accumulation of Sk under steady state conditions; and (3) levels of non-specific deuterium incorporation into HPD were low. Other explanations for the biphasic kinetics must therefore be explored.
We considered two models to account for the biphasic decay. In the E:Se model (Scheme 2), the absorbing species in E:S (λmax = 492 nm) is the HOPDA enolate. Moreover, 1/τ1, 1/τ2 and 1/τ3 reflect deprotonation (or twisting of the enolate), ketonization, and C-C cleavage, respectively. Biphasic decay of E:Se at the observed values for 1/τ2 and 1/τ3 can be appropriately modeled (48)4 assuming that ketonization is a reversible process with approximately equal forward and reverse rate constants (k2 ∼ k−2 ∼ 20 s−1), and that the subsequent C-C cleavage is irreversible (k3 ∼ 15 s−1). However, this model is inconsistent with several experimental observations. First, 1/τ2 decreased with increased solvent viscosity. This is not consistent with an intramolecular event such as ketonization, which should not be affected by viscosity. Second, we obtained no spectroscopic evidence of a ketonized intermediate. That is, the appearance of the species that absorbs maximally at 270 nm is apparently coupled to the decay of E:S. For this to be true in the E:Se model, HPD and ketonized HOPDA would have to possess essentially identical spectra, including similar molar absorptivities at 270 nm. This seems unlikely considering that the enol tautomer of HPD (analogous to enol-HOPDA) absorbs maximally at 270 nm, while the keto tautomer (analogous to keto-HOPDA) does not. Finally, this model does not readily explain the apparent ordered release of products suggested from product inhibition experiments.
Scheme 2.

In the two-conformation model (Scheme 3), 1/τ1 reflects ketonization and the biphasic kinetics result from two interconverting enzyme conformations: E, capable of catalyzing C-C cleavage/HPD release, and E', incapable of C-C cleavage/HPD release. C-C cleavage/HPD dissociation (k2) and benzoate release (k3) must occur in E (half the enzyme sites) before a conformational change converts E' (the other half of the active sites) to E, thereby allowing catalysis to proceed in the remaining active sites. Hence each subunit cycles between E and E' conformations.
Scheme 3.

Several lines of evidence are consistent with a two-conformation model. First, the amplitudes of 1/τ2 and 1/τ3 were approximately equal to each other under all experimental conditions investigated herein, suggesting that they represent events occurring in 50% of the enzyme molecules. Second, increased solvent viscosity, achieved by adding glycerol, decreased 1/τ2, but has no effect on 1/τ3. According to Scheme 3, 1/τ2 represents C-C cleavage/HPD release in 50% of the enzyme molecules, and should thus be sensitive to the effect of viscosity on diffusive HPD release occurring with the rate constant k2. In contrast, 1/τ3 is unaffected by viscosity, consistent with the proposal that this relaxation contains contributions from a rate-limiting (since 1/τ3 ∼ kcat), non-diffusive release of benzoate (k3), followed by rapid HPD formation in the remaining half sites (k2'). Third, this model more reasonably assigns the absorbance at 270 nm solely to HPD, and does not require an equivalent molar absorptivity of ketonized HOPDA. Fourth, the observed ordered product release is accounted for in this scenario. Finally, significant conformational differences are observed between the active sites of monomers A and B in several of the crystal structures, and, in the S112C+HOPDA structure, the density for the substrate is less clear in monomer B than in monomer A, suggesting lower occupancy.
The two-conformation model is intriguing in light of crystallographic evidence. First, in an MhpC:inhibitor complex, the substrate analog was bound to only one subunit of the dimer (20). Moreover, openings to the sub-sites in the MhpC structures are of insufficient diameter to permit passage of substrate, suggesting that some movement is necessary for substrate binding and product release (20). However, single turnover stopped-flow analysis of MhpC at neutral pH exhibited only a single relaxation assigned to C-C cleavage/product release (21), suggesting that the apparent half-of-sites reactivity does not influence this step. Stopped-flow analysis of HOPDA hydrolysis by His6-BphDLB400 also demonstrated a single step kinetic mechanism for substrate disappearance and HPD formation (40). However, it is possible that the histidine tag disrupts the quaternary structure that gives rise to the two distinct enzyme conformers.
Structural data further suggest at least two origins of a conformational change: the lid domain and Arg190. Thus, some conformational differences in the lid domains occur among our structures. Movement in the lid domain could be analogous to what occurs in other α/β-hydrolases such as lipases (7, 8). In BphD, the lid domains make contacts dependent on the tetramer (i.e., the AB' dimer in Figure 6). Consistent with an apparent single step for product release in MhpC, the MhpC dimer corresponds to the AB dimer of BphD, in which the lid domains do not interact directly with each other (20). Alternatively, the conformational change could involve movement of Arg190 to facilitate HPD release. This residue is directed toward the solvent in some MCP hydrolase crystal structures, greatly increasing the solvent accessibility of the P sub-site (17, 18). It is possible that conformational changes in the lid domain movement and Arg190 occur in a concerted manner to facilitate HPD release.
The possibility of an acyl-enzyme intermediate
A notable feature of the results is thatk3 is both rate-determining and unaffected by viscosity. In the two-conformation model, two mechanisms may be invoked to explain k3: 1) the conformational change accompanying the release of benzoate is rate-limiting, or 2) the conformational change occurs rapidly after a rate-limiting hydrolysis of a benzoyl-enzyme intermediate. Consideration of the product inhibition data in light of the structural data is most consistent with the second mechanism. Thus, in all three BphDLB400 structures, the benzoate-binding (NP) sub-site of the active site is freely accessible to solvent while the HPD-binding (P) sub-site appears to be partially occluded. Hence, in the absence of a covalent intermediate (e.g., the gem-diol mechanism, Figure 2), the structural data indicate that benzoate should be released first. However, the product inhibition data suggest that HPD is released from the enzyme prior to benzoate. The apparent contradiction between the product inhibition and structural data may be reconciled by invoking a conformational change.
A simpler explanation for the observed ordered release of products is that catalysis involves a more conventional role for the catalytic serine whereby it attacks Sk to liberate HPD, and the remaining covalent benzoyl-enzyme complex is hydrolyzed in a final, rate-determining step (Figure 9). Such a nucleophilic mechanism is consistent with the viscosity effects outlined above. Moreover, the crystallographic observation of a hemi-ketal adduct in the MhpC:inhibitor complex (20) demonstrates that the catalytic serine is capable of nucleophilic attack at a substrate carbonyl. In light of the uncertain role of the catalytic serine in MCP hydrolases, we are performing experiments using BphD to distinguish mechanisms involving gem-diol and acyl-enzyme intermediates.
ACKNOWLEDGEMENTS
We thank Yong Ge for purified C23O, Fred Vaillancourt for purified DHBD, Mark Okon for assistance in acquiring NMR spectra, Shouming He for the mass spectrometric analysis, Cheryl Whiting for technical assistance, and Martin Tanner for reading the manuscript. Use of the Argonne National Laboratory Structural Biology Center and BioCARS beamlines at the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Biological and Environmental Research, under Contract No. W-31-109-ENG-38. We are grateful to the staff of both sectors. Use of the BioCARS Sector 14 was also supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707.
Footnotes
This work was funded by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant) and the National Institutes of Health (GM-52381).
Abbreviations: MCP, meta-cleavage product; HPD, 2-hydroxy-penta-2,4-dienoic acid; PCB, polychlorinated biphenyl; HOPDA, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid; DHB, 2,3-dihydroxybiphenyl; DHBD, dihydroxybiphenyl dioxygenase; Sk, ketonized substrate; E:Sk, enzyme-bound ketonized substrate; P, polar; NP, non-polar; I, ionic strength.
For an instrument dead time of 1.2 ms, only ∼50% of a reaction with 1/τ = 500 s−1 may be observed. The observable fraction of the reaction (fobs) can be determined from the instrument dead time (td) and the half-life of the reaction (t1/2) as follows: fobs = (1/2)^(td/t1/2) (36).
The reaction contained 1% water (from addition of aqueous BphD), and 2% ethanol (from addition of concentrated HOPDA), thereby causing a small signal from H-5E.
1/τ2 = (p + q)/2; 1/τ3 = (p – q)/2; where p = k2 + k−2 + k3 + k−3; and q = [p2 – 4(k2k3 + k−2k−3 + k2k−3)]1/2 (48).
REFERENCES
- 1.Dagley S. Biochemical approach to some problems of environmental pollution. Essays Biochem. 1975;11:81–138. [PubMed] [Google Scholar]
- 2.Pieper DH. Aerobic degradation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 2005;67:170–191. doi: 10.1007/s00253-004-1810-4. [DOI] [PubMed] [Google Scholar]
- 3.Seah SYK, Labbé G, Nerdinger S, Johnson MR, Snieckus V, Eltis LD. Identification of a serine hydrolase as a key determinant in the microbial degradation of polychlorinated biphenyls. J. Biol. Chem. 2000;275:15701–15708. doi: 10.1074/jbc.275.21.15701. [DOI] [PubMed] [Google Scholar]
- 4.Ohtsubo Y, Kudo T, Tsuda M, Nagata Y. Strategies for bioremediation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 2004;65:250–258. doi: 10.1007/s00253-004-1654-y. [DOI] [PubMed] [Google Scholar]
- 5.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderton MC, Bhakta S, Besra GS, Jeavons P, Eltis LD, Sim E. Characterization of the putative operon containing arylamine N-acetyltransferase (nat) in Mycobacterium bovis BCG. Mol. Microbiol. 2006;59:181–192. doi: 10.1111/j.1365-2958.2005.04945.x. [DOI] [PubMed] [Google Scholar]
- 7.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verscheuren KHG, Goldman A. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 8.Holmquist M. Alpha/beta hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Prot. Pept. Sci. 2000;1:209–235. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 9.Heikinheimo P, Goldman A, Jeffries C, Ollis DL. Of barn owls and bankers: A lush variety of alpha/beta hydrolases. Structure. 1999;7:141–146. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 10.Lam WWY, Bugg TDH. Chemistry of extradiol aromatic ring cleavage: Isolation of a stable dienol ring fission intermediate and stereochemistry of its enzymatic hydrolytic cleavage. J. Chem. Soc., Chem. Comm. 1994:1163–1164. [Google Scholar]
- 11.Lam WWY, Bugg TDH. Purification, characterization, and stereochemical analysis of a C-C hydrolase: 2-Hydroxy-6-keto-nona-2,4-diene-1,9-dioic acid 5,6-hydrolase. Biochemistry. 1997;36:12242–12251. doi: 10.1021/bi971115r. [DOI] [PubMed] [Google Scholar]
- 12.Henderson IMJ, Bugg TDH. Pre-steady-state kinetic analysis of 2-hydroxy-6-keto-nona-2,4-diene-1,9-dioic acid 5,6-hydrolase: Kinetic evidence for enol/keto tautomerization. Biochemistry. 1997;36:12252–12258. doi: 10.1021/bi971116j. [DOI] [PubMed] [Google Scholar]
- 13.Fleming SM, Robertson TA, Langley GJ, Bugg TDH. Catalytic mechanism of a C-C hydrolase enzyme: Evidence for a gem-diol intermediate, not an acyl enzyme. Biochemistry. 2000;39:1522–1531. doi: 10.1021/bi9923095. [DOI] [PubMed] [Google Scholar]
- 14.Speare DM, Fleming SM, Beckett MN, Li JJ, Bugg TDH. Synthetic 6-aryl-2-hydroxy-6-ketohexa-2,4-dienoic acid substrates for C-C hydrolase BphD: Investigation of a general base catalytic mechanism. Org. Biomol. Chem. 2004;2:2942–2950. doi: 10.1039/B410322J. [DOI] [PubMed] [Google Scholar]
- 15.Speare DM, Olf P, Bugg TDH. Hammett analysis of a C-C hydrolase-catalysed reaction using synthetic 6-aryl-2-hydroxy-6-ketohexa-2,4-dienoic acid substrates. Chem. Commun. 2002:2304–2305. doi: 10.1039/b208168g. [DOI] [PubMed] [Google Scholar]
- 16.Bugg TDH. Diverse catalytic activities in the alpha beta-hydrolase family of enzymes: Activation of H2O, HCN, H2O2, and O2. Bioorg. Chem. 2004;32:367–375. doi: 10.1016/j.bioorg.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Nandhagopal N, Yamada A, Hatta T, Masai E, Fukuda M, Mitsui Y, Senda T. Crystal structure of 2-hydroxyl-6-oxo-6-phenylhexa-2,4-dienoic acid (HPDA) hydrolase (BphD enzyme) from the Rhodococcus sp. strain RHA1 of the PCB degradation pathway. J. Mol. Biol. 2001;309:1139–1151. doi: 10.1006/jmbi.2001.4737. [DOI] [PubMed] [Google Scholar]
- 18.Fushinobu S, Saku T, Hidaka M, Jun SY, Nojiri H, Yamane H, Shoun H, Omori T, Wakagi T. Crystal structures of a meta-cleavage product hydrolase from Pseudomonas fluorescens IP01 (CumD) complexed with cleavage products. Protein Sci. 2002;11:2184–2195. doi: 10.1110/ps.0209602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habe H, Morii K, Fushinobu S, Nam JW, Ayabe Y, Yoshida T, Wakagi T, Yamane H, Nojiri H, Omori T. Crystal structure of a histidine-tagged serine hydrolase involved in the carbazole degradation (CarC enzyme) Biochem. Biophys. Res. Commun. 2003;303:631–639. doi: 10.1016/s0006-291x(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 20.Dunn G, Montgomery MG, Mohammed F, Coker A, Cooper JB, Robertson T, Garcia JL, Bugg TDH, Wood SP. The structure of the C-C bond hydrolase MhpC provides insights into its catalytic mechanism. J. Mol. Biol. 2005;346:253–265. doi: 10.1016/j.jmb.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Montgomery MG, Mohammed F, Li JJ, Wood SP, Bugg TDH. Catalytic mechanism of C-C hydrolase MhpC from Escherichia coli: Kinetic analysis of His263 and Ser110 site-directed mutants. J. Mol. Biol. 2005;346:241–251. doi: 10.1016/j.jmb.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 22.Nerdinger S, Kendall C, Marchhart R, Riebel P, Johnson MR, Yin CF, Eltis LD, Snieckus V. Directed ortho metalation and Suzuki-Miyaura cross-coupling connections: Regiospecific synthesis of all isomeric chlorodihydroxybiphenyls for microbial degradation studies of PCBs. Chem. Commun. 1999:2259–2260. [Google Scholar]
- 23.Seah SYK, Terracina G, Bolin JT, Riebel P, Snieckus V, Eltis LD. Purification and preliminary characterization of a serine hydrolase involved in the microbial degradation of polychlorinated biphenyls. J. Biol. Chem. 1998;273:22943–22949. doi: 10.1074/jbc.273.36.22943. [DOI] [PubMed] [Google Scholar]
- 24.Seah SY, Terracina G, Bolin JT, Riebel P, Snieckus V, Eltis LD. Purification and preliminary characterization of a serine hydrolase involved in the microbial degradation of polychlorinated biphenyls. J. Biol. Chem. 1998;273:22943–22949. doi: 10.1074/jbc.273.36.22943. [DOI] [PubMed] [Google Scholar]
- 25.Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 26.Cornish-Bowden A. Analysis of Enzyme Kinetic Data. Oxford University Press; New York: 1995. [Google Scholar]
- 27.Lian HL, Whitman CP. Ketonization of 2-hydroxy-2,4-pentadienoate by 4-oxalocrotonate tautomerase: Implications for the stereochemical course and the mechanism. J. Am. Chem. Soc. 1993;115:7978–7984. [Google Scholar]
- 28.McPherson A. The Preparation and Analysis of Protein Crystals. John Wiley & Sons; New York: 1982. [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr., Sweet RM, editors. Methods Enzymol. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 30.O'Neill P, Stevens DL, Garman EF. Physical and chemical considerations of damage induced in protein crystals by synchrotron radiation: A radiation chemical perspective. J. Synchrotron Radiat. 2002;9:329–332. doi: 10.1107/s0909049502014553. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey S. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A 47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Seah SYK, Labbé G, Kaschabek SR, Reifenrath F, Reineke W, Eltis LD. Comparative specificities of two evolutionarily divergent hydrolases involved in microbial degradation of polychlorinated biphenyls. J. Bacteriol. 2001;183:1511–1516. doi: 10.1128/JB.183.5.1511-1516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiromi K. Kinetics of Fast Enzyme Reactions. Halsted Press; Tokyo: 1979. [Google Scholar]
- 37.Pollard JR, Henderson IMJ, Bugg TDH. Chemical and biochemical properties of 2-hydroxypentadienoic acid, a homologue of enolpyruvic acid. Chem. Commun. 1997:1885–1886. [Google Scholar]
- 38.Adams JA, Taylor SS. Energetic limits of phosphotransfer in the catalytic subunit of cAMP-dependent protein kinase as measured by viscosity experiments. Biochemistry. 1992;31:8516–8522. doi: 10.1021/bi00151a019. [DOI] [PubMed] [Google Scholar]
- 39.Cole PA, Burn P, Takacs B, Walsh CT. Evaluation of the catalytic mechanism of recombinant human Csk (C-terminal Src kinase) using nucleotide analogs and viscosity effects. J. Biol. Chem. 1994;269:30880–30887. [PubMed] [Google Scholar]
- 40.Li JJ, Bugg TDH. Stereochemistry of the reaction catalysed by 2-hydroxy-6-keto-6-phenyl-hexa-2,4-dienoic acid 5,6-hydrolase (BphD) Chem. Commun. 2005:130–132. doi: 10.1039/b415491f. [DOI] [PubMed] [Google Scholar]
- 41.Whitman CP, Aird BA, Gillespie WR, Stolowich NJ. Chemical and enzymatic ketonization of 2-hydroxymuconate, a conjugated enol. J. Am. Chem. Soc. 1991;113:3154–3162. [Google Scholar]
- 42.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. 3rd ed. Portland Press; London: 2004. [Google Scholar]
- 43.Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins: Struct., Funct., Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 44.Jocelyn PC. Biochemistry of the SH group. Academic Press; London: 1972. [Google Scholar]
- 45.Pathak D, Ollis D. Refined structure of dienelactone hydrolase at 1.8 Å. J. Mol. Biol. 1990;214:497–525. doi: 10.1016/0022-2836(90)90196-s. [DOI] [PubMed] [Google Scholar]
- 46.Kamphuis IG, Kalk KH, Swarte MB, Drenth J. Structure of papain refined at 1.65 Å resolution. J. Mol. Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 47.D'Ordine RL, Tonge PJ, Carey PR, Anderson VE. Electronic rearrangement induced by substrate-analog binding to the enoyl-CoA hydratase active site: Evidence for substrate activation. Biochemistry. 1994;33:12635–12643. doi: 10.1021/bi00208a014. [DOI] [PubMed] [Google Scholar]
- 48.Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W. H. Freeman; New York: 1999. [Google Scholar]