

Abstract
Nanotechnology-based drug delivery systems (nanoDDSs) have seen recent popularity due to their favorable physical, chemical, and biological properties, and great efforts have been made to target nanoDDSs to specific cellular receptors. CD44/chondroitin sulfate proteoglycan (CSPG) is among the receptors overexpressed in metastatic melanoma, and the sequence to which it binds within the type IV collagen triple-helix has been identified. A triple-helical “peptide-amphiphile” (α1(IV)1263-1277 PA) which binds CD44/CSPG has been constructed and incorporated into liposomes of differing lipid compositions. Liposomes containing distearoyl phosphatidylcholine (DSPC) as the major bilayer component, in combination with distearoyl phosphatidylglycerol (DSPG) and cholesterol, were more stable than analogous liposomes containing dipalmitoyl phosphatidylcholine (DPPC) instead of DSPC. When dilauroyl phosphatidylcholine (DLPC):DSPG:cholesterol liposomes were prepared, monotectic behavior was observed. The presence of the α1(IV)1263-1277 PA conferred greater stability to the DPPC liposomal systems and did not affect the stability of the DSPC liposomes. A positive correlation was observed for cellular fluorophore delivery by the α1(IV)1263-1277 PA liposomes and CD44/CSPG receptor content in metastatic melanoma and fibroblast cell lines. Conversely, non-targeted liposomes delivered minimal fluorophore to these cells regardless of the CD44/CSPG receptor content. When metastatic melanoma cells and fibroblasts were treated with exogeneous α1(IV)1263-1277, prior to incubation with α1(IV)1263-1277 PA liposomes, to potentially disrupt receptor/liposome interactions, a dose-dependent decrease in the amount of fluorophore delivered was observed. Overall, our results suggest that PA-targeted liposomes can be constructed and rationally fine-tuned for drug delivery applications based on lipid composition. The selectivity of α1(IV)1263-1277 PA liposomes for CD44/CSPG-containing cells represents a targeted-Nano-DDS with potential for further development and application.
Introduction
A major goal in drug delivery is to effectively deliver drugs to their intended biological target without deleterious side effects. In principle, targeted drug delivery would minimize toxicities while delivering an effective dose of the drug where desired. This type of delivery typically requires the chemical conjugation of drugs or drug carriers to the targeting moiety. The conjugation of drugs directly to the targeting ligand, however, can negatively affect the targeting molecule in a manner that disrupts receptor/ligand recognition1 and may alter the cytotoxicity of the drug.2,3 Drug delivery systems (DDSs) can improve the pharmacological properties of conventional drugs by altering drug pharmacokinetics and biodistribution, as well as functioning as drug reservoirs.4 Nanotechnology-based DDSs (nanoDDSs), in which the drug carriers have diameters of ~100 nm or less, have seen recent popularity due to the favorable physical, chemical, and biological properties of biomolecules of that size.4,5 NanoDDSs include liposomes, dendrimers, micelles, and polymeric and ceramic nanoparticles.6,7 These nanoDDSs have been widely studied for delivery of various drugs to cellular targets, but each does not posses inherent targeting capabilities. Micelles, liposomes, and nanoparticles can be easily modified to incorporate targeting moieties that allow for more specific or guided delivery of the drug. For example, previous liposomal targeting strategies have utilized simple peptides, proteins (including antibodies) or protein fragments, carbohydrates, or vitamins.8–17 These targeting ligands are specific for certain receptors that are overexpressed by transformed versus normal cells. However, simple unstructured peptides may be readily degraded prior to reaching their targets. Proteins increase the complexity of creating the targeting molecule, as they are subject to proteolysis and may bind to multiple receptors and/or induce immune responses. Antibody applicability is limited to a small subset of tumors, and antibody modified liposomes may be removed from circulation more rapidly than unmodified liposomes.10,18 Carbohydrates may be bound non-specifically, whereas vitamins are readily metabolized.
The topologically stabilized peptide-amphiphile (PA) construct can be utilized as a targeting ligand with high specificity, low degradability, and which can be conveniently incorporated into various delivery vehicles such as liposomes or micelles. The term peptide-amphiphile was first used in 1984, when an alanine residue was interposed between a charged head group and a double-chain pseudo-lipid tail.19 Simple PAs were found to self-associate, with ordered interactions between the peptide head groups.20–22 PAs were subsequently utilized to mimic defined topological structures by incorporating an amino acid sequence with the propensity to form a triple-helix as the polar head group and a dialkyl or monoalkyl hydrocarbon chain as the non-polar tail (Scheme 1a).23–26 The application of PAs has since broadened to include a vast array of structures, such as β-sheets based on β-amyloid, silk, or elastin sequences, as well as coiled-coils, and fibronectin-derived RGD turns.27–30 PAs are advantageous in that they represent a class of multivalent ligands31 that are chemically well-defined, avoiding loss of activity that can occur during non-specific coupling of peptides to lipids.32 The amphiphilic character of PAs allows for the control of assembled structures by manipulating their molecular composition.33–36 For example, the thermal stability of triple-helical and α-helical PA head groups can be modulated by the length of the lipophilic moiety.24,25,37–39 Desirable peptide head group melting temperature (Tm) values can be achieved for in vivo use, as both triple-helical and α-helical PAs have been constructed with Tm values ranging from 30 to 70 °C.24,25,37–43
Scheme 1.
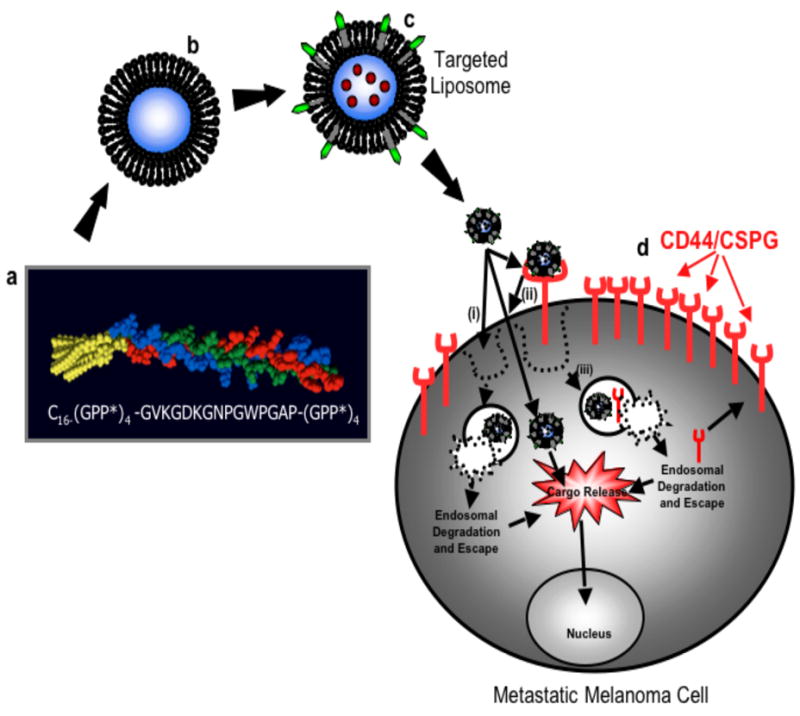
Basis for targeted drug delivery in metastatic melanoma cells. a, α1(IV)1263-1277 PA sequence and triple-helical structure. b, Fluorophore loaded liposome. c, Fluorophore loaded liposome with α1(IV)1263-1277 PA. d, Metastatic melanoma cell with overexpressed CD44/CSPG receptors. Rhodamine internalization may proceed by one of three possible routes: (i) direction fusion of the liposome with the cell bilayer and rhodamine release; (ii) liposome binding to CD44/CSPG receptors, followed by fusion of the liposome with the bilayer and rhodamine release; or (iii) liposome binding to CD44/CSPG receptors, followed by internalization of the receptor/liposome complex and rhodamine release in endosomes.
The overexpression of CD44 on a variety of tumor cells has made this receptor a potential candidate for targeted drug delivery. It has previously been shown that cells with higher expression of CD44 have a greater migratory and invasive potential on hyaluronate-coated substrates.44 Elevated CD44 expression by 4 to 6-fold is associated with tumor growth and metastasis.45 CD44 in the chondroitin sulfate proteoglycan (CSPG) modified form is among the receptors uniquely overexpressed in metastatic melanoma.46 Ligands binding to CD44 undergo endocytosis,47,48 suggesting that CD44 could be a good target for liposomal drug delivery into melanoma cells.
Melanoma CD44 targeting has been achieved previously using hyaluronan-containing liposomes.13,49–51 Hyaluronan targeted liposomes were shown to be more effective than free doxorubicin for delivery in vitro against B16F10 melanoma cells49 and in vivo in syngeneic and human xenograft mouse tumor models.50 However, the disadvantages of using hyaluronan or hyaluronic acid as targeting ligands is that they are high molecular weight species which are quickly removed from circulation by hepatic cells.52 In an attempt to circumvent this disadvantage, enzymatically degraded hyaluronic acid fragments of lower molecular weight have been used as targeting moieties in doxorubicin loaded liposomes.13 A rapid, dose dependent binding of these targeted liposomes to B16F10 melanoma cells was observed. Unfortunately, the low molecular weight hyaluronic acid fragments were also found to have lower affinity to the CD44 receptor than the intact hyaluronic acid, thus diminishing the targeting capabilities. Most importantly, an approach that employs hyaluronan or hyaluronic acid and its fragments as the targeting moiety to CD44 suffers from reduced selectivity because other cell surface receptors, such as RHAMM, have been shown to bind both hyaluronan and hyaluronic acid just as avidly as CD44.53,54 In addition, HA binds to the CD44 amino-terminal globular domain,55 and thus is not sensitive to distinct CD44 glycosylation patterns.
Metastatic melanoma cell CD44/CSPG cell surface receptors bind to a triple-helical sequence within the basement membrane (type IV) collagen. The sequence to which CD44 binds within the type IV collagen triple-helix has been identified as α1(IV)1263-1277 (gene-derived sequence Gly-Val-Lys-Gly-Asp-Lys-Gly-Asn-Pro-Gly-Trp-Pro-Gly-Ala-Pro).56–58 Interaction of CD44 with this region is strongly dependent upon ligand triple-helical conformation and CSPG modification of CD44.43,57,58 Triple-helical PA models of α1(IV)1263-1277 [general structure Cn-(Gly-Pro-Hyp)4-Gly-Val-Lys-Gly-Asp-Lys-Gly-Asn-Pro-Gly-Trp-Pro-Gly-Ala-Pro-(Gly-Pro-Hyp)4-NH2] have been previously constructed and shown to be specific for CD44/CSPG.24–26,58
In the present study, the α1(IV)1263-1277 PA was incorporated into a liposome bilayer to confer targeting capabilities to liposomes against metastatic melanoma cells that overexpress the CD44 cell surface receptor (Scheme 1). Liposomes varying in lipid composition were prepared both with and without the targeting PA, and were used to determine the effect of the PA as well as the lipid composition on liposomal stability over time, at different temperatures. The cellular internalization of rhodamine, delivered by α1(IV)1263-1277 PA liposomes, was quantified as a function of cellular expression of CD44 receptors. The specificity of α1(IV)1263-1277 PA liposomal delivery was examined by pretreating cells with increasing concentrations of exogenous α1(IV)1263-1277 triple-helical peptide (THP) followed by application of liposomes. Overall, this study has evaluated the use of the α1(IV)1263-1277 PA as a targeting moiety to create a drug delivery vehicle selective for highly metastatic melanoma cells.
Materials and Methods
Chemicals
All phospholipids and cholesterol were purchased from Avanti Polar Lipids (Birmingham, AL). The solvents used in the synthesis of the vesicles and peptides, such as methanol, chloroform, tert-butyl ether, N,N-dimethylformamide, N,N-diisopropylethylamine, and N-methylpyrrolidione, were from Fisher Scientific (Pittsburg, PA), Sigma Chemicals (St. Louis, MO), or Aldrich (Milwaukee, WI). Rhodamine 6G was obtained from ACROS Organics (Fairlawn, NJ). The appropriately protected amino acids, 1-hydroxybenzotriazole, N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide, and Rink amide MBHA resin were all obtained from Novabiochem (La Jolla, CA). All other chemicals and solvents for the triple-helical peptide (THP) and PA syntheses were obtained from ACROS Organics with the exception of phenol, which was obtained from Fisher Scientific. Palmitic acid [CH3-(CH2)14-CO2H, designated C16] was purchased from Aldrich. The preparation, purification, and characterization of α1(IV)1263-1277 THP [(Gly-Pro-Hyp)4-Gly-Val-Lys-Gly-Asp-Lys-Gly-Asn-Pro-Gly-Trp-Pro-Gly-Ala-Pro-(Gly-Pro-Hyp)4-NH2] and the α1(IV)1263-1277 PA possessing a C16 tail have been described previously.25
Liposome Preparation
The phospholipids and cholesterol were combined in fixed ratios (Table 1), and dissolved in an organic phase mixture of methanol, tert-butyl ether and chloroform (1:2:2.4) by vortexing for 0.5 h at room temperature. At this stage, if PA-targeted liposomes were the desired product (Table 1), the α1(IV)1263-1277 PA was added to the lipid organic phase mixture. The organic phase was then removed under reduced pressure by rotary evaporation, leaving a thin lipid film at the bottom of the flask which was dried over night in vacuo. The aqueous phase, consisting of rhodamine 6G (400 μM) in PBS, was then added to the lipid film and the resulting dispersion was vortexed extensively. The dispersion was then stirred for 30 min at 60 °C. The maintenance of this temperature for a sustained time was necessary as the lipid tails were mobilized and thus allowed the aqueous medium to traverse the lipid bilayers. The resulting multilamellar vesicle (MLV) suspension was then subjected to four freeze-thaw cycles followed by 10 cycles of extrusion through 100 nm double-stacked polycarbonate filters using a Lipex Extruder (Northern Lipids, Inc., Vancouver, British Columbia) at pressures typically at the lower end of the 250–700 psi range. The polycarbonate filters employed in the extrusion process were obtained from SPI Supplies (West Chester, PA). The unencapsulated fluorophore was then separated from the fluorophore-loaded liposomes by size exclusion chromatography using a G50 medium grade resin Sephadex column (Amersham Biosciences) pre-conditioned with PBS (pH 7.4). The size of liposomes was evaluated by SEC and dynamic light scattering.38,39,59,60 Dynamic light scattering analysis, using a Dawn Eos MALS, was carried out to determine the mean diameter of the liposomes from each batch prepared (Table 1). Typically, the average diameter obtained for the targeted and non-targeted liposomes was in the range of 120–140 nm (small unilamellar vesicles; SUVs). Liposomes were used within 1 day of preparation and stored at 4 °C under argon. The liposome phospholipid content was determined by the Stewart (ammonium ferrothiocyanate) assay as described previously.1,61 The presence of the α1(IV)1263–1277 PA in the liposomal bilayer was examined by MALDI-TOF mass spectrometry (MS) using an α-cyano-4-hydroxycinnaminic acid matrix. The concentration of the α1(IV)1263–1277 PA in the liposomal bilayer was determined by absorbance at λ = 280 nm following liposome disruption with ethanol, dilution with water, lyophilization, and resuspension in 500 μL ethanol.
Table 1.
Liposomal systems utilized for stability evalulation.
| Liposome Formulation | Molar Ratio | Liposome Diameter (nm) |
|---|---|---|
| Distearoyl phosphatidylglycerol (DSPG) | 1 | 126 ± 7 |
| Distearoyl phosphatidylcholine (DSPC) | 4 | |
| Cholesterol | 5 | |
| Distearoyl phosphatidylglycerol (DSPG) | 1 | 129 ± 2 |
| Dipalmitoyl phosphatidylcholine (DPPC) | 4 | |
| Cholesterol | 5 | |
| Distearoyl phosphatidylglycerol (DSPG) | 1 | 122 ± 4 |
| Distearoyl phosphatidylcholine (DSPC) | 4 | |
| Cholesterol | 5 | |
| α1(IV)1263-1277 peptide-amphiphile (PA) | 0.1 | |
| Distearoyl phosphatidylglycerol (DSPG) | 1 | 126 ± 14 |
| Dipalmitoyl phosphatidylcholine (DPPC) | 4 | |
| Cholesterol | 5 | |
| α1(IV)1263-1277 peptide-amphiphile (PA) | 0.1 |
Cell Culture Conditions
The M14#5 human metastatic melanoma cell line was generously provided by Dr. Barbara Mueller. The BJ foreskin fibroblasts and Hs895Sk fibroblasts from a melanoma patient were obtained from American Type Culture Collection (ATCC) (Manassas, VA). Cell media and trypan blue were obtained from Fisher Scientific or CellGro (Herndon, VA), and all reagents required for cell culture were purchased from Sigma Chemicals. Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine sera, 0.1 mg/mL gentamicin, 50 units/mL penicillin, and 0.05 mg/mL streptomycin. Cells were cultured with complete medium at 37 °C in a humidified atmosphere of 5% CO2 in air. For all experiments cells were harvested from sub-confluent (<80%) cultures using a trypsin-EDTA solution and then re-suspended in fresh medium. Cells with a >90% viability, as determined by trypan blue exclusion, were used.
Whole Cell ELISA
The cell surface CD44 concentration was evaluated for the BJ fibroblast, Hs895Sk fibroblast, and M14#5 melanoma cell lines by ELISA.58 Briefly, cells were diluted in PBS and plated at various concentrations in a 96-well plate. The plate was incubated at 4 °C for 2 h and the PBS was removed. The cells were then fixed with methanol and the plate blocked with BSA at 4 °C. Anti-CD44 mAb monoclonal antibody (Zymed Laboratories, Inc., South San Francisco) was diluted in PBST (PBS with 0.05% Tween-20) containing 2 mg/mL BSA and incubated at 4 °C. The plate was washed with PBST and subsequently incubated with goat anti-mouse IgG conjugated to horseradish peroxidase in PBST and 2 mg/mL PBSA. The plate was washed and horseradish peroxidase activity was detected using 3,3′,5,5′-tetramethylbenzidine (Pierce) as substrate by UV-Vis absorption spectroscopy at λ = 450 and 620 nm.
Liposome Stability
The stability of the encapsulated rhodamine 6G in the various liposome systems was initially determined by monitoring fluorophore release from the vesicles (200 μL of 3 mg/ml vesicle solution) at 4, 25, and 37 °C, over time. The fluorescence intensity for each vesicle sample at each temperature was measured at selected time points within a 1 month period using a Spectra Max Gemini EM Fluorescent Plate Reader (Molecular Devices) at λexcitation = 525 nm and λemission = 555 nm. Complete release of fluorophore from the vesicles at each time point yields 100% dequenching, and was obtained from control ethanol-treated liposome samples. The percentage release of fluorophore from the vesicles was determined from the fluorescence intensity of each sample relative to 100% dequenching, which can then be expressed in terms of percentage of fluorophore release.
Subsequently, the stability of α1(IV)1263-1277 PA SUVs versus non-targeted SUVs was determined by monitoring release of the rhodamine from the vesicles in the presence of cells or preconditioned cell media. Cells (1 × 106 cells grown overnight in complete media) were incubated in adhesion media (3 mL) for 1 h at 37 °C and then treated with the liposomes (20 μL, 0.3 mg/mL). Similarly, for the experiment with the preconditioned adhesion media, the media was decanted after 1 h incubation time with the cells and used in the liposomal stability assay. At various time points up to 1 h, 100 μL aliquots of the liposome-media supernatant were removed from each Petri dish, and dispensed into a 96-well quartz plate. The fluorescence intensity for each vesicle sample was measured as described above, and percentage fluorophore release calculated.
Cellular Fluorophore Accumulation
An Olympus IX70 inverted Fluorescence Microscope equipped with a 40x LUCPlanFl objective was used for the co-localization and fluorophore internalization studies of the rhodamine 6G loaded α1(IV)1263-1277 PA and non-targeted SUVs. The filters employed for the fluorescence images were WG, green filter for rhodamine, and WU, ultraviolet filter for DAPI. The BJ fibroblast, Hs895Sk fibroblast, and M14#5 human melanoma cells were harvested, and 5 × 105 cells were seeded in 60 mm plates, in complete medium. Cells were incubated overnight at 37 °C in a humidified atmosphere of 5% CO2 in air. The cells were then washed with adhesion media (3 mL), and fresh adhesion media containing 0.3 mL of 0.1 mg/mL 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI, a nucleus staining dye) (Pierce, Rockford, IL) was added followed by a 30 min incubation period, in the dark, at 37 °C in a humidified atmosphere of 5% CO2 in air. The DAPI containing medium was carefully removed and the cells were gently washed with adhesion media (3 × 3 mL). An additional aliquot of adhesion media (3 mL) was added to the cells at room temperature and then 20 μL (0.3 mg/mL) of either the targeted α1(IV)1263-1277 PA or non-targeted SUVs were added. Cells were monitored for fluorophore accumulation using fluorescence microscopy, at various time intervals, for a period of 35 min. Fluoresence microscopic images were captured with the Olympus IX70 inverted Fluorescence Microscope camera and intracellular rhodamine quantified with Quantity One® v.4.2.2 software. Control experiments were performed, using fluorescence and confocal fluorescence microscopy, to ascertain that the rhodamine quantified was intracellular (data not shown).
Competitive Displacement of α1(IV)1263-1277 PA SUVs Using Exogenous α1(IV)1263-1277 THP
Adhesion media containing the α1(IV)1263-1277 THP at a final concentration of 25, 50, or 100 μM was added to the cells and the cells were left at room temperature for 30 min, in the dark (to avoid bleaching of the DAPI nucleus dye). Cells were then treated with 20 μL (0.3 mg/mL) of either the targeted α1(IV)1263-1277 PA or non-targeted SUVs and the cellular accumulation of fluorophore was monitored using fluorescence microscopy, at various time intervals, for a period of 35 min.
Results
Construction and Characterization of Non-Targeted and Targeted Liposomes
We have previously constructed a triple-helical peptide-amphiphile (α1(IV)1263-1277 PA) specific for the CD44/CSPG cell surface receptor.24–26,43,58 To develop liposomes with targeting capabilities against highly metastatic melanoma cells, we have incorporated the α1(IV)1263-1277 PA within the liposomal bilayer of three lipid systems. The systems contained distearoyl phosphatidylglycerol (DSPG) and cholesterol, and either distearoyl phosphatidylcholine (DSPC), dipalmitoyl phosphatidylcholine (DPPC), or dilauroyl phosphatidylcholine (DLPC) as the major phospholipid component (Table 1). The choice of lipids and lipid ratios was based on prior studies that considered liposome clearance rates as a function of lipid phase transition temperatures and charge.10 Our aim was to evaluate the stability of these systems and therefore their appropriateness for future application against metastatic melanoma cells.
By convention the gel to liquid crystalline phase transition temperatures (Tm) of a particular liposome system is based on the lipid component with the highest Tm. However, for biological purposes, the overall stability of any liposome system must also consider the Tm values of all lipid components. Hence, we derived a net weighted average bio-transition temperature for the systems studied here by taking into account the Tm and relative abundance of all lipid components (Table 2).
Table 2.
Double phospholipid-cholesterol systems with transition temperatures for the least and most stable lipid components.70
| Lipid System | Ratio | Transition temperature for most stable lipid (°C) | Transition temperature for least stable lipid (°C) | Bio-transition temperatures for the overall liposome system (°C) |
|---|---|---|---|---|
| DSPG:DSPC:cholesterol | 1:4:5 | 57.5 | 55 | 55.5 |
| DSPG:DPPC:cholesterol | 1:4:5 | 57.5 | 41 | 44.3 |
| DSPG:DMPC:cholesterol | 1:4:5 | 57.5 | 23 | 29.9 |
| DSPG:DLPC:cholesterol | 1:4:5 | 57.5 | −1 | 10.7 |
The liposome systems were initially characterized using dynamic light scattering. All liposomes were within the same size range of 125 ± 20 nm (Table 1), allowing for valid stability comparisons between each system. This size range allows for efficacious liposomal drug delivery to tumors.10,62,63 The liposome sizes were found to be constant for at least 30 days at 4, 25, and 37 °C (Supporting Information). The phospholipid concentration of all the liposome systems was 0.3 mg/mL, as verified by the Stewart Assay.64
The alkyl chain length of the PA can be varied to control the PA triple-helical thermostability.25 The current study used a α1(IV)1263-1277 PA construct with a C16 tail. The C16 tail serves to maximize the potential hydrophobic interactions with the C18 phospholipid tails of DSPG and the C18, C16, or C12 phospholipid tails of DSPC, DPPC, or DLPC, respectively, in the lipid bilayer. Furthermore, PAs with longer tails will maintain their triple-helical structure at higher temperatures when compared to PAs with shorter tails. For example, a α1(IV)1263-1277 PA construct with a C16 tail will maintain its triple-helicity up to 69.8 °C, whereas a C4-containing PA will lose its triple-helical structure at ~35 °C.25 A higher PA thermostability is desirable as the extrusion temperature during liposomal preparation needs to be maintained above the lipid transition temperature of ~55 °C. Thus, PAs with Tm values greater than ~55 °C will maintain their triple-helical structure throughout liposomal preparation. In addition, methanol, which is present during the liposome preparation process, enhances the thermal stability of triple-helical peptides compared to aqueous environments.65,66 As previously demonstrated, if the Tm of a α1(IV)1263-1277 PA construct is higher than the lipid transition temperature of the lipids utilized in the liposomal preparation, the incorporation of the α1(IV)1263-1277 PA into liposomes does not affect the triple-helical content or stability of the PA.67,68
To confirm the incorporation of the α1(IV)1263-1277 PA within the lipid bilayer of the SUVs, targeted liposomes were treated with ethanol to liberate the α1(IV)1263-1277 PA from the lipid bilayer. MALDI-TOF mass spectral analysis of the resulting solution produced a peak corresponding to the mass of the α1(IV)1263-1277 PA ([M+H]+ = 3813.3 Da, theoretical [M+H]+ = 3813.3 Da). The incorporation efficiency for α1(IV)1263-1277 PA was 64%, based on the observed final concentration of 9 μM.
Stability Comparison of Liposomes Incorporating DSPC, DPPC, or DLPC Phospholipids
To determine the effect of the lipid tail length on overall liposomal stability, rhodamine-loaded liposomes composed of either DSPC, DPPC, or DLPC as the major constituent were monitored for leakage at 4, 25, or 37 °C for a period of 4 weeks (Figure 1a–c). Maximum fluorescence intensity was determined for each vesicle system by breaking the liposomes open with 100% ethanol. At 4 °C, the DSPC and DPPC liposome systems displayed similar stability profiles over the 4 week period, with ~25% fluorophore release after this time (Figure 1a). In contrast, at 25 °C the stability profiles for these two systems began to differ, with ~40% fluorophore escape observed for DSPC liposomes and ~70% fluorophore leakage observed for DPPC liposomes (Figure 1b). Furthermore, at 37 °C, the DSPC containing liposomes continued to be substantially more stable than the DPPC liposomes after 4 weeks, with ~45 and 90% fluorophore release, respectively (Figure 1c). The destabilization of the DPPC liposomes with increasing temperature is most likely due to the increasing movement of the DPPC phospholipid tails in the bilayer as the temperature starts to approach the DPPC transition temperature of 41 °C. This effect is minimal for the DSPC liposomes, which have a transition temperature of 55 °C.
Figure 1.

Temperature dependant stability comparison between liposomes containing either DSPC or DPPC phospholipids as the predominant bilayer components. Liposomes were stored at (a) 4 °C, (b) 25 °C, or (c) 37 °C. Fluorophore release was determined as described in Materials and Methods.
Liposomes prepared using DLPC (a lipid with a C12 tail) as the major phospholipid component proved to be the least stable of the three systems at 4 °C (data not shown). However, these liposomes became more stable with increasing temperature. Previous work has shown that liposomes containing mixtures of phospholipids varying in tail length by four or more carbon atoms exhibit monotectic behavior.69,70 Thus, in the current study the liposomes containing both the DLPC and DSPG phospholipids gave rise to two distinct liposomal populations depending on the storage temperature. At 4 °C only the DLPC liposomes are present, while at 25 and 37 °C DLPC:DSPG liposomes are present. Due to this complex behavior, further studies with DLPC-containing liposomes were not pursued.
Stability Comparison of Liposomes With and Without α1(IV)1263-1277 PA
To determine the effect that the α1(IV)1263-1277 PA has on liposomal stability, rhodamine loaded liposomes with either DSPC or DPPC as the major phospholipid component were prepared with and without the α1(IV)1263-1277 PA. Fluorescence intensity measurements for each vesicle sample at 4, 25, or 37 °C was then measured at selected time points over a 4 week period.
The presence of the α1(IV)1263-1277 PA did not serve to destabilize the liposomes used in this study (Figure 2). In fact, liposomes composed primarily of DPPC exhibited higher stability when the α1(IV)1263-1277 PA was introduced into the bilayer, particularly at physiological temperature (37 °C) (Figure 2d–f). The stabilizing effect of the α1(IV)1263-1277 PA on DSPC containing liposomes at 37 °C was not as pronounced (Figure 2a–c). This could be due to the fact that DSPC liposomes are more stable, even without the α1(IV)1263-1277 PA, than the DPPC liposomes. After 4 weeks, approximately 50% fluorophore release at 37 °C was observed with DSPC liposomes, either with or without the α1(IV)1263-1277 PA. In comparison, 90% fluorophore release was observed for the DPPC liposomes, which decreased to ~65% when the α1(IV)1263-1277 PA was incorporated into the lipid bilayer. The phase transition temperature of the DPPC liposomes is 41 °C; hence, at 37 °C the tail movement for this phospholipid system is quite significant (as discussed earlier), whereas the DSPC liposome system has a transition temperature of 55 °C and therefore minimal tail movement occurs at 37 °C. Thus, the presence of the α1(IV)1263-1277 PA in the lipid bilayer of the DPPC liposomes seems to retard the movement of the phospholipid tails, whereas it has far less of an impact on the DSPC liposomes at 37 °C. Due to the greater stability offered by the DSPC-containing liposomes compared with the DPPC-containing liposomes, all subsequent experiments were performed using only DSPC-based SUVs.
Figure 2.

Temperature dependant stability comparison between liposomes with and without the α1(IV)1263-1277 PA. Liposomes containing DSPC as the predominant bilayer component were stored at (a) 4 °C, (b) 25 °C, or (c) 37 °C. Liposomes containing DPPC as the predominant bilayer component were stored at (d) 4 °C, (e) 25 °C, or (f) 37 °C. Fluorophore release was determined as described in Materials and Methods.
Stability of Liposomes in the Presence of Cells or Preconditioned Cell Media
The stability of the α1(IV)1263-1277 PA versus non-targeted SUVs was examined by monitoring the percentage rhodamine release from each vesicle system in the presence of all three adhered cell lines or preconditioned cell media from each cell line. The goal was to monitor the release of rhodamine from each type of SUV in the biologically relevant extracellular environment, and thus establishing the stability of the SUVs in the presence of cells and prior to the delivery of their fluorescent cargo. We found that both the targeted and non-targeted SUVs showed negligible levels of leakage over a period of 60 min at room temperature (Figure 3) in the presence and absence of cells, under conditions that emulate those employed in the subsequent cellular fluorescence microscopy experiments. Thus, the vesicles used in our study are stable throughout the experimental time frame and longer.
Figure 3.

Stability comparison between DSPC-containing liposomes with and without the α1(IV)1263-1277 PA in the presence of cells and cell conditioned media at room temperature. α1(IV)1263-1277 PA liposomes were treated with (a) cells or (b) conditioned media. Under analogous conditions, non-targeted liposomes were treated with (c) cells or (d) conditioned media. Fluorophore release was determined as described in Materials and Methods.
CD44 Receptor Levels in Cultured Cells
ELISA studies were performed to determine the relative cell surface concentrations of the CD44 receptors for the M14#5, Hs895Sk, and BJ cell lines used in this study. M14#5 cells are known to express CD44 at relatively high levels, as we have assayed these cells for CD44 cell surface concentration in a previous study.58 In the current study, it was important to determine the relative levels of CD44 for the Hs895Sk and BJ fibroblasts compared with the M14#5 metastatic melanoma cells. The M14#5 cells had higher levels of CD44 than Hs895Sk, whereas the BJ cells had lower levels of CD44 than Hs895Sk (Figure 4). More precisely, BJ fibroblasts had ~60% of the CD44 content of M14#5 melanoma cells, while Hs895Sk had ~75% of the CD44 content.
Figure 4.
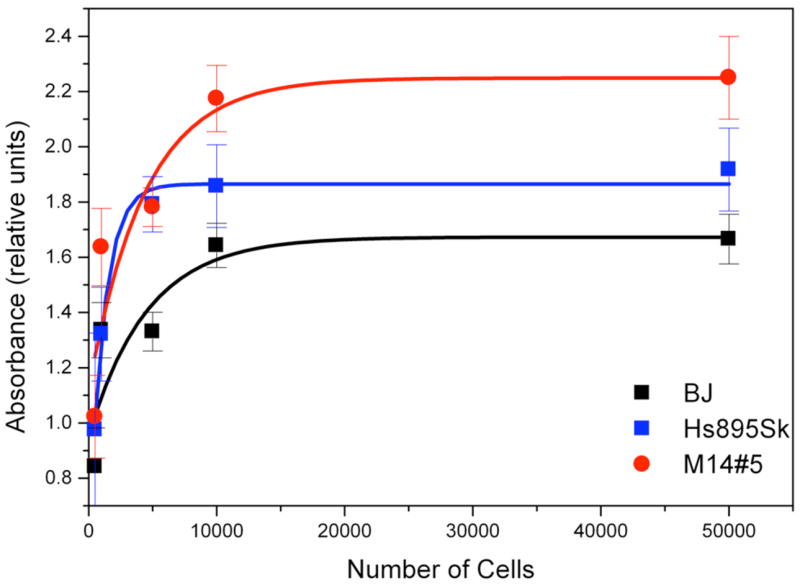
Whole cell ELISA of human metastatic melanoma cells (M14#5), normal fibroblasts from a metastatic melanoma patient (Hs895Sk), and normal foreskin fibroblasts (BJ) CD44 cell surface protein levels.
Analysis of the Cellular Fluorophore Accumulation with α1(IV)1263-1277 PA and Non-Targeted SUVs
The M14#5 metastatic melanoma, Hs895Sk fibroblast, and BJ fibroblast cell lines were treated with either the α1(IV)1263-1277 PA or non-targeted SUVs at room temperature, and monitored for rhodamine accumulation using fluorescence microscopy over a period of 35 min. The M14#5 cells treated with the α1(IV)1263-1277 PA SUVs internalized the rhodamine within 2 min in 100% of cells within the field of view (Figure 5, left hand panel). Continued (and) increasing fluorophore accumulation was observed over the 35 min experimental time period. In contrast, the Hs895Sk fibroblasts treated with α1(IV)1263-1277 PA SUVs internalized rhodamine only after 16 min, and to a much lesser degree than the M14#5 cells (Figure 5, middle panel). At the 35 min time point very small quantities of fluorophore were perceptible in ~90% of the Hs895Sk cells. The least amount of fluorophore accumulation was observed for the BJ fibroblasts, with trace amounts detected in ~90% of the cells after 35 min (Figure 5, right panel).
Figure 5.

Microscope images of cellular fluorophore accumulation based on α1(IV)1263-1277 PA liposome delivery. Delivery of rhodamine was monitored as a function of time for (left panel) human metastatic melanoma cells (M14#5), (middle panel) normal fibroblasts from a metastatic melanoma patient (Hs895Sk), and (right panel) normal foreskin fibroblasts (BJ). All liposomes utilized DSPC as the primary lipid (see Table 1).
Rhodamine incorporation was then quantified for all time points and cell lines. The highest level of rhodamine incorporation (M14#5 cells at 35 min) was designated as 100% relative incorporation (Figure 6). At early time points (2 and 6 min), M14#5 cells had ~20% of maximum rhodamine delivery, while less than 2% was seen for the other two cell lines. At the latest time point (35 min), Hs895Sk cells had 50% of the maximum rhodamine delivery, while BJ cells had ~2%. Thus, relative rhodamine incorporation was positively correlated to cell CD44 content. Regardless of the cell line, cells treated with non-targeted SUVs exhibited a similar lack of rhodamine accumulation over time (Figure 6), with the exception of the M14#5 cells which internalized trace amounts of the fluorophore in ~50% of the cells at the 35 min time point (image presented in Supporting Information).
Figure 6.
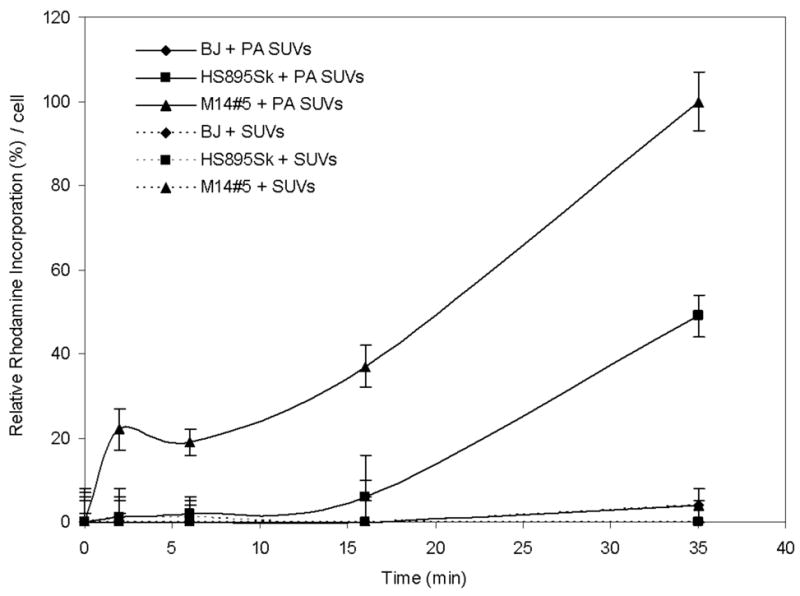
Incorporation of rhodamine as a function of time in human metastatic melanoma cells (M14#5), fibroblasts from a metastatic melanoma patient (Hs895Sk), and normal foreskin fibroblasts (BJ) following treatment with α1(IV)1263-1277 PA liposomes or non-targeted lipsomes. Relative quantification was performed as described in Materials and Methods. One hundred percent rhodamine incorporation was considered to be M14#5 melanoma cell treatment with PA liposomes for 35 min. All liposomes utilized DSPC as the primary lipid (see Table 1).
Competitive Displacement of α1(IV)1263-1277 PA SUVs Using Exogenous α1(IV)1263-1277 THP
The M14#5, Hs895Sk, and BJ cells were pretreated with increasing concentrations of exogenous α1(IV)1263-1277 THP (final concentrations of 0, 25, 50, or 100 μM) followed by incubation with α1(IV)1263-1277 PA SUVs. As described above, the M14#5 cells treated with α1(IV)1263-1277 PA SUVs, in the absence of the exogenous α1(IV)1263-1277 THP, exhibited extensive cellular fluorophore accumulation in ~90% of the cells by the 2 min time point, which dramatically increased to even more significant levels in 100% of the cells by the 35 min time point. Cellular fluorophore accumulation levels drop off proportionally at each time point when the M14#5 cells were pretreated with exogenous α1(IV)1263-1277 THP (25 μM) prior to the addition of the α1(IV)1263-1277 PA SUVs (images presented in Figure 7). The decrease in cellular internalization of rhodamine from the α1(IV)1263-1277 PA SUVs is even more pronounced with pretreatment concentrations of 50 and 100 μM exogenous α1(IV)1263-1277 THP, with the earliest fluorophore accumulation detected at 6 and 16 min, respectively (images presented in Figure 7). Overall, the M14#5 cellular fluorophore accumulation at the 35 min time point significantly decreases with increasing exogenous α1(IV)1263-1277 THP pretreatment concentrations (Figure 8). For example, the 35 min time point image of the M14#5 cells with α1(IV)1263-1277 PA SUVs and 100 μM exogenous α1(IV)1263-1277 THP is comparable in fluorophore intensity to the 2 min time point for the M14#5 cells with α1(IV)1263-1277 PA SUVs and no exogenous α1(IV)1263-1277 THP. The same overall trend was observed for the Hs895Sk and BJ fibroblasts (Figure 8), except that the fluorophore uptake decreases more dramatically, at earlier time points, and with lower pretreatment levels of the exogenous α1(IV)1263-1277 THP (images presented in Supporting Information). In fact, at an exogenous α1(IV)1263-1277 THP concentration of 100 μM, the BJ fibroblasts did not internalize any detectable rhodamine from the α1(IV)1263-1277 PA SUVs throughout the experimental time frame (Figure 8; images presented in Supporting Information), while the Hs895Sk cells internalized trace amounts in ~30% of the cells, only at the 35 min time point (image presented in Supporting Information).
Figure 7.

Microscope images of inhibition of rhodamine incorporation into human metastatic melanoma cells (M14#5). Cells were initially treated with 0 (far left panel), 25 (second from left panel), 50 (second from right panel), or 100 (far right panel) μM exogenous α1(IV)1263-1277 THP for 30 min, followed by treatment with α1(IV)1263-1277 PA liposomes. All liposomes utilized DSPC as the primary lipid (see Table 1).
Figure 8.
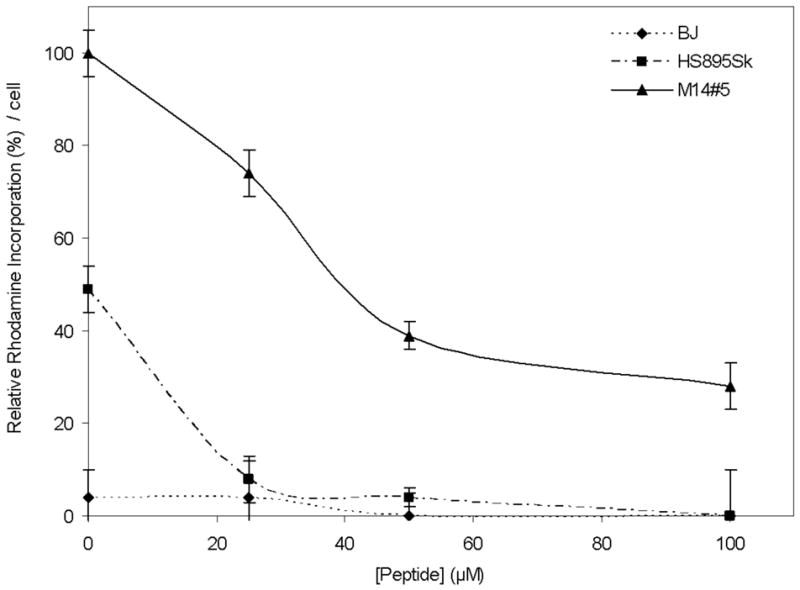
Inhibition of rhodamine incorporation into human metastatic melanoma cells (M14#5), fibroblasts from a metastatic melanoma patient (Hs895Sk), and normal foreskin fibroblasts (BJ). Cells were initially treated with 0, 25, 50, or 100 μM exogenous α1(IV)1263-1277 THP for 30 min, followed by treatment with α1(IV)1263-1277 PA liposomes for 35 min. Relative quantification was performed as described in Materials and Methods. One hundred percent rhodamine incorporation was considered to be M14#5 melanoma cell treatment with PA liposomes for 35 min. All liposomes utilized DSPC as the primary lipid (see Table 1).
Discussion
Due to a variety of innovations, liposomes have recently begun to realize their potential as drug delivery vehicles. Modification with poly(ethylene glycol) (PEG) or N-(2-hydroxypropyl)methacrylamide (HPMA) has improved liposome circulation time, achieved via decreased interaction with the reticuloendothelial system (RES).16–18,71–77 Although not targeted, PEG-stabilized liposomes are in clinical use for doxorubicin delivery to Kaposi’s sarcoma patients (DaunoXome) and ovarian carcinoma patients (Doxil).4,16,77 The potential of targeted nanoparticle delivery systems could further extend the applicability of liposomes.4,6,10,16,17 However, it is important to evaluate targeting in a variety of liposomal systems, so that efficiency of delivery can be correlated to relative liposome stability. It should also be noted that, due to the sometimes unpredictable cellular responses to specific lipids within a liposome,78 examining several liposomal compositions offers the best opportunity for developing drug delivery vehicles that minimize undesired side effects.
The present study has monitored stability of different liposomal systems, as well as quantifying the effect of a targeting moiety on these systems, as a function of temperature and time. Prior studies have typically focused on the stability of one liposomal system, and have usually not explicitly examined liposome stability over a defined time period, but rather stated “liposomes are stable up to one month.” Alternatively, the time period may have been quite short, or the vesicle system highly specialized.79–81 Furthermore, the present study has systematically accounted for an array of variables which affect overall liposomal stability, versus prior studies that examine one or two variables.
Liposomes containing DSPC phospholipid as the major bilayer component were found to be more stable than those containing DPPC or DLPC. This result was not unexpected, as DSPC has the highest transition temperature, resulting in minimal phospholipid tail movement in the bilayer relative to the DPPC- or DLPC-containing liposomes. Comparison of the present systems to prior results is somewhat difficult, given variations in liposome composition, cargos, and buffers. Nonetheless, the liposomal systems studied here exhibited comparable or better stabilities than other systems where stability has been examined in detail. For example, liposomes containing DSPC:dipalmitoyl phosphatidylglycerol (DPPG) (10:1) or DSPC:DPPG:cholesterol (10:1:10) exhibited virtually no carboxyfluorescein leakage at 4 °C after 40 days.82,83 In our case (Figure 1a), we did observe some leakage with DSPC:DSPG:cholesterol liposomes at 4 °C after 28 days, but our lipid/cholesterol distribution and fluorophore were different. The rates of doxorubicin leakage have been measured from DPPC:cholesterol (2:1) liposomes at 24 °C.84 The DPPC-containing liposomes had a leakage rate of 0.034%/min, while our DPPC-containing liposomes had a leakage rate of 0.002%/min at 25 °C (Figure 1b). The stability of dimyristoylphosphatidylcholine (DMPC):cholesterol:dihexadecyl phosphate (5:4:1) liposomes containing the hydrophobic dye tris(1,10-phenanthroline)ruthenium chloride [Ru(phen)3] was examined at 25 and 37 °C over 11 days.85 These liposomes appeared to be slightly less stable than the systems reported here, which is expected based on the Tm of 23 °C for DMPC.70
There are considerable data on the stability of liposomes incorporating egg-PC. However, liposomes containing egg-PC are typically less stable than those described here. Considering that egg-PC is a mixture of saturated and unsaturated acyl chains with a Tm ranging from −2.5 to −15 °C,10 greater stability is anticipated for the DPPC liposomes. For example, egg-PC:phosphatidylserine (PS) (10:1) liposomes showed complete loss of carboxyfluorescein at 4 °C after 27 days.82,83 Egg-PC:cholesterol (2:1) liposomes exhibited a doxorubicin leakage rate of 0.13%/min at 24 °C,84 considerably worse than DPPC-containing liposomes (see above). In similar fashion, analysis of carboxyfluorescein leakage from tissue-derived PC:cholesterol (1:1) liposomes at 37 °C over 140 h86 indicated less stability than the present liposomes. Stability of egg-PC liposomes can be enhanced by the presence of multiple components. Egg-PC:PS:cholesterol (10:1:4) liposomes exhibited a carboxyfluorescein leakage rate of ~0.00026%/min at 4 °C,82,83 better than the DPPC-containing liposome leakage rate of 0.00050%/min at 4 °C (Figure 1a).
Incorporation of 0.1 molar equivalents of the α1(IV)1263-1277 PA did not destabilize either of two liposomal systems examined here, as monitored by fluorophore leakage rates (Figures 2 and 3). Prior studies have examined the inclusion of lipids modified by proteins, peptides, or PEG into liposomes. In similar fashion to the present study, incorporation of a protein-lipid (up to 0.005 molar equivalents) into egg-PC liposomes did not result in increased leakage.87 Similarly, leakage from egg-PC:cholesterol (1:1) liposomes, examined in the presence or absence of 0.16 molar equivalents of PEG5000-phosphatidylethanolamine,71 was similar to that observed here. PEG2000-DSPE, at 5 molar %, also did not increase leakage from egg-PC:cholesterol (10:3) liposomes.88 However, in a case where a peptide-lipid (α-melanocyte stimulating hormone attached to a dialkyl C16 artificial lipid) was incorporated into egg-PC liposomes at 10 molar equivalents, substantial leakage was seen compared with egg-PC liposomes alone.89 More precisely, the liposomes exhibited 0.15%/min leakage, while the peptide-liposomes had 0.6%/min leakage. Some stability was restored by the addition of 30 molar equivalents of cholesterol. Overall, the incorporation of lipids modified with peptides, proteins, or PEG does not drastically alter liposomal stability at low (<0.2) molar equivalents.
The amphiphilic design of the α1(IV)1263-1277 PA construct facilitates the anchoring of the functional head group of the construct to the liposome surface by the insertion of the hydrophobic acyl tail into the lipid bilayer. This in turn allows ~50% (statistically) of the hydrophilic head group or targeting portion of the PA to protrude outward from the liposomal surface making it available to interact with the CD44 receptor. Our method of preparation of the α1(IV)1263-1277 PA targeted liposomes ensures incorporation of multiple copies of the PA into the liposomal bilayer, as verified by mass spectrometry and absorption spectroscopy. The presence of multiple copies of the PAs in the liposome potentially improves targeting selectivity for cells with a higher density of CD44 cell surface receptor. In addition, the triple-helical structure of the PA provides for enhanced target specificity and resistance to degradation from proteases while in circulation, as triple-helices are hydrolyzed efficiently by only a small subset of proteases.90
Our results demonstrate that, when cells are treated with α1(IV)1263-1277 PA SUVs, there is a direct correlation between the amount of rhodamine uptake and the CD44 cell surface receptor density. Conversely, treatment with the non-targeted SUVs resulted in minimal and similar rhodamine uptake in all three cell lines in this study, regardless of the CD44 receptor density. Thus, the M14#5 melanoma cells that contained the greatest CD44 receptor density exhibited the most rhodamine accumulation from the α1(IV)1263-1277 PA SUVs, and at the fastest rate. Accordingly, the Hs895Sk fibroblasts that contained the second highest CD44 receptor density internalized the targeted SUVs at a much slower rate and to a lesser extent than the M14#5 cells. The BJ fibroblasts, containing the least CD44, internalized the smallest amount of targeted SUVs at the slowest rate. However, while there is a strong qualitative correlation between CD44 content and rhodamine uptake, a quantitative correlation is absent. For example, the BJ fibroblasts contained 60% of the CD44 content compared with the M14#5 cells (Figure 4), but displayed considerably lower rhodamine accumulation at equivalent time points (Figure 6). This could be due to a combination of factors. First, since rhodamine delivery appears to be via a targeted, receptor based mechanism (Scheme 1d), fibroblasts may internalize ligand-bound CD44 slower than metastatic melanoma cells do. Ligands binding to CD44 undergo endocytosis,47,48 but correlation of rates to specific cell types has not been noted. Second, different modifications of CD44 may be present in fibroblasts compared with melanoma cells, which can affect ligand binding. We previously demonstrated that CD44 on M14#5 cells is predominantly CS modified, and CS modification modulates α1(IV)1263-1277 binding.58,91 Fibroblast CD44 is unmodified in the resting state, while cell induction by growth factors induces both CS and dermatan sulfate modification.92 Third, CD44 can be produced as a “standard” core protein of 37 kDa (CD44s or CD44H), or as isoforms with insertion domains (CD44v) that increase the core protein size to 80–250 kDa.46 Different isoforms have different potential glycosylation patterns, which in turn can affect ligand binding (see CS discussion above). Melanoma cells possess primarily the non-spliced, standard CD44s, while many “normal” cells possess spliced variants.93,94 Fourth, ligand binding to CD44 requires CD44 activation/dimerization for several normal cell types, while CD44 appears to be constitutively active in metastatic melanoma.55,95–97
To further demonstrate fluorophore delivery observations can be attributed to receptor/ligand interaction, competitive displacement experiments were carried out. From these experiments we have found that the α1(IV)1263-1277 PA SUVs binding to all cell lines, regardless of the levels of CD44, was inhibited in a dose dependent manner by exogenous α1(IV)1263-1277 THP. This competition for receptor binding was most pronounced for the M14#5 cell line that contains the highest CD44 receptor content. These results are consistent with a mechanism of internalization of rhodamine from the α1(IV)1263-1277 PA targeted SUVs that involves SUVs interacting with the cell surface CD44 receptors (Scheme 1d, path ii or iii). We have subsequently found these vehicles to be capable of targeted doxorubicin delivery, with lower doses of doxorubicin required for cytoxocity compared with non-targeted or free doxorubicin delivery.a
Other prior studies have established the use of lipids modified with cell adhesion ligands to create targeted liposomes,8,15 but these ligands do not offer the stability of the triple-helical α1(IV)1263-1277 PA, nor are they as straightforward to construct. The use of solid-phase methodology allows for the relatively easy assembly of a variety of PAs, which may differ by their fatty acid carbon chain length, level of saturation, and/or modification24,25,39 or head group sequences and/or structures.23,26,27,38,39 Insertion of the PA into liposomes is also simple, and a similar approach could be used elsewhere, such as creation of PA-targeted immunomicelles.98,99 The present results suggest that stable PA-targeted liposomes can be tailored to the unique characteristics of the drug load and different targeting moieties. Eventually, a systematic correlation of delivery efficiency to the unique physical properties of each liposome system can be performed, offering a “tunable” approach for targeted drug transport.
Furthermore, the PA-targeting moiety can also be catered to target different receptors, such as the α2β1 integrin which is upregulated in metastatic melanoma and for which the corresponding PA has also been previously constructed.39,100,101 Additional triple-helical PA constructs can be designed to target the α1β1, α2β1, α3β1, or α11β1 integrin, or the collagen-binding MSCRAMM from Staphylococcus aureus.102–112 Thus, the PA targeting ligand offers a uniquely adaptable yet stable and highly selective targeting system that can be tailored to various delivery vehicles, and is particularly applicable to liposomes.
Supplementary Material
Liposome sizes after 30 days at 4, 25, and 37 °C and microscopic images of non-targeted liposome delivery to cells and inhibition of delivery by exogenous α1(IV)1263-1277 THP. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge support of this work by the National Institutes of Health (CA 77402 and EB 000289 to GBF) and a BioFlorida “Legacy in Life Science” Scholarship (to EMR).
Footnotes
D.R. Khan, E.M. Rezler, and G.B. Fields, manuscript in preparation.
References
- 1.Backer MV, Gaynutdinov TI, Patel V, Jehning BT, Myshkin E, Backer JM. Bioconjugate Chem. 2004;15:1021–1029. doi: 10.1021/bc0499477. [DOI] [PubMed] [Google Scholar]
- 2.Chau Y, Tan FE, Langer R. Bioconjugate Chem. 2004;15:931–941. doi: 10.1021/bc0499174. [DOI] [PubMed] [Google Scholar]
- 3.Kline T, Torgov MY, Mendelsohn BA, Cerveny CG, Senter PD. Mol Pharm. 2004;1:9–22. doi: 10.1021/mp0340183. [DOI] [PubMed] [Google Scholar]
- 4.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 5.Willis RC. Mod Drug Discovery. 2004 July;:30–36. [Google Scholar]
- 6.Sahoo SK, Labhasetwar V. Drug Discov Today. 2003;8:1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- 7.Vine W, Gao K, Zegelman JL, Helsel SK. Drug Delivery Technol. 2006;6(5):34–39. [Google Scholar]
- 8.Hojo H, Kojima T, Yamauchi K, Kinoshita M. Tetrahedron Lett. 1996;37:7391–7394. [Google Scholar]
- 9.Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P, Benz CC, Papahadjopoulos D. Biochemistry. 1997;36:66–75. doi: 10.1021/bi962148u. [DOI] [PubMed] [Google Scholar]
- 10.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Pharmacol Rev. 1999;51:691–744. [PubMed] [Google Scholar]
- 11.Drummond DC, Hong K, Park JW, Benz CC, Kirpotin DB. Vitamin Horm. 2000;60:285–332. doi: 10.1016/s0083-6729(00)60022-5. [DOI] [PubMed] [Google Scholar]
- 12.Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A. Clin Cancer Res. 2000;6:1949–1957. [PubMed] [Google Scholar]
- 13.Eliaz RE, Szoka JFC. Cancer Res. 2001;61:2592–2601. [PubMed] [Google Scholar]
- 14.Backer MV, Aloise R, Przekop K, Stoletov K, Backer JM. Bioconj Chem. 2002;13:462–7. doi: 10.1021/bc0155770. [DOI] [PubMed] [Google Scholar]
- 15.Hallahan D, Geng L, Qu S, Scarfone C, Giorgio T, Donnelly E, Gao X, Clanton J. Cancer Cell. 2003;3:63–74. doi: 10.1016/s1535-6108(02)00238-6. [DOI] [PubMed] [Google Scholar]
- 16.Torchilin VP. Nature Rev Drug Disc. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 17.Gabizon AA, Shmeeda H, Zalipsky S. J Liposome Res. 2006;16:175–183. doi: 10.1080/08982100600848769. [DOI] [PubMed] [Google Scholar]
- 18.Duzgunes N, Nir S. Adv Drug Deliv Rev. 1999;40:3–18. doi: 10.1016/s0169-409x(99)00037-x. [DOI] [PubMed] [Google Scholar]
- 19.Murakami Y, Nakano A, Yoshimatsu A, Uchitomi K, Mastsuda Y. J Am Chem Soc. 1984;106:3613–3623. [Google Scholar]
- 20.Shimizu T, Mori M, Minamikawa H, Hato M. Chem Lett. 1989:1341–1344. [Google Scholar]
- 21.Shimizu T, Hato M. Biochim Biophys Acta. 1993;1147:50–52. doi: 10.1016/0005-2736(93)90315-q. [DOI] [PubMed] [Google Scholar]
- 22.Cha X, Ariga K, Kunitake T. Bull Chem Soc Jpn. 1996;69:163–167. [Google Scholar]
- 23.Berndt P, Fields GB, Tirrell M. J Am Chem Soc. 1995;117:9515–9522. [Google Scholar]
- 24.Yu YC, Berndt P, Tirrell M, Fields GB. J Am Chem Soc. 1996;118:12515–12520. [Google Scholar]
- 25.Yu YC, Tirrell M, Fields GB. J Am Chem Soc. 1998;120:9979–9987. [Google Scholar]
- 26.Yu YC, Roontga V, Daragan VA, Mayo KH, Tirrell M, Fields GB. Biochemistry. 1999;38:1659–1668. doi: 10.1021/bi982315l. [DOI] [PubMed] [Google Scholar]
- 27.Pakalns T, Haverstick KL, Fields GB, McCarthy JB, Mooradian DL, Tirrell M. Biomaterials. 1999;20:2265–2279. doi: 10.1016/s0142-9612(99)00157-x. [DOI] [PubMed] [Google Scholar]
- 28.Dillow AK, Ochsenhirt SE, McCarthy JB, Fields GB, Tirrell M. Biomaterials. 2001;22:1493–1505. doi: 10.1016/s0142-9612(00)00304-5. [DOI] [PubMed] [Google Scholar]
- 29.Rosler A, Klok HA, Hamlye IW, Castelletto V, Mykhaylyk OO. Biomacromol. 2003;4:859–863. doi: 10.1021/bm034058s. [DOI] [PubMed] [Google Scholar]
- 30.Vandermeulen GWM, Klok HA. Macromol Biosci. 2004;4:383–398. doi: 10.1002/mabi.200300079. [DOI] [PubMed] [Google Scholar]
- 31.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.García M, Alsina MA, Reig F, Haro I. Vaccine. 2000;18:276–283. doi: 10.1016/s0264-410x(99)00198-x. [DOI] [PubMed] [Google Scholar]
- 33.Gore T, Dori Y, Talmon Y, Tirrell M, Bianco-Peled H. Langmuir. 2001;17:5352–5360. [Google Scholar]
- 34.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;297:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 35.Hartgerink JD, Beniash E, Stupp SI. Proc Natl Acad Sci USA. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lauer-Fields JL, Minond D, Brew K, Fields GB. Methods Mol Biol. 2007;386:125–166. doi: 10.1007/978-1-59745-430-8_5. [DOI] [PubMed] [Google Scholar]
- 37.Fields GB, Lauer JL, Dori Y, Forns P, Yu YC, Tirrell M. Biopolymers. 1998;47:143–151. doi: 10.1002/(SICI)1097-0282(1998)47:2<143::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 38.Forns P, Lauer-Fields JL, Gao S, Fields GB. Biopolymers. 2000;54:531–546. doi: 10.1002/1097-0282(200012)54:7<531::AID-BIP60>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 39.Malkar NB, Lauer-Fields JL, Juska D, Fields GB. Biomacromolecules. 2003;4:518–528. doi: 10.1021/bm0256597. [DOI] [PubMed] [Google Scholar]
- 40.Lauer-Fields JL, Tuzinski KA, Shimokawa K, Nagase H, Fields GB. J Biol Chem. 2000;275:13282–13290. doi: 10.1074/jbc.275.18.13282. [DOI] [PubMed] [Google Scholar]
- 41.Lauer-Fields JL, Nagase H, Fields GB. J Chromatogr A. 2000;890:117–125. doi: 10.1016/s0021-9673(00)00396-4. [DOI] [PubMed] [Google Scholar]
- 42.Lauer-Fields JL, Broder T, Sritharan T, Nagase H, Fields GB. Biochemistry. 2001;40:5795–5803. doi: 10.1021/bi0101190. [DOI] [PubMed] [Google Scholar]
- 43.Malkar NB, Lauer-Fields JL, Borgia JA, Fields GB. Biochemistry. 2002;41:6054–6064. doi: 10.1021/bi012071w. [DOI] [PubMed] [Google Scholar]
- 44.Thomas L, Etoh T, Stamenkovic I, Mihm MC, Jr, Byers HR. J Invest Dermatol. 1993;100:115–20. doi: 10.1111/1523-1747.ep12462776. [DOI] [PubMed] [Google Scholar]
- 45.Goebeler M, Kaufmann D, Brocker EB, Klein CE. J Cell Sci. 1996;109:1957–64. doi: 10.1242/jcs.109.7.1957. [DOI] [PubMed] [Google Scholar]
- 46.Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. Crit Rev Clin Lab Sci. 2002;39:527–579. doi: 10.1080/10408360290795574. [DOI] [PubMed] [Google Scholar]
- 47.Tammi R, Rilla K, Pienimaki JP, MacCallum DK, Hogg M, Luukkonen M, Hascall VC, Tammi M. J Biol Chem. 2001;276:35111–35122. doi: 10.1074/jbc.M103481200. [DOI] [PubMed] [Google Scholar]
- 48.Jiang H, Peterson RS, Wang W, Bartnik E, Knudson CB, Knudson W. J Biol Chem. 2002;277:10531–10538. doi: 10.1074/jbc.M108654200. [DOI] [PubMed] [Google Scholar]
- 49.Eliaz RE, Nir S, Marty C, Szoka F., Jr Cancer Res. 2004;64:711–718. doi: 10.1158/0008-5472.can-03-0654. [DOI] [PubMed] [Google Scholar]
- 50.Peer D, Margalit R. Neoplasia. 2004;6:343–353. doi: 10.1593/neo.03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peer D, Margalit R. Int J Cancer. 2004;108:780–789. doi: 10.1002/ijc.11615. [DOI] [PubMed] [Google Scholar]
- 52.Akima K, Ito H, Iwata Y, Matsuo K, Watari N, Yanagi M, Hagi H, Oshima K, Yagita A, Atomi Y, Tatekawa I. J Drug Target. 1996;4:1–8. doi: 10.3109/10611869609046255. [DOI] [PubMed] [Google Scholar]
- 53.Lokeshwar VB, Selzer MG. J Biol Chem. 2000;275:27641–27649. doi: 10.1074/jbc.M003084200. [DOI] [PubMed] [Google Scholar]
- 54.Chen Q, Cai S, Shadrach KG, Prestwich GD, Hollyfield JG. J Biol Chem. 2004;279:23142–50. doi: 10.1074/jbc.M401584200. [DOI] [PubMed] [Google Scholar]
- 55.Naor D, Slonov RV, Ish-Shalom D. In: Adv Cancer Res. Vande Woude GF, Klein G, editors. Vol. 71. Academic Press; Orlando: 1997. pp. 241–319. [DOI] [PubMed] [Google Scholar]
- 56.Chelberg MK, McCarthy JB, Skubitz APN, Furcht LT, Tsilibary EC. J Cell Biol. 1990;111:261–270. doi: 10.1083/jcb.111.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fields CG, Mickelson DJ, Drake SL, McCarthy JB, Fields GB. J Biol Chem. 1993;268:14153–14160. [PubMed] [Google Scholar]
- 58.Lauer-Fields JL, Malkar NB, Richet G, Drauz K, Fields GB. J Biol Chem. 2003;278:14321–14330. doi: 10.1074/jbc.M212246200. [DOI] [PubMed] [Google Scholar]
- 59.Balsara NP, Stepanek P, Lodge TP, Tirrell M. Macromolecules. 1991;24:6227–6230. [Google Scholar]
- 60.Guenoun P, Delsanti M, Gazeau D, Mays JW, Cook DC, Tirrell M, Auvray L. Eur Phys J B. 1998;1:77–86. [Google Scholar]
- 61.Zuidam NJ, de Vrueh R, Crommelin DJA. In: Liposomes: A Practical Approach. 2. Torchilin VP, Weissig V, editors. Oxford University Press; 2003. pp. 31–78. [Google Scholar]
- 62.Nagayasu A, Uchiyama K, Kiwada H. Adv Drug Deliv Rev. 1999;40:75–87. doi: 10.1016/s0169-409x(99)00041-1. [DOI] [PubMed] [Google Scholar]
- 63.Charrois GJR, Allen TM. Biochim Biophys Acta. 2003;1609:102–108. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 64.Stewart JC. Anal Biochem. 1980;104:10–4. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 65.Engel J, Chen HT, Prockop DJ, Klump H. Biopolymers. 1977;16:601–622. doi: 10.1002/bip.1977.360160310. [DOI] [PubMed] [Google Scholar]
- 66.Horng JC, Kotch FW, Raines RT. Protein Sci. 2007;16:208–215. doi: 10.1110/ps.062560107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tu R, Mohanty K, Tirrell M. Am Pharm Rev. 2004;7(2):36–41. [Google Scholar]
- 68.Rezler EM, Khan DR, Tu R, Tirrell M, Fields GB. Methods Mol Biol. 2007;386:269–298. doi: 10.1007/978-1-59745-430-8_10. [DOI] [PubMed] [Google Scholar]
- 69.Mabrey S, Sturtevant JM. Proc Natl Acad Sci USA. 1976;73:3862–3866. doi: 10.1073/pnas.73.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor KMG, Craig DQM. In: Liposomes: A Practical Approach. 2. Torchilin VP, Weissig V, editors. Oxford University Press; 2003. pp. 79–103. [Google Scholar]
- 71.Klibanov AL, Maruyama K, Torchilin VP, Huang L. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 72.Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 73.Allen TM, Hansen C. Biochim Biophys Acta. 1991;1068:133–141. doi: 10.1016/0005-2736(91)90201-i. [DOI] [PubMed] [Google Scholar]
- 74.Maruyama K, Ishida O, Takizawa T, Moribe K. Adv Drug Deliv Rev. 1999;40:89–102. doi: 10.1016/s0169-409x(99)00042-3. [DOI] [PubMed] [Google Scholar]
- 75.Oku N. Adv Drug Deliv Rev. 1999;40:63–73. doi: 10.1016/s0169-409x(99)00040-x. [DOI] [PubMed] [Google Scholar]
- 76.Whiteman KR, Subr V, Ulbrich K, Torchilin VP. J Liposome Res. 2001;11:153–164. doi: 10.1081/LPR-100108459. [DOI] [PubMed] [Google Scholar]
- 77.Jamil J, Sheikh S, Ahmad I. Mod Drug Discovery. 2004;7(1):36–39. [Google Scholar]
- 78.Siegmann DW. Biochem Biophys Res Commun. 1987;145:228–233. doi: 10.1016/0006-291x(87)91310-6. [DOI] [PubMed] [Google Scholar]
- 79.Neumann R, Ringsdorf H, Patton EV, O’Brien DF. Biochim Biophys Acta. 1987;898:338–348. doi: 10.1016/0005-2736(87)90075-7. [DOI] [PubMed] [Google Scholar]
- 80.Domazou A, Luisi PL. J Liposome Res. 2002;12:205–220. doi: 10.1081/lpr-120014758. [DOI] [PubMed] [Google Scholar]
- 81.Yoshimoto M, Wang S, Fukunaga K, Walde P, Kuboi R, Nakao K. Biotechnol Bioeng. 2003;81:695–704. doi: 10.1002/bit.10514. [DOI] [PubMed] [Google Scholar]
- 82.Crommelin DJA, van Bommel EMG. Pharm Res. 1984;1:159–163. doi: 10.1023/A:1016344523988. [DOI] [PubMed] [Google Scholar]
- 83.Swarbrick J, Boylan JC. Encyclopedia of Pharmaceutical Technology: Liposomes As Pharmaceutical Dosage Forms to Microencapsulation. Marcel Dekker; 1994. [Google Scholar]
- 84.Haran G, Cohen R, Bar LK, Barenholz Y. Biochim Biophys Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 85.McNamara KP, Rosenzweig Z. Anal Chem. 1998;70:4853–4859. [Google Scholar]
- 86.Fonseca MJ, Alsina MA, Reig F. Biochim Biophys Acta. 1996;1279:259–263. doi: 10.1016/0005-2736(95)00265-0. [DOI] [PubMed] [Google Scholar]
- 87.Weissig V, Lasch J, Klibanov AL, Torchilin VP. FEBS Lett. 1986;202:86–90. doi: 10.1016/0014-5793(86)80654-8. [DOI] [PubMed] [Google Scholar]
- 88.Chou TH, Chen SC, Chu IM. J Biosci Bioeng. 2003;95:405–408. [PubMed] [Google Scholar]
- 89.Ogawa Y, Kawahara H, Yagi N, Kodaka M, Tomohiro T, Okada T, Konakahara T, Okuno H. Lipids. 1999;34:387–394. doi: 10.1007/s11745-999-0377-5. [DOI] [PubMed] [Google Scholar]
- 90.Lauer-Fields JL, Juska D, Fields GB. Biopolymers (Peptide Sci) 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]
- 91.Knutson JR, Iida J, Fields GB, McCarthy JB. Mol Biol Cell. 1996;7:383–396. doi: 10.1091/mbc.7.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Clark RAF, Lin F, Greiling D, An J, Couchman JR. J Invest Dermatol. 2004;122:266–277. doi: 10.1046/j.0022-202X.2004.22205.x. [DOI] [PubMed] [Google Scholar]
- 93.Lesley J, Hyman R, English N, Catterall JB, Turner GA. Glycoconjugate J. 1997;14:611–622. doi: 10.1023/a:1018540610858. [DOI] [PubMed] [Google Scholar]
- 94.Ranuncolo SM, Ladeda V, Gorostidy S, Morandi A, Varela M, Lastiri J, Loria D, del Aguila R, de Kier Joffé EB, Pallotta G, Puricelli L. Oncology Reports. 2002;9:51–56. [PubMed] [Google Scholar]
- 95.Kincade PW, Zheng Z, Katoh S, Hanson L. Curr Opin Cell Biol. 1997;9:635–642. doi: 10.1016/s0955-0674(97)80116-0. [DOI] [PubMed] [Google Scholar]
- 96.Liu D, Liu T, Li R, Sy MS. Frontiers Biosci. 1998;3:631–636. doi: 10.2741/a307. [DOI] [PubMed] [Google Scholar]
- 97.Lesley J, Hyman R. Frontiers Biosci. 1998;3:616–630. doi: 10.2741/a306. [DOI] [PubMed] [Google Scholar]
- 98.Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Proc Natl Acad Sci USA. 2003;100:6039–6044. doi: 10.1073/pnas.0931428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Torchilin VP. Drug Delivery Technol. 2006;4(2):30–39. [Google Scholar]
- 100.Baronas-Lowell D, Lauer-Fields JL, Fields GB. J Biol Chem. 2004;279:952–962. doi: 10.1074/jbc.M305989200. [DOI] [PubMed] [Google Scholar]
- 101.Baronas-Lowell D, Lauer-Fields JL, Borgia JA, Sferrazza GF, Al-Ghoul M, Minond D, Fields GB. J Biol Chem. 2004;279:43503–43513. doi: 10.1074/jbc.M405979200. [DOI] [PubMed] [Google Scholar]
- 102.Eble J, Golbik R, Mann K, Kuhn K. EMBO J. 1993;12:4795–4802. doi: 10.1002/j.1460-2075.1993.tb06168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Golbik R, Eble JA, Ries A, Kühn K. J Mol Biol. 2000;297:501–509. doi: 10.1006/jmbi.2000.3575. [DOI] [PubMed] [Google Scholar]
- 104.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. J Biol Chem. 2000;275:35–40. doi: 10.1074/jbc.275.1.35. [DOI] [PubMed] [Google Scholar]
- 105.Xu Y, Gurusiddappa S, Rich RL, Owens RT, Keene DR, Mayne R, Höök A, Höök M. J Biol Chem. 2000;275:38981–38989. doi: 10.1074/jbc.M007668200. [DOI] [PubMed] [Google Scholar]
- 106.Saccá B, Sinner EK, Kaiser J, Lübken C, Eble JA, Moroder L. ChemBioChem. 2002;9:904–907. doi: 10.1002/1439-7633(20020902)3:9<904::AID-CBIC904>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 107.Saccá B, Fiori S, Moroder L. Biochemistry. 2003;42:3429–3436. doi: 10.1021/bi0206762. [DOI] [PubMed] [Google Scholar]
- 108.Zhang WM, Käpylä J, Puranen JS, Knight CG, Tiger CF, Pentikäinen OT, Johnson MS, Farndale RW, Heino J, Gullberg D. J Biol Chem. 2003;278:7270–7277. doi: 10.1074/jbc.M210313200. [DOI] [PubMed] [Google Scholar]
- 109.Renner C, Saccá B, Moroder L. Biopolymers (Peptide Sci) 2004;76:34–47. doi: 10.1002/bip.10569. [DOI] [PubMed] [Google Scholar]
- 110.Kim JK, Xu Y, Xu X, Keene DR, Gurusiddappa S, Liang X, Wary KK, Hook M. J Biol Chem. 2005;280:32512–32520. doi: 10.1074/jbc.M502431200. [DOI] [PubMed] [Google Scholar]
- 111.Zong Y, Xu Y, Liang X, Keene DR, Hook A, Gurusiddappa S, Hook M, Narayana SVL. EMBO J. 2005;24:4224–4236. doi: 10.1038/sj.emboj.7600888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Raynal N, Hamaia SW, Siljander PRM, Maddox B, Peachey AR, Fernandez R, Foley LJ, Slatter DA, Jarvis GE, Farndale RW. J Biol Chem. 2006;281:3821–3831. doi: 10.1074/jbc.M509818200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Liposome sizes after 30 days at 4, 25, and 37 °C and microscopic images of non-targeted liposome delivery to cells and inhibition of delivery by exogenous α1(IV)1263-1277 THP. This material is available free of charge via the Internet at http://pubs.acs.org.