

Abstract
Over the last two decades, the rapid development of new synthetic routes for the preparation of expanded porphyrin macrocycles has allowed exploration of a new frontier consisting of “porphyrin-like” coordination chemistry. In this Account, we summarize our exploratory forays into the still relatively poorly explored area of oligopyrrolic macrocycle metalation chemistry. Specifically, we describe our successful formation of both mono- and binuclear complexes and in doing so highlight the diversity of coordination modes available to expanded porphyrin-type ligands. The nature of the inserted cation, the emerging role of tautomeric equilibria, and the importance of hydrogen-bonding interactions in regulating this chemistry are also discussed.
1. Introduction
Living systems routinely synthesize tetrapyrrolic macrocycles, either in their metal free forms (e.g., pheophorbide) or, more commonly, the corresponding metal complexes (e.g., chlorophyll, heme, coenzyme B12, etc.). The resulting species have been called “the pigments of life” because they perform a variety of fundamental biological functions that lie at the very core of life as we understand it.1
While the extraordinary chemistry of these tetrapyrrolic compounds continues to fascinate chemists, biologists and materials scientists, the last few decades have witnessed the development of a new class of ligands termed “expanded porphyrins”.2, 3 The defining feature of these oligopyrrolic macrocycles is a larger internal cavity as compared to those present in natural tetrapyrroles. More specifically, expanded porphyrins are macrocyclic compounds containing heterocyclic units (pyrrole, furan, or thiophene-like) linked together, either directly or through spacers, so that the internal ring pathway contains at least 17 atoms.
Over the past three decades, the chemistry of expanded porphyrins has brought about remarkable synthetic advances and provided new insights into the fundamental features of aromaticity.4 More recently, the scope of expanded porphyrin chemistry has grown to encompass the field of anion binding and transport.5 However, one of the main motivating forces behind the synthesis of expanded porphyrins is that they might extend the frontiers of “porphyrin-like” coordination chemistry.
To date, some of this latter promise has been realized. For instance, expanded porphyrins have been used to stabilize complexes containing typically large cations, including those of the lanthanide and actinide series. They have also allowed for the generation of complexes containing multiple cations. These synthetic findings, combined with a range of spectroscopic and physical properties, have prompted the study of expanded porphyrin metal complexes as photosensitizers in photodynamic therapy,6 contrast agents in magnetic resonance imaging,7 building blocks in nonlinear optical materials,8 and enzyme models in bioinorganic chemistry.9
While we have previously reviewed the stabilization of actinides in oligopyrrolic macrocycles,10 in this Account we wish to present our contribution to the coordination of transition metals in expanded porphyrins. With the intent to provide a critical overview of the reported metalation studies, we highlight the key factors influencing the metal coordination modes and the “degrees of freedom” of the macrocyclic cavities. As befits an article of this type, the focus is almost entirely on work from our laboratory. However, other contributions, particularly from the Chandrashekar,11 Osuka,12 and Furuta13 groups, have played a key role in developing this area of chemistry. For instance, work with N-confused and inverted porphyrins14 and expanded porphyrins13 has provided a novel mode of metal binding in porphyrin-like chemistry, namely coordination to the β-carbon atoms of the pyrrolic rings. These exciting developments are extending the range of expanded porphyrin coordination chemistry into the field of organometallic chemistry.
2. Carbon-bridged systems
The expanded porphyrins that display the greatest resemblance to natural porphyrins are those that contain either meso-like bridging carbon atoms or direct links between the heterocyclic subunits. According to the nomenclature put forward by Franck and Nonn,15 the name of these systems consists of three parts: 1) the number of π-electrons in the shortest conjugation pathway (in square brackets), 2) a core name indicating the number of pyrroles or other heterocycles (e.g., pentaphyrin, hexaphyrin), 3) the numbers of bridging carbon atoms between each pyrrole subunit (in round brackets and separated by dots). For instance, according to this nomenclature, porphyrin would be named as [18]tetraphyrin(1.1.1.1).
2.1 [22]Pentaphyrin(1.1.1.1.0) (sapphyrin)
The aromatic pentapyrrolic macrocycle 1 represents the first reported example of an expanded porphyrin and was discovered serendipitously by Woodward and coworkers in the early 1960s.16 Since this compound crystallizes as a dark blue solid, Woodward assigned it the name “sapphyrin” and thus began a trivial nomenclature for these macrocycles whose names end with the suffix “phyrin” or “rin” taken from porphyrin. Although limited to widely-recognized systems, this unorthodox nomenclature will generally be used in this Account.
The size of the cavity (ca. 5.5 Å vs. ca. 4.0 Å for porphyrins) and the presence of an additional nitrogen donor led Woodward and his group to envision the use of sapphyrins for metal coordination. Their early investigations showed that Co2+ and Zn2+ complexes could be isolated, but no structural information was obtained.16
When we reexamined this chemistry in the early 1990s, we proposed that the difficulty in the characterization of these complexes was due to the small size of the coordinating cations and thus to inherent kinetic instability. In fact, while our first metalation attempts using halides of larger cations (e.g., HgCl2, RhCl3, IrCl3) led to decomposition of the macrocycle, we were able to isolate the first fully characterized complexes of sapphyrin using rhodium and iridium dicarbonyl chloride salts in the presence of triethylamine (Scheme 1).17, 18
Scheme 1.
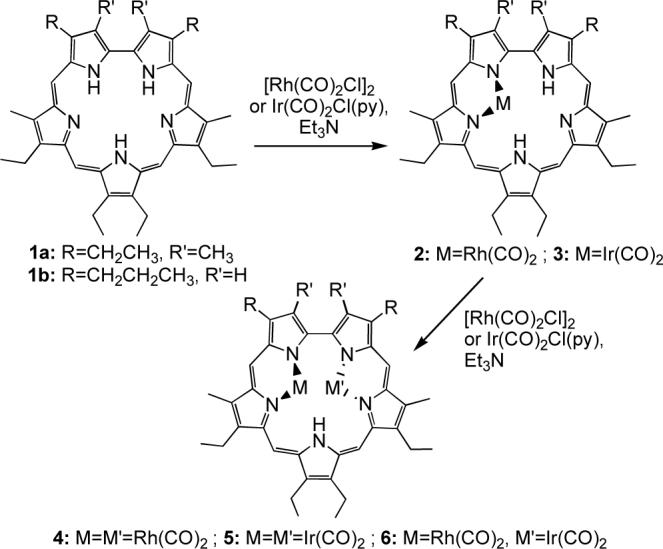
As shown in Figure 1,19 in the resulting rhodium(I) and iridium(I) complexes, the metal carbonyl units are found above or below the average macrocycle plane and each metal center is bound to a dipyrrinato moiety of the sapphyrin scaffold. This coordination mode is reminiscent of the one reported for [Rh(CO)2]2(octaethylporphyrin) and was later observed in several stable Rh(I) complexes of expanded porphyrins, such as our amethyrin,20 Chandrashekar's core-modified smaragdyrin,21 Furuta's N-confused pentaphyrin,13 and Osuka's N-fused pentaphyrin.12 Interestingly, the isolation of mono-Rh(CO)2 complex 2 and mono-Ir(CO)2 sapphyrin 3 allowed us to prepare the heterobimetallic species 6 by simple addition of Ir(CO)2(py)Cl or Rh2(CO)4Cl2, respectively (Scheme 1).
Figure 1.

Crystal structures of the mono-Ir(CO)2 complex 3 and the bis-Rh(CO)2 sapphyrin complex 4.
Analogous complexes of Rh and Ir with heterosapphyrins have been reported (Scheme 2), including the structurally characterized monothiasapphyrin-bis[dicarbonylrhodium(I)]22 8 and monoselenasapphyrin-bis[dicarbonyliridium(I)]23 9 (Figure 2).
Scheme 2.
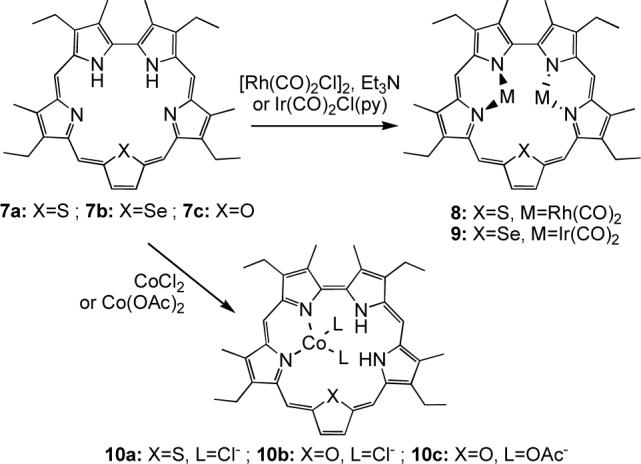
Figure 2.

Crystal structures of the bis-Ir(CO)2 selenasapphyrin complex 9 and the mono-CoCl2 thiasapphyrin complex 10a.
Our study of heterosapphyrins also led to the isolation of the mono-cobalt(II) complexes22 10a-c (Scheme 2). In contrast to what was seen in the case of Rh(I) and Ir(I), insertion of Co(II) cations was not accompanied by deprotonation of the ligand. Rather, a tautomeric rearrangement of the pyrrolic protons takes place. This allows the metal center to retain its counterions (Cl− or OAc−) and to coordinate to two iminic nitrogens on neighboring pyrrolic subunits.
To date, the metalation chemistry of sapphyrins has not displayed the diversity observed for other macrocycles (vide infra). However, the recent discovery of the biological activity of hydrophilic sapphyrins24 as potential cancer therapeutics is likely to fuel new studies of this most venerable expanded porphyrin.
2.2 [24]Hexaphyrin (1.0.0.1.0.0) (amethyrin)
Amethyrin 11, a 24 π-electron hexapyrrolic macrocycle, owes its name to the purple color (from Greek amethus) displayed by its protonated form in organic solution. This system is of considerable importance from an historic perspective since it was the first expanded porphyrin in which multiple modes of cation complexation was demonstrated and the first with which the in-plane coordination of two cations was established unequivocally.25 For instance, in an early metalation study it was found that reaction of the dichloride acid salt of amethyrin with ZnCl2 in the presence of Et3N afforded the bis-zinc complex 12 after purification by column chromatography (Scheme 3). X-ray diffraction analysis of the isolated product (Figure 3) showed that each zinc center coordinates to a dipyrrinato moiety and to two bridging counterions, namely a chloride and a hydroxyl anion (the latter presumably exchanged for chloride during chromatographic purification).
Scheme 3.
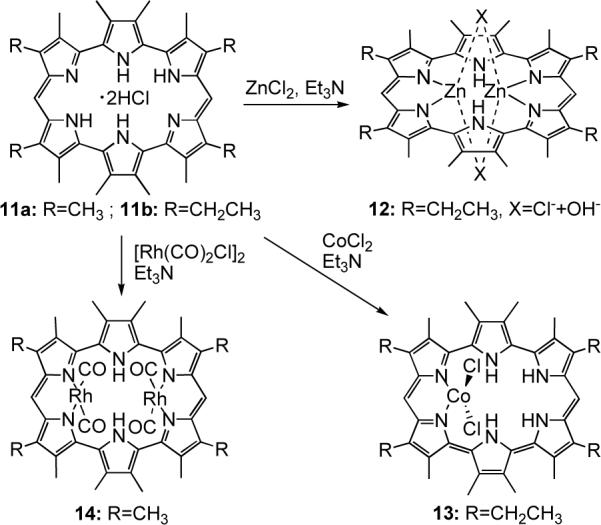
Figure 3.

Crystal structures of amethyrin complexes; from left to right: bis-Zn(II) complex 12, mono-Co(II) complex 13, bis-Rh(CO)2 complex 14, and bis-Cu(II) complex 15.
In a parallel metalation study, it was found that reaction of amethyrin with CoCl2 produced a complex wherein only a single metal cation is coordinated (13, Scheme 3). In analogy to what was observed for the cobalt complexes 10a-c of heterosapphyrins, the metal insertion proceeds with no deprotonation of the ligand and is accompanied by a tautomeric rearrangement that affects the conjugation pathway of the macrocycle. As a result, the Co(II) center appears to fill its coordination sphere with two iminic nitrogen atoms from neighboring pyrrolic units (Figure 3).
Amethyrin also forms a mono- and a bis-[Rh(CO)2] adduct (14, Scheme 3);20 however, in contrast to what is seen for the bis-[Rh(CO)2] complexes of octaethylporphyrin, sapphyrin, and various heterosapphyrins, in the case of amethyrin the two rhodium centers lie on the same side of the convex, bowl-shaped macrocycle. Presumably, this reflects the greater size of amethyrin relative to, for example, sapphyrin.
Tests carried out with other late transition metals confirmed that amethyrin can stabilize the formation of bimetallic nickel(II) and copper(II) complexes. A bis-Ni complex was obtained by treating the macrocycle free base with Ni(acetylacetonate)2; however, this complex was not structurally characterized and thus the coordination mode of the metal centers could not be deduced.26 Conversely, treatment of amethyrin with CuCl and subsequent exposure to air afforded a bis-Cu(II) complex in which the divalent oxidation state for the copper centers was confirmed by EPR and SQUID magnetometry. In the crystal structure of complex 15, the slightly bowl-shaped macrocycle hosts two copper centers held at the unusually short Cu···Cu distance of 2.761(1) Å. Each metal cation is bound to three nitrogen donors so that all of the pyrrolic nitrogen atoms are engaged in metal coordination (Figure 3).27 The required charge balance for the overall complex gave rise to two possible hypothesis regarding the conjugation pathway of the macrocycle: (i) metal insertion is accompanied by oxidation of the ligand from a 24 π-electron to a 22 π-electron system thereby giving rise to a hexadentate dianionic ligand; (ii) alternatively, the bis-[(μ-chloro)copper(II)](amethyrin) could be a dianionic species with two countercations, presumably protons, being present in the crystal structure (but not observed experimentally). While this issue is still not resolved, a comparison with what has been observed recently in the case of a bis-[Cu(II)(μ-Cl)] complex of amethyrin isomer 16 (see below; Scheme 3) leads us to favor the first of these hypotheses.
2.3 [24]Hexaphyrin(1.0.1.0.0.0) (isoamethyrin)
This quaterpyrrole-containing isomer of amethyrin (16, Scheme 4) is a 24 π-electron antiaromatic system, as judged by the strong downfield shifts of its inner NH protons (δ = 23.7, 23.9 and 24.2 in CD2Cl2 at room temperature).28 Treatment of this macrocycle with the actinide oxocations UO22+ and NpO2+ is accompanied by a dramatic color change and by oxidation of the ligand to a 22 π-electron aromatic system.28 As a consequence, isoamethyrin is currently being evaluated for its potential use as a colorimetric actinide sensor.29
Scheme 4.
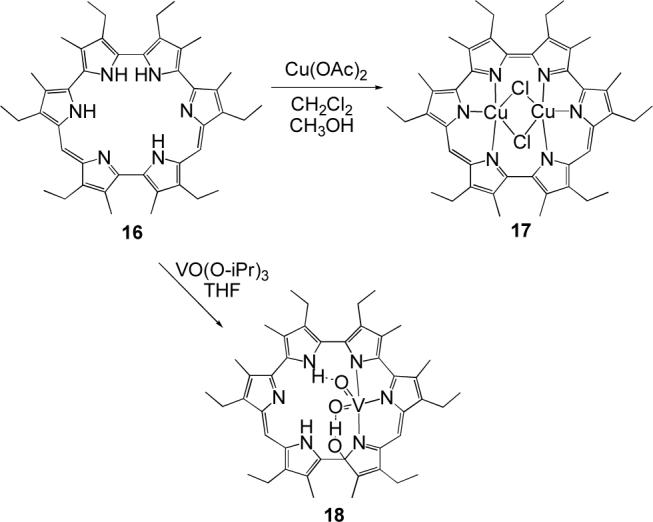
The ability of isoamethyrin to coordinate transition metals was recently examined. While exposure to most transition metal salts failed to provide evidence of complex formation,29 by using appropriate metal precursors two novel complexes could be obtained (Scheme 4).30 In particular, it was found that treatment of the free base 16 with copper(II) acetate afforded the corresponding bis-Cu(II) complex 17, which bears a remarkable resemblance to the amethyrin complex 15. In complex 17, each copper center coordinates to three pyrrolic nitrogen donors and two bridging chloride anions, and the two Cu atoms are separated by an unusually short distance (2.744(2) Å). As observed in the case of the actinyl cations, metal coordination is accompanied by ligand oxidation.
Interestingly, an analogue of complex 17 without the bridging chloride anions has not been isolated to date; rather, exposure of such putative species to any chlorinated solvent was found to give a chloro-bridged complex. However, the specific origin of the chloride anions in this complex is still under investigation.
Another structurally characterized transition metal complex of isoamethyrin, an oxovanadium(V) derivative, was prepared by addition of VO(Oi-Pr)3 to the free base macrocycle at 0°C in THF. The resulting product, 18, contains a single coordinated VO2+ cation that is ligated to three (only) of the potential six pyrrolic donor atoms. The cationic species is also hydrogen-bound to one of the pyrrolic NH protons through one of the oxo ligands (Figure 4). The result is a bimodal stabilization analogous to that seen in a different kind of oxovanadium expanded porphyrin complex (cf. Section 3.3). Separate from this, both the crystallographic and NMR spectroscopic data are consistent with the macrocyclic scaffold undergoing nucleophilic attack by an hydroxide anion at the α-carbon of a pyrrole ring during the course of metal insertion. This type of reactivity, while unusual, has been observed previously during the formation of our uranyl sapphyrin complex,31 and of Osuka's bis-Cu(II) complexes of meso-aryl-substituted hexaphyrins.32 In one extreme case reported by Osuka and coworkers, metal insertion was found to occur concurrent with hydrolytic cleavage of a pyrrole ring.33
Figure 4.

Crystal structures of the bis-Cu(II) isoamethyrin 17 and oxovanadium(V) isoamethyrin derivative 18.
3. Nitrogen-bridged systems
Imine-containing pyrrolic macrocycles, or Schiff-base expanded porphyrins,34 are systems wherein a meso carbon is formally substituted by a nitrogen atom. In many cases, the metal complexes of such systems have proved to be more stable than the metal-free forms. In other cases, the macrocycle is obtained most easily as the result of direct metal-templating procedures, as has been appreciated by workers in the area since the first seminal studies of Schiff-base pyrrolic macrocycles by Fenton and coworkers.35 As a consequence, the coordination chemistry of nitrogen-bridged expanded porphyrins has generally evolved in parallel with their synthetic development.
3.1 Texaphyrin
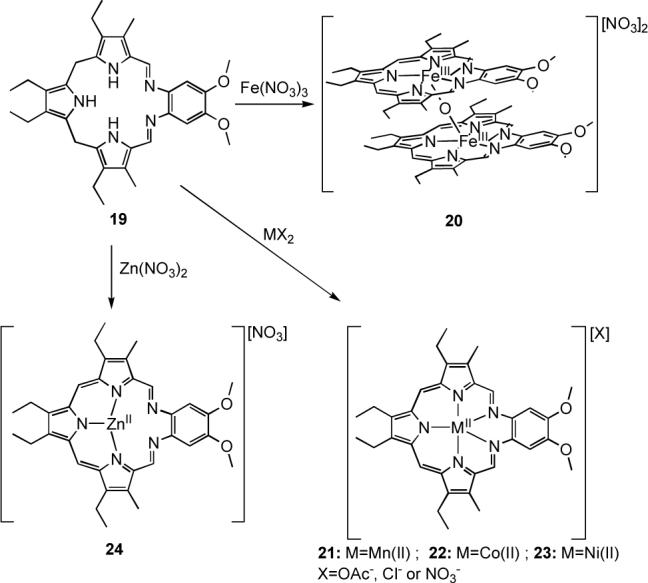
Arguably the best studied of Schiff-base oligopyrrolic macrocycles is texaphyrin (19, Scheme 5). This system represents the first expanded porphyrin for which a rich coordination chemistry was described. It thus played an important historical role in terms of showcasing some of the advantages of “expanding” the porphyrin core.
Scheme 5.
The lutetium(III) and gadolinium(III) complexes of a water-soluble texaphyrin have been the subject of clinical study as potential photosensitizers and radiation enhancers for arteriosclerotic disease and cancer, respectively. The chemistry involved in these medical application lies outside the scope of this Account and has been reviewed elsewhere.6
The pentaaza core of texaphyrin is 20% larger than that of porphyrin and offers a “lone-star” pentagonal array of nitrogen donors that provide a single negative charge when the ligand is deprotonated. In the early 1990s this cavity was found to be suitable for the coordination of all non-radioactive lanthanide(III) cations;36 however, it was not until 2002 that a series of first-row transition metal complexes (Mn(II), Fe(III), Co(II), Ni(II), and Zn(II)) of texaphyrin was reported.37 In the case of these latter cations, it was found that the metal insertion proceeds through a simultaneous oxidation/metalation analogous to the one previously observed in the preparation of lanthanide complexes. Specifically, the non-aromatic texaphyrin macrocycle 19, termed “sp3-texaphyrin” due to the hybridization state of the meso-like carbon bridges, undergoes oxidation to the aromatic form upon complexation of the metal cation (Scheme 5).
The Fe(III) complex 20 of texaphyrin was isolated as a μ-oxo dimer, a common product in iron porphyrin chemistry.38 Conversely, the corresponding manganese and cobalt texaphyrin complexes, 21 and 22, feature Mn(II) and Co(II) centers, respectively, even though these cations are typically stabilized in the +3 oxidation state in porphyrins. Presumably, the increased cavity size, as well as the decreased ligand charge, favors stabilization of the lower oxidation states, which are found in rather uncommon six- or seven-coordinate geometries (cf., e.g., the crystal structure of 21 in Figure 5).
Figure 5.

Crystal structures of texaphyrin complexes 21 and 24.
Finally, the solid-state structure of the Zn(II) complex 24 provided the first example of texaphyrin acting as a tridentate ligand (Figure 5). In fact, the relatively small Zn(II) cation is seen to coordinate only to the tripyrrolyldimethene fragment. This stands in contrast to what is observed in the case of the largest lanthanide cations (e.g., La(III)), where the metal centers are found to sit above the plane of the pentadentate macrocycle. These two kinds of complexes thus represent opposite ends of the texaphyrin coordination “spectrum”.
The availability of texaphyrin transition metal complexes provided an incentive to investigate their potential biomedical applications. In the context of this work, a water-soluble Mn(II) texaphyrin complex was prepared; it was found to act in vitro as a catalyst for the decomposition of peroxynitrite,39 a reactive oxygen species whose cytotoxic effects have been implicated in numerous disorders, including cancer, amyotrophic lateral sclerosis, and atherosclerosis.
3.2 Dipyrromethane-based macrocycles
The use of dipyrromethane derivatives for the synthesis of expanded porphyrins dates back to the mid-1980s, specifically to the seminal work of Mertes (now Bowman-James) and coworkers who reported the preparation of the so-called “accordion porphyrins”. These macrocycles, with two accordion-like aliphatic chains bridging two dipyrromethane moieties, were initially isolated as binuclear lead and copper complexes.40 Later, a bis-Mn(II) accordion porphyrin complex was found to act as a functional mimic for binuclear enzymes such as monooxygenases9 and catalases.41
More recently, we reported the preparation of the dipyrromethane-based macrocycle 25, which proved to be an efficient receptor for the chloride anion.42 However, the wide nitrogen-rich cavity of this ligand prompted us to investigate its potential as a binucleating cation receptor. While our first metalation attempts using simple iron salts (e.g., FeCl3, Fe(acetylacetonate)3, [Fe(CH3CN)6][AlCl4]2) failed to afford identifiable metal complexes, the organometallic Fe2Mes6 reagent proved to be an excellent iron source.43 This organometallic species is a convenient deprotonating agent and, further, does not introduce any anionic counterion into the reaction mixture. The reactions of 25 (both as the free base and as the dihydrochloride salt) with iron and copper mesityl reagents are summarized in Scheme 6; the structures of the resulting complexes are depicted in Figure 6.
Scheme 6.

Figure 6.

Crystal structures of the bis-Fe(III) complexes 26 and 27, bis-Cu(II) complex 28, and bis-Cu(I) complex 29.
The μ-oxo bis-Fe(III) complexes 26 and 27 were the first examples of iron complexes in which a Fe(III)-O-Fe(III) bridge is stabilized within a single macrocyclic framework. Interestingly, however, the structures of the complexes obtained during the metal insertion reaction depended on the protonation state of the starting macrocycle. In complex 26, obtained from the free base 25, the macrocycle behaves as a bis-tetradentate ligand. On the other hand, when the same reaction conditions are applied to the corresponding acid salt, 25·2HCl, the resulting complex 27 features a bis-tridentate ligand and retains two chloride anions.
The dramatic effect of the starting form of the ligand on the coordination modes within the macrocycle cavity was further established by a related study involving formation of copper complexes from 25.44 Reaction of Cu5Mes5 with 25·2HCl gave the bis-Cu(I) complex 29, which retains two chloride anions of the starting material and does not involve the pyrrolic nitrogen atoms in metal coordination. Conversely, treatment of the free base 25 with Cu5Mes5 at room temperature and subsequent air oxidation of the resulting putative Cu(I) species afforded the bimetallic Cu(II) complex 28, wherein the macrocycle behaves as a bis-tetradentate ligand for two copper centers constrained within what is essentially a square-planar geometry. The coordination mode of the copper atoms in this binuclear complex is reminiscent of the bis-Cu(II) accordion porphyrin reported by Mertes (vide supra)9 and of other copper complexes of oligopyrrolic Schiff-base macrocycles as described by Brooker.45
The versatility of this oligopyrrolic Schiff-base macrocycle was further highlighted by the work of Love and Arnold, who reported the preparation of a bis-Pd(II) complex46 and a mono-UO22+ adduct47 of 25, as well as an unusual metal-directed expansion of the macrocycle scaffold.48 Interestingly, the monouranyl complex could be used as a substrate for the further coordination of Mn(II), Fe(II), and Co(II) cations to afford three different heterobimetallic complexes.49
3.3 Bipyrrole-based macrocycles
The appearance of the bipyrrole subunit in a Schiff-base expanded porphyrin dates back to the early 1990s, namely to the synthesis of the tetrapyrrolic octaaza macrocycle 30. Our first investigations revealed that 30 binds methanol within its cavity, thus providing the first example of neutral substrate complexation by an expanded porphyrin.50
Early metalation studies revealed that macrocycle 30 reacts readily with simple metal salts to give binuclear Ni(II), Cu(II) and Zn(II) complexes (Scheme 7). The crystal structure of the bis-Ni(II) complex 31 showed that the two metal centers coordinate to the macrocycle via the iminic nitrogen atoms, whereas the pyrrolic NH protons are engaged in hydrogen-bonding interactions with the acetate counterions (Figure 7).51
Scheme 7.
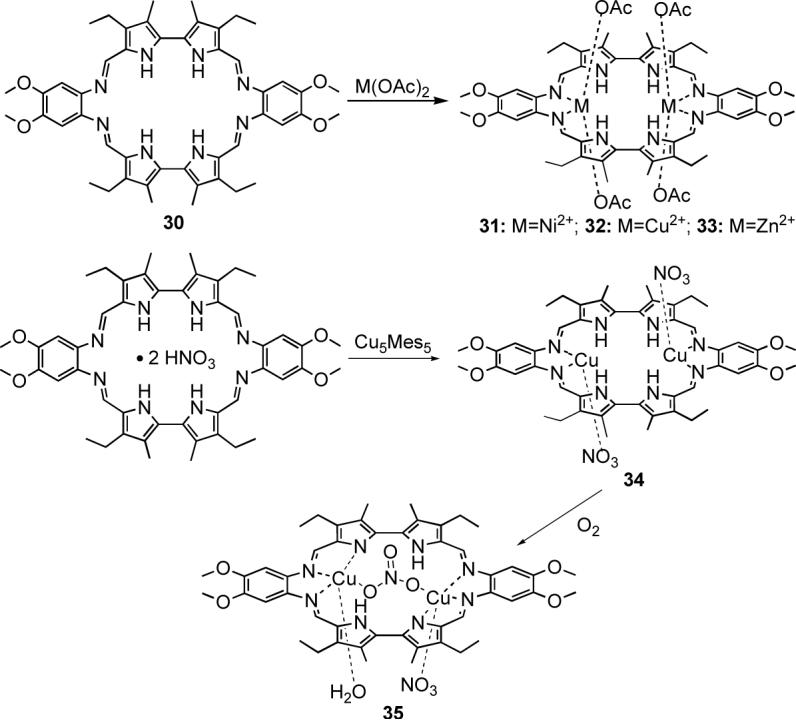
Figure 7.

Crystal structures of complexes 31, 34, 35, and 36 (side view) derived from macrocycle 30. The methoxy groups on the phenyl rings and the alkyl substituents on the pyrrole rings have been removed for clarity.
A recent reexamination of this chemistry indicated that 30 can also be used for the coordination of monovalent cations, such as Cu(I)51 and Ag(I).52 As previously observed for the bis-Ni(II) complex 31, the X-ray crystal structures of the copper(I) and silver(I) complexes, 34 and 36, respectively, revealed that the monovalent metal cations coordinate to 30 through the iminic nitrogens and not the pyrrolic nitrogen atoms.
A new Cu(II) complex (35, Scheme 7) was obtained through air-oxidation of the bis-Cu(I) complex 34.51 An X-ray structural analysis (Figure 7) revealed that the macrocycle adjusts to the increased metal charges through a dramatic conformational change that allows the coordination of an anionic pyrrolic nitrogen donor to each metal center. Presumably, this rearrangement reflects the fact that in the absence of other available anionic counterions, the coordination mode is driven by the change in the oxidation state of the metal centers.
More recent studies of the coordination chemistry of 30 revealed that the complexation of silver cations is subject to strong cooperative behavior.52 This latter conclusion was supported by a combination of NMR and UV-vis spectroscopic experiments, which served to demonstrate that coordination of a first silver(I) cation activates the macrocycle strongly towards the complexation of a second silver ion. Such positive homotropic allosterism was unprecedented in expanded porphyrin chemistry and hence receptor 30 could be the first example of a new (and potentially large) class of hosts for cooperative metal coordination.
The bipyrrolic subunit is also featured in the smaller Schiff-base macrocycle 37, which was shown to bind the uranyl and neptunyl cations.53 We recently found that this system acts as a receptor for a lighter congener of the actinide oxocations, namely the VO2+ cation.54 Interestingly, NMR spectroscopic data and X-ray diffraction analysis of the oxovanadium(V) adduct 38 revealed that the metal ion does not react with the free base macrocycle 37 but rather with enaminic tautomer 37’ (Scheme 8). As a consequence of this enamine-imine tautomeric rearrangement, the nonspherical cation VO2+ is accommodated through metal-ligand interactions involving three pyrrolic nitrogens, as well as via hydrogen-bonding interactions involving a pyrrolic NH and an enaminic NH (Figure 8). The coordination of the VO2+ cation by 37’ is important not only because it provides the first structurally characterized example of early transition metal coordination in an expanded porphyrin, it also represents an interesting case of bimodal (i.e., covalent and noncovalent) recognition of a nonspherical guest within an oligopyrrolic receptor.
Scheme 8.
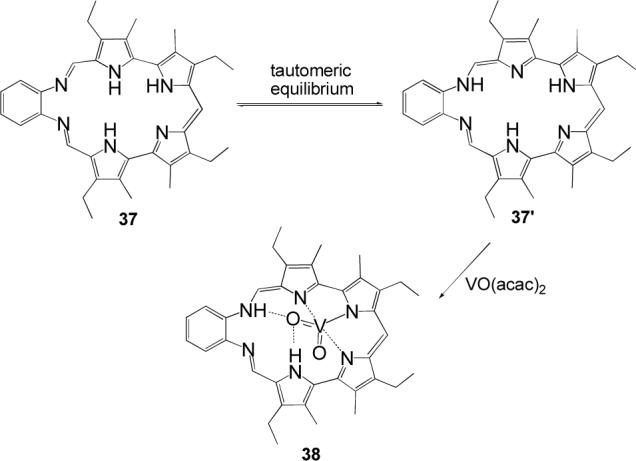
Figure 8.
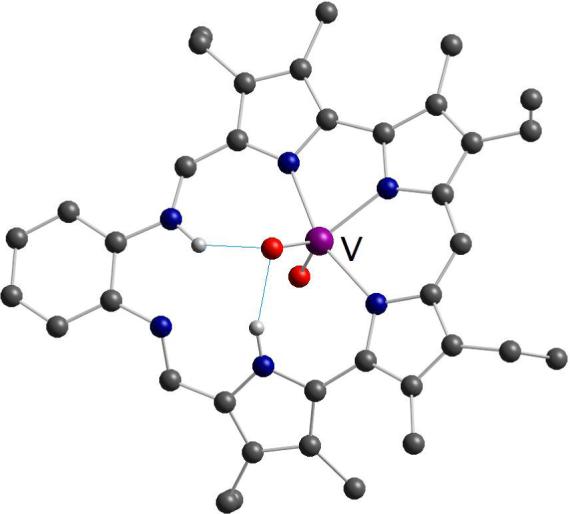
Crystal structure of the oxovanadium(V) complex 38 showing two hydrogen-bonding interactions with one of the oxo ligands.
3.4 Other systems
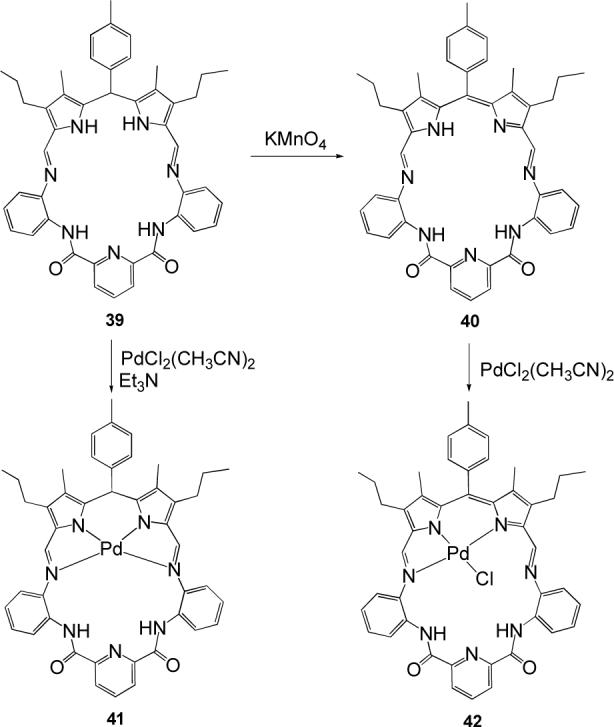
A dipyrromethane subunit, combined with a diamidopyridine fragment, is featured in the Schiff-base hybrid macrocycle 39. This ligand, which proved to be an effective anion binding receptor,55 can be selectively oxidized to the conjugated diiminodipyrromethene system 40 (Scheme 9). A first metalation study of this macrocycle has shown that the Pd(II) cation binds preferentially to the pyrrolic moiety, both in the dipyrromethane system 41 and in the oxidized dipyrromethene system 42.56
Scheme 9.
Concluding remarks
A major thrust of modern coordination chemistry is the engineering of nano-scaled spaces for the stabilization, activation and/or recognition of metal ions. In the expanded porphyrin field, this has inspired the preparation of a number of novel macrocyclic receptors and the subsequent exploration of their cavities as novel coordination environments. In the case of expanded porphyrins, initial explorations have begun to reveal what promises to be a very diverse coordination chemistry have been revealed. For instance, the metalation chemistry of texaphyrin was found to run the gamut from the large lanthanide cations to the small zinc(II) ion. Moreover, the cobalt complexes of sapphyrin and amethyrin, as well as the vanadium complex of the Schiff-base macrocycle 37, have served to showcase the role of tautomeric equilibria in the metalation of conjugated expanded porphyrin. Studies of congeners 25 and 30 have illustrated the importance of macrocycle protonation and counterion effects in terms of defining the final structure of a given metal-containing complex. Finally, the role played by hydrogen-bonding interactions in the cooperative binding of Ag(I) cations in 30 and in the bimodal recognition of the nonspherical VO2+ cation in the Schiff-base system 38 and isoamethyrin 18 reveal a supramolecular aspect to the coordination chemistry of oligopyrrolic macrocycles that could provide a starting point for future investigations.
Given the large number of expanded porphyrin systems currently being reported in the literature, we expect that this area of research will grow rapidly in the near future. It has the potential to produce metal complexes with properties very different from those stabilized in more-common tetrapyrrolic macrocycles, including porphyrins. The chemistry of bimetallic expanded porphyrin complexes is particularly promising, as it will likely provide new opportunities to investigate magnetic interactions and to engineer binuclear catalytic centers wherein two metal ions act in a concerted way on a given reaction substrate. In the event, expanded porphyrin complexes have already demonstrated potential utility in fields as diverse as drug discovery, bio-inspired catalysis, optical data storage, and nuclear waste remediation and it is likely that this will continue to be the case.
Acknowledgments
The work reviewed in this Account was supported by grants to J.L.S. from the National Science Foundation, the National Institute of Health and the U.S. Department of Energy. E.T. thanks the Robert A. Welch Foundation for a Professional Development Award.
The authors are grateful to a number of talented graduate students, postdoctoral fellows and collaborators whose names are cited in the references and whose considerable efforts have made expanded porphyrin coordination chemistry a reality.
Biographical Information
Jonathan L. Sessler received his B.S. degree in 1977 from the University of California at Berkeley. In 1982, he earned a Ph.D. from Stanford University while working with Prof. J. P. Collman. After postdoctoral work with Profs. J.-M. Lehn in Strasbourg, France, and I. Tabushi in Kyoto, Japan, he joined the faculty at The University of Texas at Austin, where he is now the Roland K. Pettit Centennial Professor of Chemistry.
Elisa Tomat received her B.S. degree in 2001 from the University of Trieste, Italy. In 2002, she joined the research group of Prof. J. L. Sessler at the University of Texas at Austin, where she is currently completing her Ph.D. degree. Following this, she plans to carry out postdoctoral work with Prof. S. J. Lippard at MIT.
References
- 1.Battersby AR, Fookes CJR, Matcham GWJ, McDonald E. Biosynthesis of the Pigments of Life - Formation of the Macrocycle. Nature. 1980;285:17–21. doi: 10.1038/285017a0. [DOI] [PubMed] [Google Scholar]
- 2.Jasat A, Dolphin D. Expanded Porphyrins and Their Heterologs. Chem. Rev. 1997;97:2267–2340. doi: 10.1021/cr950078b. [DOI] [PubMed] [Google Scholar]
- 3.Sessler JL, Weghorn SJ. Expanded, Contracted and Isomeric Porphyrins. Pergamon; New York: 1997. [Google Scholar]
- 4.Sessler JL, Seidel D. Synthetic expanded porphyrin chemistry. Angew. Chem., Int. Ed. 2003;42:5134–5175. doi: 10.1002/anie.200200561. [DOI] [PubMed] [Google Scholar]
- 5.Sessler JL, Gale PA, Cho WS. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 6.Sessler JL, Miller RA. Texaphyrins - New drugs with diverse clinical applications in radiation and photodynamic therapy. Biochem. Pharmacol. 2000;59:733–739. doi: 10.1016/s0006-2952(99)00314-7. [DOI] [PubMed] [Google Scholar]
- 7.Young SW, Qing F, Harriman A, Sessler JL, Dow WC, Mody TD, Hemmi GW, Hao YP, Miller RA. Gadolinium(III) texaphyrin: A tumor selective radiation sensitizer that is detectable by MRI. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6610–6615. doi: 10.1073/pnas.93.13.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rath H, Sankar J, PrabhuRaja V, Chandrashekar TK, Nag A, Goswami D. Core-modified expanded porphyrins with large third-order nonlinear optical response. J. Am. Chem. Soc. 2005;127:11608–11609. doi: 10.1021/ja0537575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiter WA, Gerges A, Lee S, Deffo T, Clifford T, Danby A, Bowman-James K. Accordion porphyrins - Hybrid models for heme and binuclear monooxygenases. Coord. Chem. Rev. 1998;174:343–359. [Google Scholar]
- 10.Sessler JL, Vivian AE, Seidel D, Burrell AK, Hoehner M, Mody TD, Gebauer A, Weghorn SJ, Lynch V. Actinide expanded porphyrin complexes. Coord. Chem. Rev. 2001;216:411–434. [Google Scholar]
- 11.Chandrashekar TK, Venkatraman S. Core-modified expanded porphyrins: New generation organic materials. Acc. Chem. Res. 2003;36:676–691. doi: 10.1021/ar020284n. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu S, Osuka A. Metalation chemistry of meso-aryl-substituted expanded porphyrins. Eur. J. Inorg. Chem. 2006:1319–1335. [Google Scholar]
- 13.Srinivasan A, Furuta H. Confusion approach to porphyrinoid chemistry. Acc. Chem. Res. 2005;38:10–20. doi: 10.1021/ar0302686. [DOI] [PubMed] [Google Scholar]
- 14.Chmielewski PJ, Latos-Grazynski L. Core-modified porphyrins - a macrocyclic platform for organometallic chemistry. Coord. Chem. Rev. 2005;249:2510–2533. [Google Scholar]
- 15.Franck B, Nonn A. Novel Porphyrinoids for Chemistry and Medicine by Biomimetic Syntheses. Angew. Chem., Int. Ed. 1995;34:1795–1811. [Google Scholar]
- 16.Bauer VJ, Clive DLJ, Dolphin D, Paine JB, Harris FL, King MM, Loder J, Wang SWC, Woodward RB. Sapphyrins - Novel Aromatic Pentapyrrolic Macrocycles. J. Am. Chem. Soc. 1983;105:6429–6436. [Google Scholar]
- 17.Sessler JL, Cyr MJ, Burrell AK. Sapphyrins - New Life for an Old Expanded Porphyrin. Synlett. 1991:127–134. [Google Scholar]
- 18.Burrell AK, Sessler JL, Cyr MJ, McGhee E, Ibers JA. Metal-Carbonyl-Complexes of Sapphyrins. Angew. Chem., Int. Ed. 1991;30:91–93. [Google Scholar]
- 19.In all the crystal structures depicted in this Account, the hydrogens bound to carbon atoms have been removed for clarity.
- 20.Sessler JL, Gebauer A, Guba A, Scherer M, Lynch V. Synthesis and X-ray crystallography of Rh(I) carbonyl complexes of amethyrin. Inorg. Chem. 1998;37:2073–2076. [Google Scholar]
- 21.Sridevi B, Narayanan SJ, Rao R, Chandrashekar TK, Englich U, Ruhlandt-Senge K. meso-aryl smaragdyrins: Novel anion and metal receptors. Inorg. Chem. 2000;39:3669–3677. doi: 10.1021/ic000031v. [DOI] [PubMed] [Google Scholar]
- 22.Sessler JL, Burrell AK, Lisowski J, Gebauer A, Cyr MJ, Lynch V. Cobalt(II) and rhodium(I) metal complexes of thia- and oxasapphyrins. Bull. Soc. Chim. Fr. 1996;133:725–734. [Google Scholar]
- 23.Lisowski J, Sessler JL, Lynch V. Synthesis and X-Ray Structure of Selenasapphyrin. Inorg. Chem. 1995;34:3567–3572. [Google Scholar]
- 24.Magda D, Lecane P, Wang Z, Cortez C, Thiemann P, Ma X, Boswell G, Hemmi G, Naumovski L, Miller RA, Cho DG, Sessler J, Karaman MW, Hacia JG. In vitro and in vivo antitumor activity of hydrophilic sapphyrins, a new class of tumor selective drugs. Clin. Cancer Res. 2005;11:9156S–9156S. [Google Scholar]
- 25.Sessler JL, Weghorn SJ, Hiseada Y, Lynch V. Hexaalkyl Terpyrrole - a New Building-Block for the Preparation of Expanded Porphyrins. Chem.-Eur. J. 1995;1:56–67. [Google Scholar]
- 26.Hannah S, Seidel D, Sessler JL, Lynch V. New chemistry of amethyrin. Inorg. Chim. Acta. 2001;317:211–217. [Google Scholar]
- 27.Weghorn SJ, Sessler JL, Lynch V, Baumann TF, Sibert JW. Bis[(μ-chloro)copper(II)]amethyrin: A bimetallic copper(II) complex of an expanded porphyrin. Inorg. Chem. 1996;35:1089–1090. doi: 10.1021/ic9509692. [DOI] [PubMed] [Google Scholar]
- 28.Sessler JL, Seidel D, Vivian AE, Lynch V, Scott BL, Keogh DW. Hexaphyrin(1.0.1.0.0.0): An expanded porphyrin ligand for the actinide cations uranyl (UO22+) and neptunyl (NpO2+). Angew. Chem., Int. Ed. 2001;40:591–594. doi: 10.1002/1521-3773(20010202)40:3<591::AID-ANIE591>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Sessler JL, Melfi PJ, Seidel D, Gorden AEV, Ford DK, Palmer PD, Tait CD. Hexaphyrin(1.0.1.0.0.0). A new colorimetric actinide sensor. Tetrahedron. 2004;60:11089–11097. [Google Scholar]
- 30.Sessler JL, Melfi PJ, Tomat E, Lynch VM. Copper(II) and oxovanadium(V) complexes of hexaphyrin(1.0.1.0.0.0). Dalton Trans. 2007 doi: 10.1039/b617620h. DOI: 10.1039/b617620h. [DOI] [PubMed] [Google Scholar]
- 31.Burrell AK, Cyr MJ, Lynch V, Sessler JL. Nucleophilic-Attack at the Meso Position of a Uranyl Sapphyrin Complex. J. Chem. Soc., Chem. Comm. 1991:1710–1713. [Google Scholar]
- 32.Shimizu S, Anand VG, Taniguchi R, Furukawa K, Kato T, Yokoyama T, Osuka A. Biscopper complexes of meso-aryl-substituted hexaphyrin: Gable structures and varying antiferromagnetic coupling. J. Am. Chem. Soc. 2004;126:12280–12281. doi: 10.1021/ja046102x. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu S, Tanaka Y, Youfu K, Osuka A. Dicopper and disilver complexes of octaphyrin(1.1.1.1.1.1.1.1): Reversible hydrolytic cleavage of the pyrrolic ring to a keto-imine. Angew. Chem., Int. Ed. 2005;44:3726–3729. doi: 10.1002/anie.200500676. [DOI] [PubMed] [Google Scholar]
- 34.Callaway WB, Veauthier JM, Sessler JL. Schiff-base porphyrin and expanded porphyrin analogs. J. Porphyr. Phthalocy. 2004;8:1–25. [Google Scholar]
- 35.Adams H, Bailey NA, Fenton DE, Moss S, Jones G. Monobinuclear and Homobinuclear Copper(II) Complexes of Pyrrole-Containing Schiff-Base Macrocycles. Inorg. Chim. Acta. 1984;83:L79–L80. [Google Scholar]
- 36.Sessler JL, Mody TD, Hemmi GW, Lynch V. Synthesis and Structural Characterization of Lanthanide(III) Texaphyrins. Inorg. Chem. 1993;32:3175–3187. [Google Scholar]
- 37.Hannah S, Lynch V, Guldi DM, Gerasimchuk N, MacDonald CLB, Magda D, Sessler JL. Late first-row transition-metal complexes of texaphyrin. J. Am. Chem. Soc. 2002;124:8416–8427. doi: 10.1021/ja012747a. [DOI] [PubMed] [Google Scholar]
- 38.Scheidt WR. In: The Porphyrin Handbook. Kadish KM, K. M. S., Guilard R, editors. Vol. 3. Academic Press; San Diego: 2000. pp. 70–82. [Google Scholar]
- 39.Shimanovich R, Hannah S, Lynch V, Gerasimchuk N, Mody TD, Magda D, Sessler J, Groves JT. Mn(II)-texaphyrin as a catalyst for the decomposition of peroxynitrite. J. Am. Chem. Soc. 2001;123:3613–3614. doi: 10.1021/ja005856i. [DOI] [PubMed] [Google Scholar]
- 40.Acholla FV, Mertes KB. A Binucleating Accordion Tetrapyrrole Macrocycle. Tetrahedron Lett. 1984;25:3269–3270. [Google Scholar]
- 41.Gerasimchuk NN, Gerges A, Clifford T, Danby A, Bowman-James K. Dimanganese(II) accordion porphyrin as a functional model for catalases. Inorg. Chem. 1999;38:5633–5636. doi: 10.1021/ic990576t. [DOI] [PubMed] [Google Scholar]
- 42.Sessler JL, Cho WS, Dudek SP, Hicks L, Lynch VM, Huggins MT. Synthesis and study of a calixpyrrole-texaphyrin chimera. A new oligopyrrolic chloride anion receptor. J. Porphyr. Phthalocy. 2003;7:97–104. [Google Scholar]
- 43.Veauthier JM, Cho WS, Lynch VM, Sessler JL. Calix[4]pyrrole Schiff-base macrocycles. Novel binucleating ligands for μ-oxo iron complexes. Inorg. Chem. 2004;43:1220–1228. doi: 10.1021/ic0352001. [DOI] [PubMed] [Google Scholar]
- 44.Veauthier JM, Tomat E, Lynch VM, Sessler JL, Mirsaidov U, Markert JT. Calix[4]pyrrole Schiff-base macrocycles: Novel binucleating ligands for Cu(I) and Cu(II). Inorg. Chem. 2005;44:6736–6743. doi: 10.1021/ic050690d. [DOI] [PubMed] [Google Scholar]
- 45.Li RQ, Mulder TA, Beckmann U, Boyd PDW, Brooker S. Dicopper(II) and dinickel(II) complexes of Schiff-base macrocycles derived from 5,5-dimethyl-1,9-diformyldipyrromethane. Inorg. Chim. Acta. 2004;357:3360–3368. [Google Scholar]
- 46.Givaja G, Blake AJ, Wilson C, Schroder M, Love JB. Macrocyclic diiminodipyrromethane complexes: structural analogues of Pac-Man porphyrins. Chem. Commun. 2003:2508–2509. doi: 10.1039/b308443d. [DOI] [PubMed] [Google Scholar]
- 47.Arnold PL, Blake AJ, Wilson C, Love JB. Uranyl complexation by a Schiff-base polypyrrolic macrocycle. Inorg. Chem. 2004;43:8206–8208. doi: 10.1021/ic0487070. [DOI] [PubMed] [Google Scholar]
- 48.Givaja G, Blake AJ, Wilson C, Schroder M, Love JB. Metal-directed ring-expansion in Schiff-base polypyrrolic macrocycles. Chem. Commun. 2005:4423–4425. doi: 10.1039/b507729j. [DOI] [PubMed] [Google Scholar]
- 49.Arnold PL, Patel D, Blake AJ, Wilson C, Love JB. Selective oxo functionalization of the uranyl ion with 3d metal cations. J. Am. Chem. Soc. 2006;128:9610–9611. doi: 10.1021/ja0634167. [DOI] [PubMed] [Google Scholar]
- 50.Sessler JL, Mody TD, Lynch V. Neutral Substrate Complexation by an Expanded Porphyrin. J. Am. Chem. Soc. 1993;115:3346–3347. [Google Scholar]
- 51.Sessler JL, Tomat E, Mody TD, Lynch VM, Veauthier JM, Mirsaidov U, Markert JT. A Schiff-base expanded porphyrin macrocycle that acts as a versatile binucleating ligand for late first-row transition metals. Inorg. Chem. 2005;44:2125–2127. doi: 10.1021/ic048412m. [DOI] [PubMed] [Google Scholar]
- 52.Sessler JL, Tomat E, Lynch VM. Positive homotropic allosteric binding of silver(I) cations in a Schiff base oligopyrrolic macrocycle. J. Am. Chem. Soc. 2006;128:4184–4185. doi: 10.1021/ja0582004. [DOI] [PubMed] [Google Scholar]
- 53.Sessler JL, Gorden AEV, Seidel D, Hannah S, Lynch V, Gordon PL, Donohoe RJ, Tait CD, Keogh DW. Characterization of the interactions between neptunyl and plutonyl cations and expanded porphyrins. Inorg. Chim. Acta. 2002;341:54–70. [Google Scholar]
- 54.Sessler JL, Tomat E, Lynch VM. Coordination of oxovanadium(V) in an expanded porphyrin macrocycle. Chem. Commun. 2006:4486–4488. doi: 10.1039/b608143f. [DOI] [PubMed] [Google Scholar]
- 55.Sessler JL, Katayev E, Pantos GD, Scherbakov P, Reshetova MD, Khrustalev VN, Lynch VM, Ustynyuk YA. Fine-Tuning the Anion Binding Properties of 2,6-Diamidopyridine Dipyrromethane Hybrid Macrocycles. J. Am. Chem. Soc. 2005;127:11442–11446. doi: 10.1021/ja0522938. [DOI] [PubMed] [Google Scholar]
- 56.Katayev EA, Ustynyuk YA, Lynch VM, Sessler JL. Mono-palladium(II) complexes of diamidopyridine-dipyrromethane hybrid macrocycles. Chem. Commun. 2006:4682–4684. doi: 10.1039/b611946h. [DOI] [PubMed] [Google Scholar]