

Abstract
In a search for estrogen receptor (ER) ligands to be radiolabeled with fluorine-18 for imaging of ER-positive breast tumors with positron emission tomography (PET), we investigated cyclofenil analogs substituted at the C3 or C4 position of the cyclohexyl group. McMurry coupling of 4,4′-dihydroxybenzophenone with various ketones produced key cyclofenil intermediates, from which C3 and C4 substituents containing alkyl and various oxygen or fluorine-substituted alkyl groups were elaborated. Binding assays to both ERα and ERβ revealed that the C3 site is more tolerant of steric bulk and polar groups than the C4 site, consistent with a computational model of the ERα ligand binding pocket. Fluorine substitution is tolerated very well at some sites, giving some compounds having affinities comparable to or higher than that of estradiol. These fluoro and fluoroalkyl cyclofenils merit further consideration as fluorine-18 labeled ER ligands for PET imaging of ERs in breast tumors.
Keywords: cyclofenil, estrogen receptor, PET agent, subtype selectivity
Introduction
The estrogen receptor (ER)**1,2 is a ligand-dependent transcription factor, whose conformation and activity are modulated by the binding of endogenous steroid hormones, such as 17β-estradiol (E2). Estrogens, acting through the ER, regulate many important physiological processes, such as the development and function of the female reproductive system, the maintenance of bone mineral density,3,4 cardiovascular health,5,6 and neuroprotection.7,8
The discovery that there are two ER subtypes, ERα and ERβ,11,12 provided impetus to develop novel estrogen pharmaceuticals able to activate or inhibit these subtypes selectively. Most estrogen target tissues have both ERα and ERβ, though in varying ratios; ERα is the principal subtype expressed in breast and uterine tissue, while ERβ is found at higher levels in ovary, prostate, bone, vascular epithelium, and certain brain regions;13–15 While difficult to generalize, ERβ is usually less active as a transcription factor and exerts a restraining effect on the more active ERα.16–20 It is notable that in breast cancer, the level of ERβ relative to ERα declines with disease progression,21 as one might expect with the transition to a more proliferative and invasive tumor phenotype. Thus, if the independent quantification of ERα and ERβ levels in breast cancer could be achieved by imaging—using fluorine-18 labeled ER subtype-selective ligands with positron emission tomography (PET)—it might provide information predictive of disease staging and tumor response to hormone therapies. Differential imaging of the two ER subtypes, however, has proved to be a major challenge (see below).
The ligand binding domains of ERα and ERβ have only 58% sequence identity,22 and the volume of the ERβ ligand binding pocket is smaller than that of ERα.23, 24 Otherwise, the pockets are very similar, with only two out of 24 residues being different, and these being conservative substitutions (Leu384 and Met421 in ERα corresponding to Met and Ile in ERβ, respectively). Despite this similarity, it has proved possible to develop ER ligands that bind to and activate either ERα or ERβ with a high level of selectivity (as has been reviewed25–27), and a reasonable pharmacophore model has been advanced to guide the design of subtype-selective ligands (Figure 1).28,29 ER ligands with varying core structures have been reported: pyrazoles,30 pyrroles,31 and furans,31 as ERα-selective ligands, and tetrahydrochrysenes,32 diarylpropionitriles,33 cyclofenils,34 and benzofused heterocyclic analogues35–37 as ERβ-selective ligands. In prior studies, we have investigated some fluorine-18 labeled analogs of these ligands as PET imaging agents selective for ERα38 and ERβ,39 but thus far our success has been limited.
Figure 1.
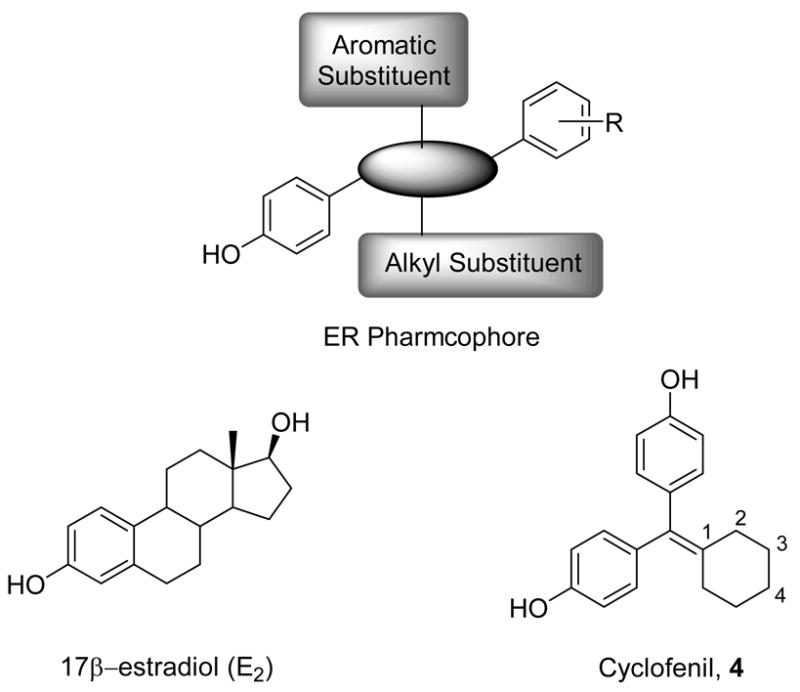
ER ligands and a pharmacophore model
Cyclofenil (bis-(4-hydroxyphenyl)methylidenecyclohexane, 4) and its analogues, while only slightly ERβ-selective, have very high binding affinity, often comparable to or greater than that of estradiol.34 After cyclofenil was reported,40 its conformation was investigated by x-ray crystallography, and its binding to uterine receptors41 was compared to that of estradiol (E2).42 Notably, cyclofenil has mixed agonist antagonist activity typical for a non-steroidal selective estrogen receptor modulator (SERM), such as tamoxifen or raloxifene, and it has been used for ovulation induction.43
Recently, we prepared cyclofenil derivatives that have bicyclic core units to examine the degree to which a three-dimensional topology in the central hydrophobic core of a ligand would be tolerated by the ERs.34 It was encouraging to find that several of the 1,1-diarylethylene motif bridged bicyclic or tricyclic core cyclofenil analogs had very high binding affinities, up to 3–5 times greater than that of estradiol in both ER subtypes. The torsion angles between the two hydroxyphenyl groups seem to correlate with the binding affinity, although other factors such as non-optimal orientation of the phenyl ring and lack of ligand steric complementarity with the binding pocket also play important roles.44 These factors were not investigated in the SAR and x-ray conformational studies of cyclofenil derivatives reported previously.41,42
Thus, in our search for agents to image ERs in breast cancer by PET, we undertook a further investigation of substituted cyclofenil ligands to establish where it might be feasible to introduce polar substituents such as fluorine, as would be needed for labeling with the positron emitting radionuclide, fluorine-18. Because we wanted to establish a structure-activity relationship independent of effects that would result from changes in the critical torsion angle of the dihydroxyphenyl group, we investigated substituents only on the C3 and C4 positions of the cyclohexane ring (Figure 1), sites where substitution is not expected to affect this torsion angle. We thought that such C3- and C4-substituted cyclofenil derivatives would help probe for differences in the largely similar hydrophobic ligand binding pockets of the ERα and ERβ. We hoped, as well, to identify ERβ-selective ligands of high affinity33,45 that are tolerant of the fluorine substitution needed for F-18 labeled PET imaging agents.
Herein, we report the syntheses of 24 cyclofenil derivatives, many of which are substituted at the C3 or C4 positions, as well as ones with different ring sizes, and an evaluation of their relative binding affinities (RBAs) to ERα and ERβ. Through this study, we have established a useful structure-activity relationship relating ERα and ERβ binding affinities to the size and polarity of the C3 or C4 substituent in the cyclofenil series. In addition, we have identified a few fluoro- or fluoroalkyl cyclofenil analogs that have binding affinity for ERα and ERβ comparable to or greater than that of the endogenous ligand, estradiol, though their ER subtype selectivity is limited. These compounds are being considered for further investigation as PET imaging agents for ER in breast cancer.
Results
Synthesis of Cyclofenil Analogs
The compounds we have synthesized can be divided into two types (Figure 2). One series of analogues (type I, compounds 2–5) has different cycloalkyl core units, ranging from cyclobutane to cycloheptane. The other series (type II) has a fixed cyclohexyl moiety onto which we have introduced various substituents at the C3 or C4 position. All of the cyclofenil derivatives or their precursors were prepared by a McMurry coupling reaction between 4,4′-dihydroxybenzophenone and a cyclic ketone. Some of the cyclic ketones are commercially available or can be readily obtained in a few steps from commercially available material. In many cases, the substituents were elaborated from functional precursors after the cyclofenil system was prepared by McMurray coupling.
Figure 2.
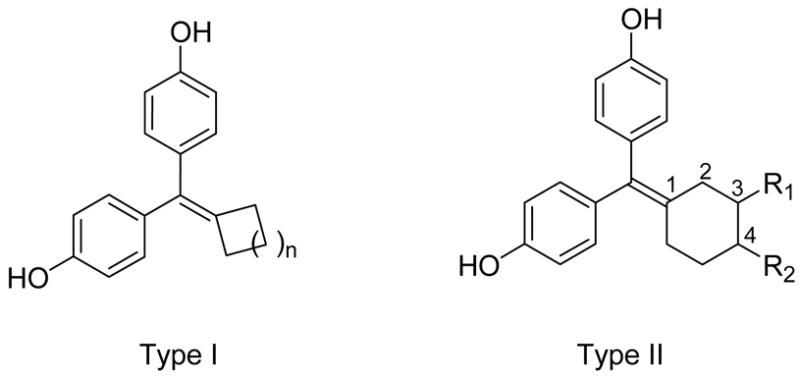
Two types of cyclofenil analogues
Synthesis of Type I Cyclofenil Analogs
McMurry coupling reaction with low valent titanium activated by zinc was used to prepare the various ring sized cyclofenil analogues2–5 (Scheme 1), according to a previously reported procedure.34,46,47 It is of note that this coupling is tolerant of free phenols. All of the reactions gave reasonable yields (62–83%), except the cyclobutyl compound 2 (8%).
Scheme 1.
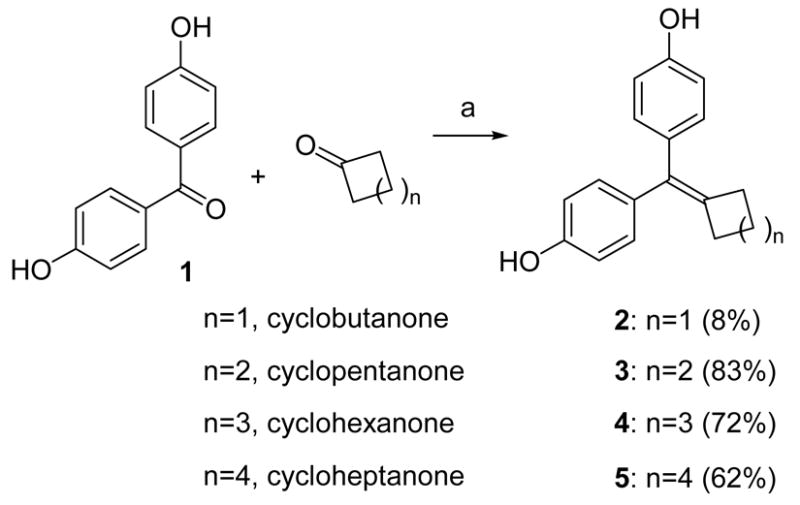
Reagents: (a) TiCl4, Zn, THF, reflux, 4 h.
Synthesis of Cyclohexanone Precursors for Type II Cyclofenil Analogs
Scheme 2 shows how various cyclohexanone precursors, functionalized at C4 or C3 position, were prepared. 1,4-Dihydroxycyclohexane (6) was oxidized to 4-hydroxycyclohexanone (7) using freshly prepared Jones reagent, and was acetylated to give 4-acetoxycyclohexanone (8). Wittig reaction of cyclohexan-1,4-dione (9) with methyl (triphenylphosphoranylidene)acetate produced methyl (4-oxo-cyclohexylidene)acetate, which, following hydrogenation over a palladium catalyst, yielded the cyclohexyl acetate 10 that was used as a source of two-carbon substituents at the C4 position.48 The C3-alkylated cyclohexanone precursor with one carbon atom (12) was prepared by esterification of the commercially available acid (11). The two-carbon substituent precursor (14) was prepared by Michael addition of dimethyl malonate to cyclohexanone (13).49 The synthesis of the cyclohexanone with a linear three-carbon unit on the C3 position (15) involved copper-catalyzed 1,4-addition to cyclohexenone of the organomagnesium reagent generated in two steps from 3-chloropropanol (via the 3-chloropropyl magnesium alkoxide), with subsequently acetylation.50 All compounds functionalized at C3 position are chiral and were used as racemates.
Scheme 2. Syntheses of cyclohexanones.
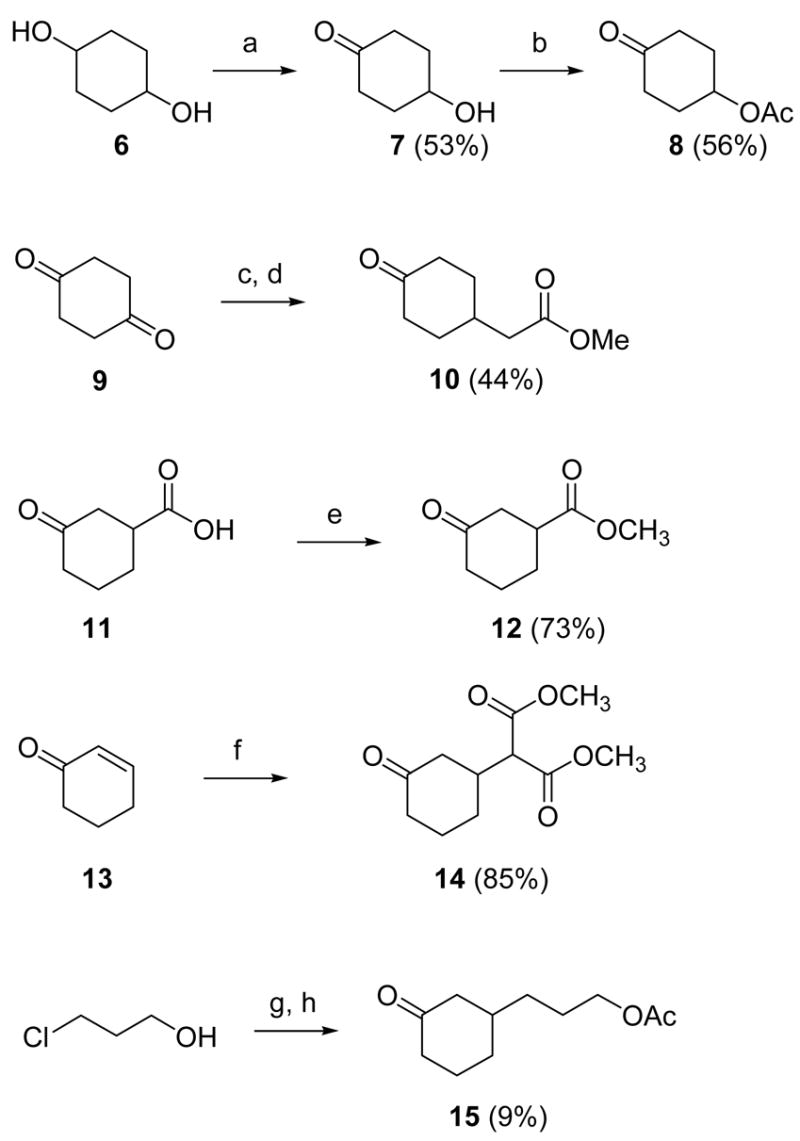
Reagents: (a) Jones reagent, acetone, 0 °C-rt, 45 min; (b) Ac2O, pyridine, rt, 24 h; (c) methyl (triphenylphosphoranylidene)acetate, toluene, 110 °C, 8 h; (d) Pd/C, H2, EtOH, rt, 6 h; (e) SOCl2, MeOH, rt, 3 h; (f) dimethyl malonate, NaOEt (cat.), rt, 2 h; (g) i. MeMgCl, THF, −78 °C-rt, 20 min; ii. Mg, 1,2-dibromoethane, THF, rt-90 °C, 2 h; iii. 13, CuBr, THF, −78 °C-0 °C, 1 h; (h) Ac2O, pyridine, CH2Cl2, rt, 18 h.
Synthesis of C4-Substituted Cyclofenil Analogs
Scheme 3 shows the preparation of the C4-functionalized cyclofenil derivatives. McMurry coupling of 4,4′-dihydroxybenzophenone with 4-acetoxycyclohexanone (8) yielded the C4 acetoxy analog 16; the phenolic hydroxy groups were then protected as MOM ethers. The C4-hydroxy cyclofenil 17b, obtained after alkaline hydrolysis of the acetate, was converted to the protected 4-fluorocyclofenil derivative 19 in 32% yield by treatment with (diethylamino)sulfur trifluoride (DAST). Acid cleavage of the MOM groups in both 19 and 17b produced the C4-hydroxy and fluoro cyclofenil analogs, 18 and 20, in 71% and 85% yield, respectively.
Scheme 3.
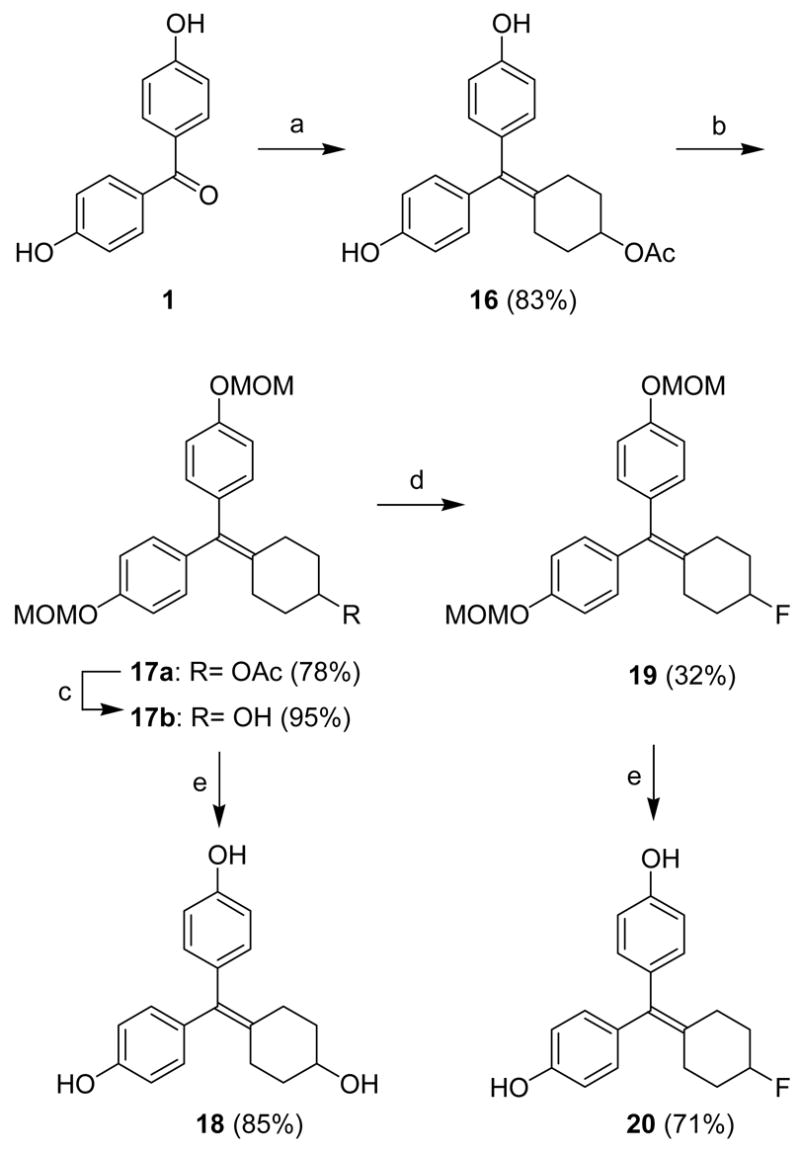
Reagents: (a) 8, TiCl4, Zn, THF, reflux, 4 h; (b) methoxymethyl chloride, NaH, DMF, 0 °C-rt, 1 h; (c) K2CO3, MeOH:H2O (5:1), rt, 12 h; (d) DAST, CH2Cl2, −78 °C-rt, 1 h; (e) HCl, MeOH, rt, 12 h.
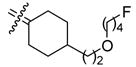
The preparation of cyclofenil derivatives with carbon chains at the C4 position is shown in Scheme 4. Ethyl 4-oxocyclohexanecarboxylate, a commercially available ketone that has a one-carbon unit at the C4 position, or methyl (4-oxo-cyclohexyl)acetate (10), which has a two-carbon unit at the C4 position (prepared as in Scheme 2), were reacted with 4,4-dihydroxybenzophenone (1) using the same McMurry coupling procedure. In this manner, we obtained cyclofenil 4-carboxylate 21a and acetate 21b in around 70% yield. The phenols in both compounds were protected as MOM ethers, and the carboxylic esters in both 22a,b were reduced to the corresponding primary alcohols with lithium aluminum hydride. These alcohols, 23a and 23b, served as intermediates for further synthesis. After MOM deprotection, they gave the C4 hydroxymethyl and 2-hydroxyethyl cyclofenils (24a,b). As the MOM ethers, they served as precursors of the C4-ethoxymethyl compound 27 and the fluorobutyloxymethyl compound 28. They were also converted to the methanesulfonates 25a,b. Fluoride ion substitution to produce the fluoromethyl and fluoroethyl analogs (26a,b) from these methanesulfonates was accomplished by reaction with cesium fluoride in an ionic liquid.51 The use of this medium for fluoride ion substitutions minimizes side reactions, such as elimination, that would produce various alkenes, which are sometimes difficult to separate from the desired fluorine-substituted products. Based on precedent in other systems, this ionic liquid fluorination would likely be effective for labeling of these compounds with F-18, even in the presence of small amounts water which typically remain after reagent drying by azeotrope distillation.52 Reductive cleavage of the methanesulfonates, followed by deprotection of the MOM ethers, yielded the C4 methyl and ethyl cyclofenil analogs (29a,b).
Scheme 4.

Reagents: (a) TiCl4, Zn, THF, reflux, 4 h; (b) methoxymethyl chloride, NaH, DMF, 0 °C-rt, 1 h; (c) LAH (1 M solution in THF), THF, 0 °C-rt, 1 h; (d) methanesulfonyl anhydride, TEA, CH2Cl2, 0 °C-rt, 1 h; (e) CsF, H2O, 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]), acetonitrile, 100 °C, 2 h; (f) HCl, MeOH, rt, 12 h; (g) bromoethane, NaH, DMF, 100 °C, 24 h; (h) 1-bromo-4-fluorobutane, NaH, DMF, 0–100 °C, 24 h.
Synthesis of C3-Substituted Cyclofenil Analogs
We used similar procedures to introduce substituents and functional groups at the C3 position of cyclofenil (Scheme 5). McMurry reaction with the cyclohexanones 12, 14 and 15 produced coupled ester compounds 30a–c, respectively. Hydrolysis of 30b and subsequent decarboxylation and esterification produced the C3 carboxymethyl compound 32. The phenols of all three cyclofenil derivatives (30a, 30c and 32) were protected as MOM ethers. Reduction of 33a and 33b with lithium aluminum hydride and hydrolysis of 33c with potassium carbonate gave hydroxymethyl, hydroxyethyl and hydroxypropyl cyclofenil derivatives (34a–c), respectively.
Scheme 5.

Reagents: (a) TiCl4, Zn, THF, reflux, 4 h; (b) 2 M NaOH, MeOH, reflux, 2 h; (c) diglyme, 160 °C, 1 h; (d) SOCl2, MeOH, rt, 90 min; (e) methoxymethyl chloride, NaH, DMF, 0 °C-rt, 1 h; (f) i. in case of 33a,b, LAH (1 M solution in THF), THF, 0 °C-rt, 1 h, ii. in case of 30c and 33c, K2CO3, MeOH:H2O (5:1), rt, 12 h; (g) methanesulfonyl anhydride, TEA, CH2Cl2, 0 °C-rt, 1 h; (h) CsF, H2O,1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]), acetonitrile, 100 °C, 2 h; (i) HCl, MeOH, rt, 12 h; (j) DAST, CH2Cl2, −78 °C-rt, 1 h.
Fluoride substitution was accomplished by two different procedures: in case of 34a and 34c, the free hydroxyl group was directly converted to fluoride by treatment with DAST, and in case of 34b, the hydroxylethyl group was converted to fluoroethyl group by fluoride ion substitution on the methanesulfonate intermediate 36b. Removal of MOM-protective group to obtain the free fluoro compounds was attempted in acidic media, but strangely the color of all three fluorinated products (37a–c) slowly changed to red. Even after the deprotected fluoro compounds were purified again by column chromatography, they developed color upon standing. When each compound was recrystallized, however, their stability increased quite a bit, but they still decomposed slowly. It proved to be especially difficult to obtain 37a in pure form, and because of its instability it was not evaluated for binding.
Estrogen Receptor Binding Assay
The relative binding affinities (RBAs) of cyclofenil analogues were determined using a competitive radiometric binding assay with purified full-length human ERα and ERβ, according to published procedures.53 The RBA value was expressed as binding affinity relative to that of estradiol (100%). RBA values of the 24 cyclofenil analogues evaluated are listed in Table 1. It is notable that the tracer and standard, estradiol, has a modest binding preference for ERα (Kd (ERα) = 0.2 nM and Kd (ERβ) = 0.5 nM).
Table 1.
Relative Binding Affinity (RBA)a of Cyclofenil Derivatives for the Estrogen Receptors α and β.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | structure | RBA (E2 = 100)a | β/αb | # | Structure | RBA (E2 = 100)a | β/αb | ||
| ERα | ERβ | ERα | ERβ | ||||||
| 2 |
|
5.61 | 20.5 | 3.7 | 27 |
|
4.88 | 2.82 | 0.58 |
| 3 |
|
17.8 | 137 | 7.7 | 28 |
|
3.77 | 1.45 | 0.38 |
| 4 |
|
124 | 285 | 2.3 | 21a |
|
6.77 | 3.02 | 0.45 |
| 5 |
|
110 | 354 | 3.2 | 21b |
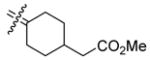
|
10.7 | 1.72 | 0.16 |
| 29a |
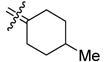
|
66.5 | 274 | 4.1 | 37b |
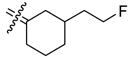
|
122 | 129 | 1.1 |
| 29b |
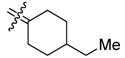
|
62.7 | 75.6 | 1.2 | 37c |
|
160 | 91.4 | 0.57 |
| 20 |
|
27.0 | 61.8 | 2.3 | 35a |
|
24.8 | 39.6 | 1.6 |
| 26a |
|
18.1 | 41.5 | 2.3 | 35b |
|
33.9 | 57.6 | 1.7 |
| 26b |
|
15.9 | 18.9 | 1.2 | 35c |
|
47.2 | 46.1 | 0.98 |
| 18 |
|
0.327 | 0.307 | 0.94 | 30a |
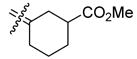
|
23.7 | 36.5 | 1.5 |
| 24a |
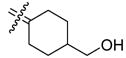
|
0.846 | 0.841 | 0.99 | 32 |
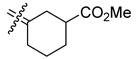
|
58.5 | 10.3 | 0.18 |
| 24b |
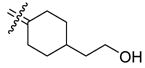
|
2.61 | 0.372 | 0.14 | 30b |
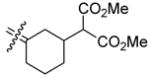
|
19.5 | 0.234 | 0.012 |
Relative Binding Affinity (RBA) values are determined by a competitive radiometric binding assay with [3H]estradiol and full length human ERα and ERβ, using methods described in the Experimental Section. The RBA of estradiol (E2) is defined as 100, and the measured RBA values represent the mean of two or more independent determinations (CV <0.3).
The binding affinity of the tracer estradiol to the ERs is Kd(ERα) = 0.2 nM and Kd(ERβ) = 0.5 nM. Thus, on an absolute scale of binding affinities, the β/α ratios would be reduced by a factor of 2.5.
Binding Affinity of Type I Compounds
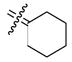
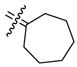
We have previously reported a structure-activity relationship (SAR) of 1,1-diarylethylene derivatives that have bridged bicyclic cores, but from this earlier study, it was not obvious what effect the bicyclic core size had on binding affinity.34 Thus, we prepared the first type of cyclofenil analogues (2–5, type I) to evaluate the RBA values of cyclofenil compounds having different ring size core units (Figure 1 and Table 1). In general, the RBA values range from 5.6 to 124 for ERα and from 20 to 354 for ERβ. Interestingly, when the ring size is increased from cyclobutyl 2 to cycloheptyl 5, binding affinities for both ERα and ERβ increase, and all compounds (2–5) show selectivity for ERβ of ca. 3-fold in terms of the RBA β/α ratio (which is comparable to that of estradiol, 2.5; see Table 1, footnote b), except for the cyclopentyl compound 3, which on this scale was 7.7-fold ERβ selective (the most ERβ selective of all compounds prepared). These data suggest that an increase in lipophilic character in the cyclic core could lead to high binding affinity for both ERα and ERβ but not a large increase in ERβ selectivity.
Binding Affinity of Type II Compounds with C4 Substituents
Twelve cyclofenil derivatives with different C4 substituents, such as alkyl (29a,b), hydroxyalkyl (18, 24a,b), alkyl carboxylate (21a,b, 27, 28) and fluoroalkyl substituents (20, 26a,b), were investigated to compare the effects on ER binding affinity of introducing alkyl groups of different carbon lengths and substitution of these groups with electronegative and/or polar units (fluoride and hydroxide).
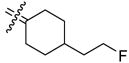
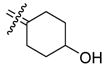
In all cases (except the C4 methyl compound 29a with ERβ), any substitution at C4 reduced binding affinity to both ERα and ERβ, but the degree to which binding was lowered depended on substituent size and polarity. In general, groups that were small and/or non polar had the highest binding affinity. Thus, the C4 methyl and ethyl compounds (29a,b) still had good binding affinity compared to the cyclofenil parent (4). Direct substitution at C4 with fluorine (20) caused some reduction in ER binding, but much less than direct substitution with hydroxyl (18); the latter (hydroxy) substitution dropped ERα binding 380 fold and ERβ binding 930 fold. Similar trends are noted for fluorine and hydroxy substitution in the C4 methyl series (29a, 26a, and 24a) and the C4 ethyl series (29b, 26b, and 24b): both substituents lower binding, but fluorine, which is less polar, is tolerated much better than hydroxy. These trends are consistent with computational models of the C4 substituted compounds in the ligand binding pocket of ERα (see below and Figure 3), in which these C4 substituents are shown to project into a constrained, hydrophobic environment. The other four analogs (27, 28, 21a, and 21b) are all of intermediate polarity and all have relatively low affinity. It is of note that the highest binding C4-substitued cyclofenil containing fluorine is the direct 4-fluorocyclofenil (20), with ERα and ERβ RBA values of 27 and 62, respectively.
Figure 3.

Schematic and computational model of substituted cyclofenil interaction with the ligand binding pocket of the estrogen receptor, ERα. Panel A. Schematic picture of the interactions of the phenolic hydroxyls and the C3 and C4 cyclofenil substituents with residues in the ligand binding pocket. ERα residue numbers precede ERβ residue numbers. Panel B. Representative computational model of 4-fluorocyclofenil (20) in the ligand binding pocket of ERα. The hydrophobic residues near the C4 position are indicated. The surface shown is the ERα binding pocket displayed as dots mapped with the lipophilic potential. Residues identities are for ERα. Panel C. Representative computational model of 3-(2-fluoropropyl)cyclofenil (37c) in the ligand binding pocket of ERα. The surface is the same as Panel B. Residues identities are for ERα.
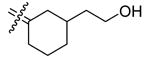
It is worthwhile to consider not just the ERα and ERβ binding affinities but also the ERα/ERβ binding selectivities of these cyclofenil analogs. One notes an overall trend that when a more polar group is attached to a C4-alkyl group, the β/α RBA ratio decreases. This is clearly evident in the C4 methyl series: methyl 29a (β/α : 4.1), fluoromethyl 26a (β/α : 2.3) and hydroxylmethyl 24a (β/α : 0.99), and reasonably evident also in the C4-ethyl series: ethyl 29b (β/α 1.2), fluoroethyl 26b (β/α : 1.2), and hydroxyethyl 24b (β/α : 0.14). Despite this shift in ER-subtype binding preference from ERβ to ERα, the overall RBA values for both ERs decrease considerably, consistent with the generally poor tolerance of polarity in the core of ER ligands. If one looks at β/α RBA ratios as a function of the length of homologous substituents (alkanes 4, 29a,b; fluoroalkanes 20, 26a,b; hydroxyalkanes 18, 24a,b), in each case, the selectivity for ERβ seems to be highest with the two-carbon species. This preference for substituents of intermediate length is also apparent, to some degree, in the other C4 analogs, (27, 28, 21a,b). Of greatest interest to this study, however, is the fact that among the C4-substituted cyclofenils, there is one fluorine-substituted analog, 4-fluorocyclofenil (20), that has a binding affinity for ERβ that is nearly comparable to that of estradiol.
Binding Affinity of Type II Compounds with C3 Substituents
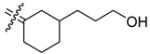
In those cases where the RBA values of C3-substituted cyclofenils can be directly compared their C4-substituted analogs having the same substituents (37b with 26b, 35a with 24a, 35b with 24b, and 32 with 21b), the C3-substituted cyclofenils have significantly greater binding affinity and a greater tolerance for polar substituents. It is notable that the homologous C3 fluoroethyl and propyl compounds (37b,c) have rather similar ERα and ERβ relative binding affinities; this is true to a somewhat lesser degree with the C4 fluoroalkyl compounds (20, 26a,b), where increasing size causes a somewhat greater decrease in affinity. In the C3-substituted cyclofenil series, both fluorine-substituted compounds (37b,c) are high affinity ligands for both ERs.
A particularly dramatic difference between the C3 and C4 series is noted in the ERs binding affinity of the C3 hydroxyethyl (35b; RBA α 34, β 58) and the C4 hydroxyethyl (24b; RBA α 2.6, β 0.37) compounds; the affinity ratio is 154 fold in case of ERβ in favor of the C3-substituted series. A similar, though somewhat less dramatic comparison can be made with the hydroxymethyl C3 and C4 analogs (35a; RBA α 25, β 40 vs. 24a; RBA α 0.85, β 0.84), again with the C3-substituted systems having higher affinities. These trends are consistent with computational models of the ERα ligand binding pocket (see below and Figure 3) in which it can be seen that the C3 substituent encounters less steric hindrance and has an opportunity to extend towards and interact with a key histidine residue (H524 in ERα and H474 in ERβ). Again, of principal interest to this study is the fact that we have identified two cyclofenil analogs, compounds, 37b and 37c, that bind better to ERα, and in the case of 37b also to ERβ, better than does estradiol.
Molecular Modeling of with C4 and C3 Substituted Cyclofenils in ER Ligand Binding Pocket of ERα
To understand why the estrogen receptor differed so greatly in its tolerance of the bulk and polarity of substituents at the C4 and C3 positions of cyclofenil, we examined how some of these compounds fit into the ligand binding pocket of ERα by computational modeling (Figure 3). A model for the ERα ligand binding domain was generated from the ERα-estradiol structure (1ERE in the RCSB) by deleting the ligand and then placing various C3- and C4-substituted cyclofenils into this pocket using the FlexiDoc routine within the Sybyl 7.1 molecular modeling software (Tripos Inc.). The initial docking orientation of substituted cyclofenil was based on initial models with unsubstituted cyclofenil (data not shown), where one phenol occupies a position equivalent to that of the A-ring of estradiol, the second projects in roughly a “steroid 11β direction”, and the cyclohexyl ring occupies roughly the “steroid 7α sub-pocket” (See Figure 3A).
To challenge this ligand orientation, the cyclofenil analogues were initially modeled with one phenol as the steroid A-ring mimic, but intentionally “misorienting” the cyclohexyl ring in the “steroid 11β direction” of the binding pocket (opposite of what was found on cyclofenil), so that the other phenol occupied the “steroid 7α sub-pocket”. Despite this deliberate initial misorientation of the ligand, the FlexiDoc routine reoriented the cyclofenil core in a characteristic manner. In all cases, one of the phenol rings of cyclofenil remained in place of the A-ring of estradiol, where that the hydroxyl group could engage in the canonical hydrogen bonds with the Glu and Arg residues (353 and 394 in ERα and 305 and 346 in ERβ, respectively). In all but a few cases, the cyclofenil core was reoriented to place other phenol in a roughly “steroid 11β direction”; in this orientation, the second hydroxyl is within 3Å of a threonine residue (T347 in ERα and T299 in ERβ). Final optimization of the model included a three-step minimization process that has been reported elsewhere (see Experimental section).
Modeling of the achiral C4-substituted cyclofenils was relatively straightforward; in these, the cyclohexane ring can adopt two chair conformations on each of which the substituent can be either axial or equatorial. Many of these cases were modeled to give stable structures. Docking always revealed one phenol as the steroidal A-ring mimic, and typically placed the second pendant phenol in the “steroid 11β direction.” In most cases, the C4 substituents project towards a region of the ligand binding pocket that is somewhat flexible but essentially hydrophobic (I424 and L428 in ERα or I376 and L380 in ERβ) (See Figure 3B). This view is consistent with the behavior of these analogs in which alkyl groups (29a,b) were very well tolerated, the electronegative fluoro (20) and fluoroalkyl groups (26a,b) were reasonably well tolerated, but the polar hydroxy (18) and hydroxyalkyl groups (24a,b) bind very poorly.
The chiral C3-substituted cyclofenils present a greater challenge for modeling, because each enantiomer can adopt two chair conformations, each of which can have the substituent disposed in an axial or equatorial position. Modeling was done with some analogs in most of these orientations, from which a consistent pattern also emerged, as is illustrated in Figure 3C. By alternate choice of the phenols and chair conformation, each C3-substituted enantiomer can adopt an orientation in which the substituent can be extended towards His 524, the residue in ERα with which the 17β-hydroxyl group of estradiol forms a hydrogen bond. Thus, this model provides a satisfying rationale for the fact that at this site the ER can tolerate both bulky and electronegative fluoroalkyl (37b,c) as well as the more polar hydroxyalkyl (35a–c) groups. It is of note that substituents originating from the C4 carbon cannot access His524. Attempts were not made to explain the relatively modest ERα vs. ERβ binding selectivities of the compounds in this series by modeling.
Conclusions
In summary, in our search for PET imaging agents for the estrogen receptors, we have focused on the preparation of C3- and C4-substituted cyclofenil derivatives so that we could explore the introduction of alkyl and functionalized alkyl substituents in a manner that would not affect the rotational mode of both bis-hydroxyphenyl groups, a conformational parameter that is very important for ER binding. We hoped, thereby, to discover ER ligands in which an electronegative substituent—fluorine in particular—might be well tolerated, perhaps even ones in which the fluorine-substituted analog would retain the “higher than estradiol” binding affinity of the cyclofenil parent, so that they might be useful as PET imaging agents for ER in breast tumors.
From the binding affinities of the 24 compounds we have investigated, we find that cyclofenils with 6 and 7 membered rings bind best to ER. Except for one case, introduction of the various C3 and C4 substituents into the cyclohexane ring of cyclofenil decreases binding affinity, particularly when the substituents bear polar and/or electronegative groups, but less so with fluorine than with hydroxyl. In general, groups that are small or of intermediate size are better tolerated, and those substituted at C3 bind better than those substituted at C4, in some cases dramatically better. The pattern of affinity dependence on the site of substitution and the size and polarity of the substituent can be nicely rationalized by computational modeling of the ligand binding pocket. Overall, however, there appears to be relatively little binding selectivity for either ERα or ERβ.
The principal motivation for this study was to identify cyclofenil analogs in which a fluorine substituent could be introduced with retention of high ERα and/or ERβ binding affinity, as would be needed to develop F-18 labeled ligands for PET imaging of the ERs in breast tumors. In terms of binding affinity, our study was successful, because we have found three fluorine-substituted compounds, 4-fluorocyclfenil (20), and 3-fluoroethyl- and 3-fluoropropylcyclofenil (37b and 37c), that bind to the ERs with an affinity that is comparable to or greater than that of estradiol. We did not find compounds having marked affinity preference or either ERα or ERβ, however. Further work on the radiolabeling and in vivo evaluation of some of these compounds will be reported elsewhere.
Experimental Section
All reagents and solvents were purchased from Aldrich, Acros, or fisher. Anhydrous THF, Et2O and CH2Cl2 were collected using a solvent dispensing system built by J. C. Meyer based on a design developed by Pangborn.54 Reaction progress was followed by TLC on 0.25 mm silica gel glass plates containing F-254 indicator (Merck). Visualization on TLC was monitored by UV light or phosphomolybdic acid indicator. The chemical shifts were reported in parts per million and were referenced to the internal solvent peaks. Coupling constants were reported in hertz. 1H spectra were obtained on 400 or 500 MHz spectrometers. 13C NMR spectra were acquired at 100 or 125 MHz. Melting points were checked using a Thomas-Hoover capillary melting point apparatus and are uncorrected. Low- and high-resolution electron impact (EI, 70 eV) spectra were obtained on a Micromass 70-VSE spectrometer. Elemental analyses were performed by the Microanalytical Service Laboratory of the University of Illinois or the Center for Collaborative Instruments at Inha University. Reactions were performed under a nitrogen atmosphere unless noted otherwise.
General Procedure for McMurry Coupling Reaction
A two-necked round-bottomed flask, containing zinc powder (1.2 g, 18 mmol) was fitted with a reflux condenser, evacuated for 10 min, and charged with nitrogen gas. After THF (15 mL) was added, the reaction mixture was cooled in an acetone-water bath (−10 °C), and then titanium(IV) chloride (1.64 g, 8.62 mmol) was added slowly. Addition of titanium(IV) chloride released a yellow fume, and the reaction mixture turned a yellow-green color. The reaction was refluxed for 2 h at 100 °C and then cooled to RT. A solution of dihydroxybenzophenone (1, 500 mg, 2.33 mmol) and each ketone (2.33 mmol) dissolved in THF (15 mL) was injected by syringe, and the reaction was refluxed for 2 h. The cooled reaction mixture was slowly poured into a NaHCO3 solution (200 mL), and Et2O (200 mL) was added with vigorous stirring. The heterogeneous solution was filtered through Celite. After the organic layer was decanted and saved, the aqueous layer was extracted with additional Et2O (100 mL). The combined organic layer was dried over MgSO4 and concentrated. Flash Column chromatography (Et2OAc/hexane; 3:7 or 4:6) gave each coupled compound. Compounds for bioassay were recrystallized from Et2O and hexane, or Et2OAc and hexane.
General Procedure for MOM Protection
To a solution of bisdihydroxyphenyl (4.4 mmol) in anhydrous DMF (15 mL) was added sodium hydride (10 mmol) in an ice-bath with N2. The cooling was removed, and methoxymethyl chloride (9.7 mmol) was slowly added dropwise by syringe with stirring. Almost at the end of the addition, the mixture turned into an off-white solution and was stirred 1 h more. After replacing the ice-bath, the reaction was quenched by water (1.0 mL) and transferred to a separatory funnel. Saturated ammonium chloride solution (100 mL) was added to remove the DMF, and the organic layer was extracted with Et2OAc (100 mL). This was washed with brine (2 × 100 mL), dried over MgSO4 and concentrated under reduced pressure. Flash column chromatography (Et2OAc/hexane; 2:8) gave the MOM protected compound.
General Procedure for Reduction
A mixture of LiAlH4 solution in THF (1 M, 5.7 mL) and THF (15 mL) was slowly added to each ester compound (5.7 mmol) under ice-bath cooling. After addition, the cooling was removed, and the reaction was stirred for about 1 h. The ice bath was replaced, and the reaction was quenched carefully by water (100 μL), 2 M sodium hydroxide solution (100 μL) and water (100 μL) in turn. Additional stirring for about 20 min gave a white precipitate that was filtered through Celite. The filtrate was concentrated, and flash column chromatography (Et2OAc/hexane; 3:7) of the residue gave each alcohol product
General Procedure for Methanesulfonylation
A two-necked round-bottomed flask, which contained the alcohol compound (0.23 mmol), was charged with nitrogen. Freshly distilled CH2Cl2 (5 mL) was added to the round bottom flask, and the reaction was stirred in an ice-bath. Methanesulfonic anhydride (94 mg, 0.54 mmol) was added in several portions, followed by small portions of triethylamine (68 mg, 0.68 mmol) slowly. The reaction mixture was stirred at RT about 1 h. The reaction was quenched by water (10 mL) with vigorous stirring, which was extracted with additional CH2Cl2 (50 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure. Flash column chromatography (Et2OAc/hexane; 4:6) gave each alcohol product.
General Procedure for Fluorination with Cesium Fluoride51
To methanesulfonate (0.35 mmol) and cesium fluoride (1.75 mmol) in a 10 mL vial was added N-butyl-N′-methylimidazolium tetrafluoroborate (1.5 mL), acetonitrile (1.5 mL) and H2O (50 μL). After the vial was tightly capped, the reaction mixture was stirred for 2 h at 100 °C. After cooling, Et2O was added to the reaction mixture, which was shaken and decanted 5 times. The combined organic layer was dried over MgSO4 and concentrated. Flash column chromatography (Et2OAc/hexane; 1:9) gave each fluorinated compound.
General Procedure for MOM Deprotection
Concentrated HCl (200 μL) was added to a solution of MOM protected compound (0.31 mmol) in methanol (10 mL). In the case of some compounds, Et2O (5 mL) was added to dissolve the compounds. The reaction mixture was stirred for 12 h, then quenched by water (10 mL) and extracted by Et2OAc (50 mL). After removal of the solvent, short flash column chromatography (Et2OAc/hexane; 1:1) gave bis(dihydroxyphenyl)methylene compounds. Compounds for bioassay were recrystallized from Et2O and hexane or Et2OAc and hexane.
Bis(4-hydroxyphenyl)methylenecyclobutane (2)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 500 mg, 2.33 mmol), cyclobutanone (163 mg, 2.33 mmol), Zn (1.2 g, 18 mmol) and titanium(IV) chloride (1.64 g, 8.62 mmol), 2 (45 mg, 8%) was obtained as a white solid: mp 177–179 °C; 1H NMR (400 MHz, acetone-d6) δ 6.97 (d, J = 8.8 Hz, 4H), 6.76 (d, J = 8.8 Hz, 4H), 2.86 (t, J = 8.0 Hz, 4H), 1.99 (quintet, J = 8.0 Hz, 2H); 13C NMR (100 MHz, acetone-d6) δ 156.6, 137.7, 133.4, 133.1, 130.7, 115.6, 32.5, 17.7; MS (EI) m/z 252 (M+, 100), 223, 107. HRMS (EI) m/z calcd for C17H16O2 252.1150, found 252.1152. Anal. (C17H16O2·0.1 H2O) C, H.
Bis(4-hydroxyphenyl)methylenecyclopentane (3)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 500 mg, 2.33 mmol), cyclopentanone (196 mg, 2.33 mmol), Zn (1.2 g, 18 mmol) and titanium(IV) chloride (1.64 g, 8.62 mmol), 3 (518 mg, 83%) was obtained as a white solid: mp 194–195 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.90 (d, J = 8.4 Hz, 4H), 6.66 (d, J = 8.4 Hz, 4H), 2.26 (bt, J = 6.4 Hz, 4H), 1.60 (bt, J = 6.4 Hz, 4H); 13C NMR (100 MHz, DMSO-d6) δ 155.4, 139.9, 134.2, 132.2, 129.9, 114.7, 32.7, 26.5; MS (EI) m/z 266 (M+, 100), 223, 168, 141, 115. Anal. (C18H18O2) C, H.
Bis(4-hydroxyphenyl)methylenecyclohexane (4)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 500 mg, 2.33 mmol), cyclohexanone (229 mg, 2.33 mmol), Zn (1.2 g, 18 mmol) and titanium(IV) chloride (1.64 g, 8.62 mmol), 4 was obtained as a white solid (518 mg, 83%): mp 235–237 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.83 (d, J = 8.4 Hz, 4H), 6.66 (d, J = 8.8 Hz, 4H), 2.10–2.21 (m, 4H), 1.42–1.60 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ 155.6, 136.3, 133.9, 133.7, 130.4, 114.7, 31.9, 28.2, 26.3; MS (EI) m/z 280 (M+), 199 (100), 107. Anal. (C19H20O2) C, H.
Bis(4-hydroxyphenyl)methylenecycloheptane (5)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 500 mg, 2.33 mmol), cyclohexanone (261 mg, 2.33 mmol), Zn (1.2 g, 18 mmol) and titanium(IV) chloride (1.64 g, 8.62 mmol), 5 (518 mg, 83%) was obtained as a white solid: mp 198–199 °C; 1H NMR (400 MHz, acetone-d6) δ 6.96 (d, J = 8.8 Hz, 4H), 6.74 (d, J = 8.8 Hz, 4H), 2.26–2.34 (m, 4H), 1.50–1.61 (m, 8H); 13C NMR (100 MHz, acetone-d6) δ 156.3, 139.0, 138.2, 136.1, 131.0, 115.5, 33.9, 29.9, 28.8; MS (EI) m/z 294 (M+, 100), 237, 199, 107. HRMS (EI) m/z calcd for C20H22O2 294.1620, found 294.1618. Anal. (C20H22O2) C, H.
4-Hydroxyhexanone (7)
In a three-necked round bottomed-flask (500 mL) fitted with a mechanical stirrer, cyclohexane-1,4-diol (5.8 g, 50 mmol) was dissolved to 200 mL of acetone. The solution was cooled in an ice bath, and freshly prepared Jones reagent (1.6 M in acetone) was added over 25 min. The green-blue solution was allowed to warm to RT over 15 min. The reaction mixture was filtered through Celite and then evaporated. Flash column chromatography (Et2OAc/hexane; 7:3) gave 7 (3.0 g, 53 %) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.17–4.23 (m, 1H), 2.55–2.66 (m, 2H), 2.25–2.36 (m, 2H), 1.91–2.11 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 211.0, 66.3, 37.1, 33.7. Registry No. 13482-22-9.
4-Acetoxycyclohexanone (8)
To a solution of 7 (2.0 g, 18 mmol) in CH2Cl2 (20 mL) was added acetic anhydride (2.1 g, 21 mmol). Pyridine was added dropwise to the reaction mixture, which was stirred for 24 h at RT. The solvent was evaporated under reduced pressure and the residual oil was dissolved in Et2OAc (100 mL). The Et2OAc layer was washed with 2 M HCl solution (100 mL), saturated NaHCO3 solution (100 mL) and brine (100 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. Flash column chromatography (Et2OAc/hexane; 4:6) gave 8 (1.5 g, 56%) as an oil: 1H NMR (400 MHz, CDCl3) δ 5.15 (m, 1H), 2.49–2.57 (m, 2H), 2.32–2.37 (m, 2H), 2.09 (s, 3H), 2.03–2.08 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 21.2, 30.7, 37.2, 170.4, 209.8. Registry No. 41043-88-3.
Methyl (4-Oxocyclohexylidene)acetate
A mixture of 1,4-cyclohexanedione (4.7 g, 42 mmol) and methyl (triphenylphosphoranylidene)acetate(7.0 g, 21 mmol) in toluene (100 mL) was heated to 110 °C for 8 h. After the reaction was cooled to RT, the solvent was evaporated. Et2O (200 mL) was added and the precipitate was filtered. The filtered organic layer was concentrated, and the residual oil was absorbed on silica gel. Flash column chromatography (Et2OAc/hexane; 4:6) gave methyl (4-oxocyclohexylidene)acetate (3.1 g, 88 %) as a white solid: 1H NMR (500 MHz, CDCl3) δ 5.85 (bt, J = 1.5 Hz, 1H), 3.72 (s, 3H), 3.21 (td, J = 7.5, 2.0 Hz, 2H), 2.66 (td, J = 6.0, 1.5 Hz, 2H), 2.46–2.53 (m, 2H). Registry No. 91158-10-0
Methyl (4-Oxocyclohexyl)acetate (10)
To a solution of methyl (4-oxocyclohexylidene)acetate (2.97 g, 1.77 mmol) in ethanol (10 mL) was added 10% palladium charcoal (300 mg), and it was placed in a hydrogenation system. The reaction mixture was stirred at RT for 6 h. The solution was filtered through Celite and concentrated under reduced pressure. Flash column chromatography (Et2OAc/hexane; 2:8) gave 10 (1.5 g, 50 %) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 3H), 2.42–2.35 (m, 4H), 2.22–2.35 (m, 2H), 2.04–2.13 (m, 2H), 1.04–1.54 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 211.2, 172.8, 51.6, 40.5, 40.0, 33.1, 32.4. Registry No. 66405-41-2
Methyl 3-Oxocyclohexanecarboxylate (12)
To a solution of 3-oxo-1-cyclohexanecarboxylic acid (1.0 g, 7.03 mmol) in methanol (12 mL) was added thionyl chloride (1.0 g, 8.4 mmol), slowly, at RT. The reaction mixture was stirred for 3 h and then quenched slowly by three drops of H2O in an ice bath. The reaction was neutralized by saturated NaHCO3 solution and the organic layer was extracted with Et2OAc (30 mL). The organic layer was washed by H2O (10 mL) and dried over Na2SO4. After removal of solvent, flash column chromatography (Et2OAc/hexane; 2:8) gave 12 (0.803 g, 73%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 3H), 2.75–2.84 (m, 1H), 2.53 (d, J = 8.0 Hz, 2H), 2.25–2.40 (m, 2H), 2.00–2.14 (m, 2H), 1.76–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 209.1, 174.1, 52.0, 43.1, 43.0, 40.9, 27.7, 24.4. Registry No. 13148-83-9
Dimethyl [2-(3-Oxocyclohexyl)]malonate (14)
To a solution of 2-cyclohexen-1-one (2.9 g, 30 mmol) and dimethyl malonate (4.4 g, 33 mmol) in THF (20 mL) was added potassium tert-butoxide (330 mg, 3.0 mmol). The reaction mixture was stirred at RT for 2 h. Water (5 mL) was added to the reaction mixture, and then more water (200 mL) was added. The organic layer was extracted with Et2OAc (100 mL) and dried over MgSO4. Et2OAc was evaporated under reduced pressure, and flash column chromatography (Et2OAc/hexane; 3:7) gave 14 (5.8 g, 85%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.75 (s, 3H), 3.74 (s, 3H), 3.34 (d, J = 8.0 Hz, 1H), 2.48–2.59 (m, 1H), 2.36–2.46 (m, 2H), 2.20–2.30 (m, 2H), 2.02–2.12 (m, 1H), 1.89–1.97 (m, 1H), 1.60–1.75 (m, 1H), 1.44–1.56 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 209.5, 168.25, 168.16, 56.6, 53.0, 45.1, 41.0, 38.1, 28.8, 24.5. Registry No. 33646-18-3
3-(3-Acetoxy-n-propyl)cyclohexanone (15)
3-Chloropropanol (1.9 g, 20 mmol) was added to a two-necked round-bottomed flask containing freshly distilled THF (40 mL) and the reaction was cooled down to −78 °C under nitrogen. Methyl magnesium chloride (6.8 mL, 20 mmol, 22 w/w % in THF) solution was added slowly by syringe. The cooling was removed and stirring was continued at RT for 20 min. After addition of Mg (583 mg, 24 mmol), the reaction mixture was heated at 90 °C and 1,2-dibromoethane (37 μL, 0.48 mmol) was added. The solution was refluxed for 1 h. After cooling, the reaction mixture was slowly added dropwise over 30 min by cannulus to a one-necked flask containing 2-cyclohexen-1-one (1.94 mL, 20 mmol) and copper(I) bromide (286 mg, 2 mmol) in THF at −78 °C. As soon as the addition was finished, the reaction temperature was increased to 0 °C and decreased again to −78 °C. The reaction was quenched by HCl, dissolved in ether (1.0 M, 40 mL) and saturated ammonium chloride (15 mL). The reaction temperature was increased to RT. The mixture was filtered and the solvent was evaporated under reduced pressure. The residual oil was dissolved with EtOAc (100 mL) and washed with ammonium chloride solution (100 mL) and brine (100 mL). The organic layer was dried by sodium sulfate and flash column chromatography (Et2OAc/hexane; 6:4) gave 3-(3-hydroxy-n-propyl)cyclohexanone (590 mg, 19%) as on oil: 1H NMR (400 MHz, CDCl3) δ 3.57 (d, J = 6.0 Hz, 2H), 2.15–2.52 (m, 4H), 1.92–2.10(m, 2H), 1.82–1,92(m, 1H), 1.69–1.82 (m, 1H), 1.46–1.68 (m, 3H), 1.24–1.67 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 212.2, 62.5, 48.0, 41.3, 38.8, 32.5, 31.1, 29.6, 25.1. Registry No.69441-81-2. Purified 3-(3-hydroxy-n-propyl)cyclohexanone (580 mg, 3.71 mmol) was stirred with acetic anhydride (450 μL, 4.83 mmol) and pyridine (780 mL, 9.65 mmol) in CH2Cl2 (15 mL) at RT. After stirring for 18 h, CH2Cl2 (50 mL) was added and the reaction was placed in a separatory funnel. The organic layer was washed with a saturated NaHCO3 solution and saturated ammonium chloride solution, dried over Na2SO4, filtered and concentrated. Flash column chromatography (Et2OAc/hexane; 3:7) gave 15 (329 mg, 45%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.03 (t, J = 6.4 Hz, 2H), 2.38–2.46 (m, 1H), 2.31–2.38 (m, 1H), 2.18–2.29 (m, 1H), 2.18–2.09 (m, 2H), 2.03 (s, 3H), 1.85–1.94 (m, 1H), 1.71–1.83(m, 1H), 1.57–1.70 (m, 3H), 1.27–1.45 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 211.5, 171.1, 64.3, 48.0, 41.2, 38.7, 32.7, 31.1, 25.8, 25.1, 20.9; MS (EI) m/z 198 (M+), 155, 138, 110, 97 (100), 84, 67. HRMS (EI) m/z calcd for C11H18O3 198.1256, found 198.1253.
4-Acetoxy-1-[bis(4-hydroxyphenyl)methylene]cyclohexane (16)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 1.17 g, 5.4 mmol), 8 (850 mg, 5.4 mmol), Zn (2.7 g, 41 mmol) and titanium(IV) chloride (3.8 g, 20.1 mmol), 16 (1.5 g, 83%) was obtained as a white solid: mp 208–210 °C; 1H NMR (400 MHz, acetone-d6) δ 6.93 (d, J = 8.4 Hz, 4H), 6.75 (d, J = 8.4 Hz, 4H), 4.90 (septet, J = 4.0 Hz, 1H), 2.55–2.45 (m, 2H), 2.11–2.21 (m, 2H), 1.93 (s, 3H) 1.92–1.83 (m, 2H), 1.62–1.51 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 169.9, 155.8, 135.2, 133.8, 133.4, 130.3, 114.7, 72.2, 32.2, 28.1, 21.1; MS m/z (EI) 338 (M+), 278 (100). HRMS (EI) m/z calcd for C21H22O4 338.1518, found 338.1517.
4-Acetoxy-1-[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (17a)
According to the general procedure for MOM protection with 16 (1.5 g, 4.4 mmol), methoxymethyl chloride (779 mg, 9.7 mmol), sodium hydride (380 mg, 10 mmol) and DMF (15 mL), 17a (1.5 g, 78%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 9.0 Hz, 4H), 6.95 (d, J = 9.0 Hz, 4H), 5.15–5.18 (m, 4H), 5.03–4.93 (m, 1H), 3.46–3.50 (m, 6H), 2.51–2.56 (m, 2H), 2.13–2.18 (m, 2H), 2.04–2.07 (m, 3H), 1.89–1.92 (m, 2H), 1.61–1.64 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 170.6, 155.7, 136.5, 135.5, 135.0, 130.7, 115.6, 94.4, 71.9, 56.0, 32.6, 28.5, 21.4; MS (EI) m/z 426 (M+), 366 (100). HRMS (EI) m/z calcd for C25H30O6 426.2042, found 426.2036.
4-[Bis(4-methoxymethoxyphenyl)methylene]cyclohexanol (17b)
Compound 16 (1.4 g, 3.3 mmol) was dissolved in methanol (15 mL) and H2O (3 mL). After addition of potassium carbonate (2.3 g, 16 mmol), the reaction mixture was stirred at RT for 12 h. The reaction mixture was slowly added to a two-phase solution of Et2OAc (100 mL) and H2O (100 mL) with stirring, and neutralized by 2 M HCl solution. The organic layer was collected and dried over sodium sulfate. After removal of the solvent, flash column chromatography (Et2OAc/hexane; 4:6) gave 17b (1.2 g, 95 %) as a white solid: mp 74–76 °C; 1H NMR (500 MHz, CDCl3) δ 7.02 (d, J = 9.0 Hz, 4H), 6.94 (d, J = 9.0 Hz, 4H), 5.15 (s, 4H), 3.89 (septet, J = 4.5 Hz, 1H), 3.48 (s, 6H), 2.57 (dt, J = 11.2, 4.3 Hz, 2H), 2.01–2.11 (m, 2H), 1.98–1.88 (m, 2H), 1.54–1.42 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.7, 136.7, 136.1, 134.7, 130.7, 115.6, 94.4, 69.7, 56.0, 36.3, 28.7; MS m/z (EI) 384 (M+, 100), 366. HRMS (EI) m/z calcd for C23H28O5 384.1937, found 384.1928.
4-[Bis(4-hydroxyphenyl)methylene]cyclohexanol (18)
According to the general procedure for MOM deprotection with 17b (120 mg, 0.31 mmol), HCl (200 μL) and methanol (10 mL), 18 (78 mg, 85%) was obtained as an off-white solid: mp 209 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.82 (d, J = 8.4 Hz, 4H), 6.66 (d, J = 8.8 Hz, 4H), 3.60–3.70 (m, 1H), 2.34–2.44 (m, 2H), 1.90–2.00 (m, 2H), 1.70–1.82 (m, 2H), 1.24–1.36 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 155.6, 135.3, 134.2, 133.7, 130.4, 114.7, 67.6, 36.3, 28.5; MS (EI) m/z 296 (M+, 100), 278, 237, 199, 107. HRMS (EI) m/z calcd for C19H20O3 296.1412 found 296.1419. Anal. (C19H20O3·0.2 H2O) C, H.
4-Fluoro[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (19)
In a one-necked round-bottomed flask, which contained 17b (200 mg, 0.52 mmol), CH2Cl2 (5 mL) was added under nitrogen, and the reaction mixture was cooled to −78 °C. Diethylaminosulfur trifluoride (120 mg, 0.62 mmol) was dropped very slowly by syringe at −78 °C. The cooling was removed, and the reaction mixture was stirred for 1 h at RT. The reaction mixture was cooled again to −78 °C and methanol (200 μL) was added. Additional stirring was followed for 30 min after removal of the Dry-ice bath, the reaction was quenched by saturated NaHCO3 solution and extracted with excess Et2OAc. The organic layer was dried over sodium sulfate and concentrated. Flash column chromatography (Et2OAc/hexane; 1:9) gave 19 (65 mg, 32%) as a slightly yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.5 Hz, 4H), 6.94 (d, J = 8.5 Hz, 4H), 5.16 (s, 4H), 4.81 (dm, 2JHF = 48.5 Hz, 1H), 3.48 (s, 6H), 2.45–2.52 (m, 2H), 2.16–2.25 (m, 2H), 1.78–1.93 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 155.7, 136.5, 135.4, 135.1, 130.7, 115.7, 94.4, 90.5 (d, 1J CF = 168.6 Hz), 56.0, 33.3 (d, 2JCF = 19.3 Hz), 27.3 (d, 3JCF = 6.4 Hz); MS (EI) m/z 386 (M+, 100). HRMS (EI) m/z calcd for C23H27O4F 386.1893, found 386.1889.
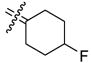
4-Fluoro[bis(4-hydroxyphenyl)methylene]cyclohexane (20)
According to the general procedure for MOM deprotection with 19 (45 mg, 0.12 mmol), HCl (100 μL) and methanol (4 mL), 20(25 mg, 71%) was obtained as an off-white solid: mp 202–203 °C; 1H NMR (500 MHz, acetone-d6) δ 6.93 (d, J = 9.0 Hz, 4H), 6.75 (d, J = 8.5 Hz, 4H), 4.78 (dm, 2JHF = 49.0 Hz, 1H), 2.41–2.48 (m, 2H), 2.16–2.24 (m, 2H), 1.82–1.95 (m, 2H), 1.69–1.79 (m, 2H); 13C NMR (125 MHz, acetone-d6) δ 156.79, 136.72, 135.23, 135.05, 131.48, 115.55, 90.53 (d, 1J CF = 168.6 Hz), 34.05 (d, 2JCF = 19.3 Hz), 28.11 (d, 3JCF = 6.38 Hz); MS (EI) m/z 298 (M+), 184, 141, 128, 115, 77 (100). HRMS (EI) m/z calcd for C19H19O2F 298.1369, found 298.1362. Anal. (C19H19O2F·0.1 H2O) C, H.
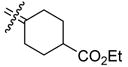
Ethyl 4-[Bis(4-hydroxyphenyl)methylene]cyclohexanecarboxylate (21a)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 2.1 g, 10 mmol), ethyl 4-oxocyclohexanecarboxylate (1.7 g, 10 mmol), Zn (4.9 g, 75 mmol) and titanium(IV) chloride (7.0 g, 37 mmol), 21a (2.7 g, 77%) was obtained as a white solid: mp 198–200 °C; 1H NMR (400 MHz, acetone-d6) δ 7.01 (d, J = 8.5 Hz, 4H), 6.94 (d, J = 8.5 Hz, 4H), 4.08 (q, J = 7.2 Hz, 2H), 2.49–2.64 (m, 3H), 1.90–2.10 (m, 4H), 1.50–1.64 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, acetone-d6) δ 206.2, 175.4, 156.7, 136.2, 135.9, 135.2, 131.5, 115.5, 60.5, 43.6, 31.3, 29.6,14.5; MS (EI) m/z 352 (M+, 100), 278, 237, 199, 107. HRMS (EI) m/z calcd for C22H24O4 352.1675, found 352.1677. Anal. (C22H24O4·0.2H2O) C, H.
Ethyl 4-[Bis(4-methoxymethylphenyl)methylene]cyclohexanecarboxylate (22a)
According to the general procedure for MOM protection with 21a (2.1 g, 6.0 mmol), methoxymethyl chloride (1.6 g, 20 mmol), sodium hydride (440 mg, 13 mmol) and DMF (15 mL), 22a (2.5 g, 95% yield) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 4H), 6.93 (d, J = 8.4 Hz, 4H), 5.15 (s, 4H), 4.13 (q, J = 7.2 Hz, 2H), 3.48 (s, 6H), 2.60 (bd, J = 12.8 Hz, 2H), 2.48–2.68 (m, 1H), 1.92–2.06 (m, 4H), 1.54–1.69 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.6, 155.6, 136.5, 136.1, 134.7, 130.7, 115.6, 94.4, 60.2, 56.0, 43.1, 30.7, 30.5, 14.2; MS (EI) m/z 440 (M+, 100). HRMS (EI) m/z calcd for C26H32O6 440.2199, found 440.2204.
4-Hydroxymethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (23a)
According to the general procedure for reduction with 22a (2.5 g, 5.7 mmol), LiAlH4 in THF solution (1 M, 5.7 mL) and THF (15 mL), 23a (2.0 g, 89%) was obtained as a white foam: mp 72 °C; 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.8 Hz, 4H), 6.94 (d, J = 8.8 Hz, 4H), 5.15 (s, 4H), 3.49 (d, J = 6.4 Hz, 2H), 3.48 (s, 6H), 2.65 (bd, J = 13.6 Hz, 2H), 1.90–2.01 (m, 2H), 1.81–1.90 (m, 2H), 1.63–1.80 (m, 1H), 1.03–1.18 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.5, 137.7, 136.8, 133.9, 130.8, 115.5, 94.4, 68.0, 56.0, 40.4, 31.1, 31.0; MS (EI) m/z 398 (M+), 303 (100), 247, 111. HRMS (EI) m/z calcd for C24H30O5 398.2093, found 398.2101.
4-Hydroxymethyl[bis(4-hydroxyphenyl)methylene]cyclohexane (24a)
According to the general procedure for MOM deprotection with 23a (100 mg, 0.25 mmol), HCl (300 μL) and methanol (4 mL), 24a (69 mg, 90%) was obtained as a white solid: mp 211–212 °C; 1H NMR (400 MHz, acetone-d6) δ 6.91 (d, J = 8.8 Hz, 4H), 6.74 (d, J = 8.8 Hz, 4H), 3.38 (d, J = 6.0 Hz, 2H), 2.62 (bd, J= 13.6 Hz, 2H), 1.80–1.98 (m, 4H), 1.61–1.73 (m, 1H), 1.02–1.15 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ 156.6, 137.7, 135.6, 135.4, 131.5, 115.4, 67.7, 41.6, 32.1, 32.0; MS (EI) m/z 310 (M+), 256 (100), 225, 199, 107, 64. HRMS (EI) m/z calcd for C20H22O3 310.1569, found 310.1562. Anal. (C20H22O3) C, H.
4-Methanesulfonyloxymethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (25a)
According to the general procedure for methanesulfonylation with 24a (90 mg, 0.23 mmol), methanesulfonic anhydride (94 mg, 0.54 mmol), triethylamine (68 mg, 0.68 mmol) and CH2Cl2 (5 mL), 25a (102 mg, 95%) was obtained as an off-white solid: mp 79–80 °C; 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 9.0 Hz, 4H), 6.94 (d, J = 9.5 Hz, 4H), 5.15 (s, 4H), 4.08 (d, J = 6.5 Hz, 2H), 3.48 (s, 6H), 3.00 (s, 3H), 2.66 (bd, J = 13.5 Hz, 2H), 1.97–2.04 (m, 3H), 1.84–1.96 (m, 2H), 1.14–1.24 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.6, 136.5, 136.4, 134.7, 130.7, 115.6, 94.4, 74.1, 55.9, 37.4, 37.1, 30.7, 30.5; MS (EI) m/z 476 (M+, 100), 446, 41, 380, 335. HRMS (EI) m/z calcd for C25H32O7S 476.1869, found 476.1873.
4-Fluoromethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
According to the general procedure for fluorination with 25a (167 mg, 0.35 mmol), cesium fluoride (266 mg, 1.8 mmol), H2O (50 μL) in 1-butyl-3-methyl-imidazolium tetrafluoroboronate (1.5 mL) and acetonitrile (1.5 mL), 4-fluoromethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (89 mg, 63%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.02 (d, J = 9.0 Hz, 4H), 6.94 (d, J = 9.0 Hz, 4H), 5.16 (s, 4H), 4.26 (dd, 2JHF = 47.5 Hz, J = 6.0 Hz, 2H) 3.48 (s, 6H), 2.66 (bd, J = 13.5 Hz, 2H), 1.89–2.02 (m, 3H), 1.81–1.89 (m, 2H), 1.12–1.24 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.6, 137.1, 136.7, 134.4, 130.8, 115.6, 94.5, 88.0 (d, 1J CF = 166.6 Hz), 56.0, 38.5 (d, 2JCF = 18.3 Hz), 30.9, 30.0 (d, 3JCF = 5.5 Hz); MS (EI) m/z 400 (M+, 100), 355, 149. HRMS (EI) m/z calcd for C24H29O4F 400.2050, found 400.2045.
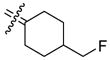
4-Fluoromethyl[bis(4-hydroxyphenyl)methylene]cyclohexane (26a)
According to the general procedure for MOM deprotection with 4-fluoromethyl[bis(4-methoxymethoxyphenyl)methylene]-cyclohexane (89 mg, 0.22 mmol), HCl (100 μL) and methanol (8 mL), 26a (38 mg, 55%) was obtained as a white solid: mp 201–202 °C; 1H NMR (500 MHz, acetone-d6) δ 6.92 (d, J = 8.5 Hz, 4H), 6.75 (d, J = 8.5 Hz, 4H), 4.28 (dd, 2JHF = 47.5 Hz, J = 6.0 Hz, 2H), 2.64 (bd, J = 13.5 Hz, 2H), 1.86–2.01 (m, 3H), 1.78–1.85 (m, 2H), 1.12–1.22 (m, 2H); 13C NMR (125 MHz, acetone-d6) δ 156.7, 136.8, 135.9, 135.4, 131.5, 115.5, 88.5 (d, 1J CF = 165.8 Hz), 39.4 (d, 2JCF = 18.3 Hz), 31.6, 30.7 (d, 3JCF = 5.5 Hz); MS (EI) m/z 312 (M+, 100), 199, 149, 107. HRMS (EI) m/z calcd for C20H21O2F 312.1526, found 312.1524. Anal. (C20H21O2F) C, H.
4-Ethoxymethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
To a solution of 23a (200 mg, 0.49 mmol) in DMF (7 mL), which was contained in a pressure tube, was added sodium hydride (35 mg, 0.97 mmol) under N2. After stirring for 10 min, bromoethane (158 mg, 0.15 mmol) was added. The pressure tube was tightly capped, and the temperature of the reaction mixture was increased to 100 °C. The mixture was stirred for 24 h and cooled down. The mixture was poured into H2O (50 mL) and Et2OAc (100 mL) solution. The extracted organic layer was washed with saturated ammonium chloride (100 mL x 2) solution and saturated brine (100 mL), and dried (MgSO4). The organic layer was concentrated and flash column chromatography (Et2OAc/hexane; 15:85) gave 4-ethoxymethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (74 mg, 36%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.8 Hz, 4H), 6.93 (d, J = 8.8 Hz, 4H), 5.15 (s, 4H), 3.44–3.52 (m, 7H), 3.27 (d, J = 6.0 Hz, 2H), 2.62 (bd, J = 13.6 Hz, 2H), 1.76–2.01 (m, 5H), 1.20 (t, J = 6.8 Hz, 3H), 1.14–1.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.5, 138.0, 136.9, 133.8, 130.8, 115.5, 94.4, 75.9, 66.4, 56.0, 38.1 31.7, 31.2, 15.2; MS (EI) m/z 426 (M+, 100), 302, 287, 149; HRMS (EI) m/z calcd for C26H34O5 426.2406, found 426.2400.
4-Ethoxymethyl[bis(4-hydroxyphenyl)methylene]cyclohexane (27)
According to the general procedure for MOM deprotection with 4-ethoxymethyl[bis(4-methoxymethoxyphenyl)-methylene]cyclohexane. (74 mg, 0.17 mmol), HCl (200 μL) and methanol (5 mL), 27 (43 mg, 73%) was obtained as a white crystal: mp 209–210 °C; 1H NMR (400 MHz, acetone-d6) δ 6.91 (d, J = 8.8 Hz, 4H), 6.74 (d, J = 8.8 Hz, 4H), 3.41 (q, J = 6.8Hz, 2H), 3.23 (d, J = 6.0 Hz, 2H), 2.61 (bd, J = 13.6 Hz, 2H), 1.87–1.98 (m, 2H), 1.71–1.86 (m, 3H), 1.03–1.16 (m, 5H); 13C NMR (100 MHz, acetone-d6) δ 156.5, 137.5, 135.5, 135.4, 131.5, 115.4, 76.3, 66.7, 39.1, 32.4, 31.9, 15.5; MS (EI) m/z 338 (M+, 100), 199, 149. HRMS (EI) m/z calcd for C22H26O3 338.1882, found 338.1887. Anal. (C22H26O3) C, H.
4-Methyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
According to the general procedure for reduction with 25a (300 mg, 0.63 mmol), LiAlH4 in THF solution (1 M, 900 μL) and THF (8 mL), 4-methyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (240 mg, 99%) was obtained as a oil: 1H NMR (400 MHz, CDCl3) δ 7.03 (d, J = 8.8 Hz, 4H), 6.94 (d, J = 8.8 Hz, 4H), 5.16 (s, 4H), 3.49 (s, 6H), 2.59 (bd, J = 13.6 Hz, 2H), 1.94 (td, J = 12.8, 4.0 Hz, 2H), 1.72–1.80 (m, 2H), 1.55–1.68 (m, 1H), 1.01–1.14 (m, 2H), 0.93 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 155.4, 138.2, 137.0, 133.4, 130.8, 115.5, 94.4, 56.0, 36.8, 32.7, 31.7, 22.0; MS (EI) m/z 382 (M+,100). HRMS (EI) m/z calcd for C24H30O4 382.2144, found 382.2135.
4-Methyl[bis(4-hydroxyphenyl)methylene]cyclohexane (29a)
According to the general procedure for MOM deprotection with 4-methyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (240 mg, 0.63 mmol), HCl (200 μL), methanol (5 mL) and Et2O (2.5 mL), 29a (152 mg, 82%) was obtained as an off-white solid: mp 183–184 °C; 1H NMR (400 MHz, methanol-d4) δ 6.86 (d, J = 8.8 Hz, 4H), 6.66 (d, J = 8.8 Hz, 4H), 2.57 (bd, J = 14.0 Hz, 2H), 1.94 (td, J = 13.4, 3.6 Hz, 2H), 1.69–1.79 (m, 2H), 1.53–1.67 (m, 1H), 0.97–1.10 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 156.7, 137.9, 136.2, 135.9, 131.9, 115.5, 38.0, 34.1, 32.8, 22.5; MS (EI) m/z 294 (M+, 100), 199, 107. Anal. (C20H22O2·0.1 H2O) C, H.
Methyl {4-[Bis(4-hydroxyphenyl)methylene]cyclohexyl}acetate (21b)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 957 mg, 4.5 mmol), 10 (760 mg, 4.5 mmol), Zn (2.2 g, 34 mmol) and titanium(IV) chloride (3.1 g, 17 mmol), 21b (1.1 g, 71%) was obtained as a white solid: mp 201–203 °C; 1H NMR (400 MHz, acetone-d6) δ 6.91 (d, J = 8.8 Hz, 4H), 6.74 (d, J = 8.8 Hz, 4H), 3.60 (s, 3H), 2.59 (bd, J = 13.6 Hz, 2H), 2.23 (d, J = 6.8 Hz, 2H), 1.89–2.02 (m, 3H), 1.76–1.85 (m, 2H), 1.06–1.18 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ 173.3, 156.6, 136.8, 135.7, 135.4, 131.5, 115.4, 51.4, 41.5, 35.7, 35.0, 32.0; MS (EI) m/z 352 (M+), 166, 107 (100), 77. HRMS (EI) m/z calcd for C22H24O4 352.1675, found 352.1670. Anal. (C22H24O4) C, H.
Methyl {4-[Bis(4-methoxymethoxyphenyl)methylene]cyclohexyl}acetate (22b)
According to the general procedure for MOM protection with 21b (980 mg, 2.8 mmol), methoxymethyl chloride (736 mg, 9.1 mmol), sodium hydride (309 mg, 9.1 mmol) and DMF (15 mL), 22b (1.2 g, 97%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.00 (d, J = 9.0 Hz, 4H), 6.92 (d, J = 8.5 Hz, 4H), 5.15 (s, 4H), 3.66 (s, 3H), 3.48 (s, 6H), 2.60 (bd, J = 14.0 Hz, 2H), 2.24 (d, J = 7.5 Hz, 2H), 1.92–2.08 (m, 3H), 1.78–1.86 (m, 2H), 1.08–1.19 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 173.5, 155.6, 137.2, 136.8, 134.1, 130.8, 115.6, 94.4, 56.0, 51.4, 41.2, 34.8, 34.4, 31.3; MS (EI) m/z 440 (M+), 320, 210, 180, 151, 121 (100), 84. HRMS (EI) m/z calcd for C26H32O6 440.2199, found 440.2196.
4-(2-Hydroxyethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (23b)
According to the general procedure for reduction with 22b (1.15 g, 2.6 mmol), LiAlH4 in THF solution (1 M, 2.6 mL) and THF (10 mL), 23b (1.0 g, 96%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.5 Hz, 4H), 6.93 (d, J = 9.0 Hz, 4H), 5.15 (s, 4H), 3.70 (t, J = 6.5 Hz, 2H), 3.48 (s, 6H), 2.60 (bd, J = 14.0 Hz, 2H), 1.89–1.99 (m, 2H), 1.79–1.85 (m, 2H), 1.62–1.71 (m, 1H), 1.52 (q, J = 6.5 Hz, 2H), 1.05–1.15 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.5, 138.0, 136.9, 136.7, 130.8, 115.6, 94.5, 60.9, 56.00, 55.99, 39.6, 34.8, 34.2, 31.6; MS (EI) m/z 412 (M+, 100). HRMS (EI) m/z calcd for C25H32O5 412.2250, found 412.2246.
4-(2-Hydroxyethyl)[bis(4-hydroxyphenyl)methylene]cyclohexane (24b)
According to the general procedure for MOM deprotection with 23b (45 mg, 0.11 mmol), HCl (200 μL) and methanol (5 mL), 24b (30 mg, 84%) was obtained as a white solid: mp 183 °C; 1H NMR (400 MHz, acetone-d6) δ 6.91 (d, J = 8.8 Hz, 4H), 6.74 (d, J = 6.8 Hz, 4H), 3.58 (bt, J = 6.4 Hz, 2H), 2.59 (bd, J = 13.2 Hz, 2H), 1.94 (td, J = 13.2, 4.4 Hz, 2H), 1.77–1.86 (m, 2H), 1.61–1.75 (m, 1H), 1.44 (q, J = 6.8 Hz, 2H), 1.00–1.13 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ 156.5, 137.7, 135.6, 135.2, 131.6, 115.4, 60.3, 40.6, 35.6, 35.1, 32.3; MS (EI) m/z 324 (M+, 100), 137, 199, 107. HRMS (EI) m/z calcd for C21H24O3 324.1725, found 324.1726. Anal. (C21H24O3) C, H.
4-(2-Methanesulfonyloxyethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (25b)
According to the general procedure for methanesulfonylation with 24b (448 mg, 1.1 mmol), methanesulfonic anhydride (284 mg, 1.6 mmol), triethylamine (331 mg, 3.3 mmol) and CH2Cl2 (6 mL), 25b (500 mg, 93%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.00 (d, J = 8.8 Hz, 4H), 6.93 (d, J = 8.8 Hz, 4H), 5.15 (s, 4H), 4.28 (t, J = 6.4 Hz, 2H), 3.48 (s, 6H), 3.30 (s, 3H), 2.61 (bd, J = 13.6 Hz, 2H), 1.95 (td, J = 13.2, 3.6 Hz, 2H), 1.78–1.87 (m, 2H), 1.65–1.75 (m, 3H), 1.05–1.17 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.6, 137.2, 137.0, 134.1, 130.8, 115.6, 94.4, 68.2, 56.0, 37.4, 35.7, 34.3, 34.0, 31.3; MS (EI) m/z 490 (M+), 446, 427 (100). HRMS (EI) m/z calcd for C26H34O7S 490.2025, found 490.2024.
4-(2-Fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
According to the general procedure for fluorination with 25b (150 mg, 0.31 mmol), cesium fluoride (235 mg, 1.6 mmol), H2O (20 μL), 1-butyl-3-methyl-imidazolium tetrafluoroboronate (1.0 mL) and acetonitrile (1.0 mL), 4-(2-fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (106 mg, 82%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.03 (d, J = 8.5 Hz, 4H), 6.95 (d, J = 8.5 Hz, 4H), 5.16 (s, 4H), 4.51 (dt, 2JHF = 47 Hz, J = 6.0 Hz, 2H), 3.49 (s, 6H), 2.63 (bd, J = 13.5 Hz, 2H), 1.96 (td, J = 13.0, 3.5 Hz, 2H), 1.81–1.90 (m, 2H), 1.60–1.78 (m, 3H), 1.07–1.18 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.5, 137.7, 136.8, 133.8, 130.8, 115.5, 94.4, 82.4 (d, 1J CF = 163.0 Hz), 55.94, 55.91, 37.0 (d, 2JCF = 19.3 Hz), 34.5, 34.0 (d, 3JCF = 4.6 Hz), 31.4; MS (EI) m/z 414 (M+, 100), 149. HRMS (EI) m/z calcd for C25H31O4F 414.2206, found 414.2202.
4-(2-Fluoroethyl)[bis(4-hydroxyphenyl)methylene]cyclohexane (26b)
According to the general procedure for MOM deprotection with 4-(2-fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (100 mg, 0.24 mmol), HCl (200 μL) and methanol (5 mL), 26b (66 mg, 84%) was obtained as a white solid: mp 192–193 °C; 1H NMR (500 MHz, acetone-d6) δ 6.91 (d, J = 8.5 Hz, 4H), 6.74 (d, J = 8.5 Hz, 4H), 4.49 (dt, 2JHF = 48.0 Hz, J = 6.0 Hz, 2H), 2.60 (bd, J = 14.0 Hz, 2H), 1.94 (td, J = 13.5, 3.5 Hz, 2H), 1.80–1.87 (m, 2H), 1.64–1.74 (m, 1H), 1.62 (dq, 3JHF = 25.5 Hz, J = 6.5 Hz, 2H), 1.05–1.17 (m, 2H); 13C NMR (125 MHz, acetone-d6) δ 156.6, 137.3, 135.54, 135.46, 131.6, 115.5, 82.9 (d, 1J CF = 162.0 Hz), 37.8 (d, 2JCF = 19.3 Hz), 35.3, 34.96 (d, 3JCF = 4.6 Hz), 32.2; MS (EI) m/z 326 (M+), 168, 141, 115, 77, 57 (100). HRMS (EI) m/z calcd for C21H23O2F 326.1682, found 326.1680. Anal. (C21H23O2F·0.1 H2O) C, H.
4-[2-(4-Fluorobutoxy)ethyl][bis(4-methoxymethoxyphenyl)methylene]cyclohexane
To a solution of 23b (120 mg, 0.29 mmol) in DMF (4 mL) was added sodium hydride (15 mg, 0.44 mmol) at 0 °C under nitrogen. After addition of 1-bromo-4-fluorobutane (54 mg, 0.35 mmol), the reaction mixture was stirred at 100 °C for 24 h and cooled to RT. The mixture was poured into H2O (50 mL) and Et2OAc (100 mL) solution, and the organic layer was extracted. This was washed with saturated ammonium chloride (100 mL x 2) solution and saturated brine (100 mL), and dried over MgSO4. After removal of the solvent, flash column chromatography (Et2OAc/hexane; 3:7) gave 4-[2-(4-fluorobutoxy)ethyl][bis(4-methoxymethoxyphenyl)methylene]cyclohexane (37 mg, 26%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.01(d, J = 9.0 Hz, 4H), 6.93 (d, J = 9.0 Hz, 4H), 5.15 (s, 4H), 4.45 (dt, 2JHF = 47.0 Hz, J = 6.0 Hz, 2H) 3.48 (s, 6H), 3.46 (t, J = 6.0 Hz, 2H), 3.44 (t, J = 6.0 Hz, 2H), 2.59 (bd, J = 13.5 Hz, 2H), 1.94 (td, J = 13.0, 3.5 Hz, 2H), 1.58–1.86 (m, 7H), 1.52 (q, J = 7.0 Hz, 2H), 1.04–1.14 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 155.5, 138.2, 136.9, 133.5, 130.8, 115.5, 94.4, 84.0 (d, 1J CF = 163.9 Hz), 70.2, 68.8, 55.97, 55.96, 36.4, 34.8, 34.6, 31.6, 27.3 (d, 2JCF = 20.1 Hz), 25.5 (d, 3JCF = 5.5 Hz); MS (EI) m/z 486 (M+, 100), 426, 287, 149, 121. HRMS (EI) m/z calcd for C29H39O5F 486.2781, found 486.2778.
4-[2-(4-Fluorobutoxy)ethyl][bis(4-hydroxyphenyl)methylene]cyclohexane (28)
According to the general procedure for MOM deprotection with 4-[2-(4-fluorobutoxy)ethyl][bis(4-methoxymethoxyphenyl)methylene]cyclohexane (32 mg, 0.066 mmol), HCl (200 μL) and methanol (3 mL), 28 (20 mg, 76%) was obtained as a white solid: mp 129 °C; 1H NMR (500 MHz, acetone-d6) δ 6.91 (d, J = 8.5 Hz, 4H), 6.74 (d, J = 9.0 Hz, 4H), 4.44 (dt, 2JHF = 47.5 Hz, J = 6.0 Hz, 2H), 3.44 (t, J = 6.5 Hz, 2H), 3.41 (t, J = 6.0 Hz, 2H), 2.59 (bd, J = 13.5 Hz, 2H), 1.92 (td, J = 13.0, 4.0 Hz, 2H), 1.77–1.85 (m, 2H), 1.59–1,77 (m, 5H), 1.48 (q, J = 7.0 Hz, 2H), 1.03–1.13 (m, 2H); 13C NMR (125 MHz, acetone-d6) δ 156.6, 137.6, 135.6, 135.3, 131.6, 115.4, 84.5 (d, 1J CF = 162.1 Hz), 70.7, 69.3, 37.3, 35.60, 35.57, 32.3, 28.2 (d, 2JCF = 20.1 Hz), 26.3 (d, 3JCF = 5.5 Hz); MS (EI) m/z 446 (M+, 100), 398, 199, 107. HRMS (EI) m/z calcd for C25H31O3F 398.2257, found 398.2265. Anal. (C21H24O3·0.1 H2O) C, H.
4-Ethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
According to the general procedure for reduction with 25b (175 g, 0.35 mmol), LiAlH4 in THF solution (1 M, 700 μL) and THF (10 mL), 4-ethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (130 mg, 92%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.5 Hz, 4H), 6.93 (d, J = 9.0 Hz, 4H), 5.15 (s, 4H), 3.48 (s, 6H), 2.59 (bd, J = 13.5 Hz, 2H), 1.91 (td, J = 13.0, 4.0 Hz, 2H), 1.77–1.84 (m, 2H), 1.32–1.43 (m, 1H), 1.19–1.30 (m, 3H), 0.98–1.08 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 155.5, 138.7, 137.1, 133.3, 130.9, 115.5, 94.5, 56.0, 39.4, 34.5, 31.7, 29.3, 11.6; MS (EI) m/z 396 (M+, 100), 251, 287. HRMS (EI) m/z calcd for C25H32O4 396.2301, found 396.2307.
4-Ethyl[bis(4-hydroxyphenyl)methylene]cyclohexane (29b)
According to the general procedure for MOM deprotection with 4-ethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (130 mg, 0.33 mmol), HCl (200 μL), methanol (2.5 mL) and THF (2.5 mL), 29b (85 mg, 84%) was obtained as a white solid: mp 188 °C; 1H NMR (500 MHz, CDCl3) δ 6.97 (d, J = 8.5 Hz, 4H), 6.73 (d, J = 7.5 Hz, 4H), 2.58 (bd, J = 13.0 Hz, 2H), 1.91 (td, J = 13.5, 3.5 Hz, 2H), 1.33–1.42 (m, 1H), 1.25 (quintet, J = 7.5 Hz, 2H), 0.98–1.08 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 153.7, 138.5, 136.1, 133.2, 114.7, 39.4, 34.5, 31.7, 29.4, 11.6; MS (EI) m/z 308 (M+, 100), 199. Anal. (C21H24O2) C, H.
Methyl 3-[Bis(4-hydroxyphenyl)methylene]cyclohexanecarboxylate (30a)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 1.1 g, 5.1 mmol), 12 (0.8 g, 5.1 mmol), Zn (2.5 g, 38 mmol) and titanium(IV) chloride (3.6 g, 19 mmol), 30a (1.4 g, 81%) was obtained as a white solid: mp 144–146 °C; 1H NMR (400 MHz, acetone-d6) δ 6.83 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 6.65 (d, J = 8.0 Hz, 2H), 3.53 (s, 3H), 2.63 (bd, J = 10.4 Hz, 1H), 2.34–2.46 (m, 2H), 2.02 (t, J = 12.4 Hz, 1H), 1.72–1.96 (m, 3H), 1.45–1.61 (m, 1H), 1.28–1.42 (m, 3H); 13C NMR (100 MHz, acetone-d6) δ 174.7, 155.7, 135.7, 133.4, 133.3, 130.29, 130.26, 114.7, 114.6, 51.3, 43.6, 33.9, 30.9, 28.7, 26.2; MS (EI) m/z 338 (M+, 100), 278, 199. HRMS (EI) m/z calcd for C21H22O4 338.1518, found 338.1518. Anal. (C21H22O4) C, H.
Methyl 3-[Bis(4-methoxymethoxyphenyl)methylene]cyclohexanecarboxylate (33a)
According to the general procedure for MOM protection with 30a (1.2 g, 3.6 mmol), methoxymethyl chloride (0.66 g, 8.2 mmol), sodium hydride (0.4 g, 8.9 mmol) and DMF (15 mL), 33a (1.3 g, 84%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 9.2 Hz, 2H), 6.93 (d, J = 8.8 Hz, 4H), 5.153 (s, 2H), 5.15 (s, 2H), 3.62 (s, 3H), 3.481 (s, 3H), 3.476 (s, 3H), 2.73–2.81 (m, 1H), 2.51–2.61 (m, 1H), 2.40–2.49 (m, 1H), 2.15 (t, J = 12.4 Hz, 1H), 1.97–2.07 (m, 1H), 1.82–1.96 (m, 2H), 1.58–1.70 (m, 1H), 1.34–1.48 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 175.8, 155.73, 155.69, 136.6, 136.3, 135.6, 135.2, 130.73, 130.72, 115.7, 115.6, 94.5, 77.2, 56.0, 51.6, 44.6, 34.1, 31.5, 29.4, 26.9; MS (EI) m/z 426 (M+), 302, 286 (100), 241, 165. HRMS (EI) m/z calcd for C25H30O6 426.2042, found 426.2041.
3-Hydroxymethyl[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (34a)
According to the general procedure for reduction with 33a (0.65 g, 1.5 mmol), LiAlH4 in THF solution (1 M, 1.5 mL) and THF (10 mL), 34a (0.59 g, 98%) was obtained as an colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 4H), 5.149 (s, 2H), 5.148 (s, 2H), 3.48 (s, 6H), 3.64–3.67 (m, 2H), 2.61 (d, J = 10 Hz, 1H), 2.55 (d, J = 13.6 Hz, 1H), 1.78–1.98 (m, 3H), 1.62–1.75 (m, 2H), 1.35–1.48 (m, 2H), 1.17–1.29 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.60, 155.57, 136.9, 136.73, 136.70, 134.4, 130.7, 115.7, 115.6, 94.5, 68.0, 56.0, 42.1, 34.9, 32.1, 29.4, 27.0; MS (EI) m/z 398 (M+), 302 (100). HRMS (EI) m/z calcd for C24H30O5 398.2093, found 398.2956.
3-Hydroxymethyl[bis(4-hydroxyphenyl)methylene]cyclohexane (35a)
According to the general procedure for MOM deprotection with 34a (0.34 g, 0.84 mmol), HCl (200 μL), methanol (7.0 mL), 35a (178 mg, 68%) was obtained as a white solid: mp 219–221 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.82 (d, J = 8.4 Hz, 4H), 6.66 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 3.11–3.26 (m, 2H), 2.54 (bd, J = 11.6 Hz, 1H), 2.44 (bd, J = 12.8 Hz, 1H), 1.68–1.87 (m, 3H), 1.42–1.60 (m, 2H), 1.21–1.35 (m, 1H), 1.10–1.20 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 155.50, 155.49, 135.5, 134.2, 133.9, 133.7, 130.43, 130.36, 114.6, 66.2, 42.0, 35.0, 31.9, 29.3, 26.8; MS (EI) m/z 310 (M+, 100). HRMS (EI) m/z calcd for C20H22O3 310.1569, found 310.1573. Anal. (C20H22O3) C, H.
Dimethyl 2-{3-[Bis(4-hydroxyphenyl)methylene]cyclohexyl}malonate (30b)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 4.3 g, 20 mmol), 14 (4.6 g, 20 mmol), Zn (9.9 g, 150 mmol) and titanium(IV) chloride (14 g, 74 mmol), 30b (6.1 g, 74%) was obtained as a white solid: mp 148–149 °C; 1H NMR (400 MHz, methanol-d4) δ 6.87(d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.67 (s, 3H), 3.49 (s, 3H), 3.26 (d, J = 9.6 Hz), 2.45–2.56 (m, 2H), 2.13–2.25 (m, 1H), 1.86–1.98 (m, 1H), 1.72–1.85 (m, 3H), 1.32–1.46 (m, 1H), 1.21–1.32 (m, 1H); 13C NMR (100 MHz, methanol-d4) δ 170.5, 170.3, 156.9, 156.8, 137.5, 135.8, 135.7, 135.6, 131.87, 131.81, 115.60, 115.58, 58.4, 52.8, 52.7, 40.9, 37.0, 32.7, 31.8, 30.0; MS (ESI) m/z 411 (M++1), 279 (100). HRMS (ESI) m/z calcd for C24H26O6 441.1808, found 441.1812. Anal. (C24H26O6) C, H.
2-{3-[Bis(4-hydroxyphenyl)methylene]cyclohexyl}malonic acid (31)
To a solution of 30b (4.5 g, 11 mmol) in methanol (20 mL) was added NaOH (2 M, 25 mL), and then the reaction mixture was refluxed at 100 °C for 2 h. After the reaction was cooled to RT, it was acidified with HCl (2 M, 40 mL) in an ice bath. Water (10 mL) was added, and a precipitate appeared which was collected by filtration. The white powder obtained was dried under vacuum, providing 31 (3.8 g, 90%) as white solid: mp 207–208 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.81 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.62 (d, J = 8.8 Hz, 2H), 3.99 (d, J = 9.2 Hz, 1H), 2.57 (bd, J = 12.0 Hz, 1H), 2.40 (bd, J = 12.8 Hz, 1H), 1.96–2.11 (m, 1H), 1.62–1.88 (m, 4H), 1.24–1.32 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 169.9, 169.7, 155.6, 155.5, 135.1, 134.4, 133.8, 133.2, 130.5, 114.7, 114.6, 57.8, 38.5, 35.4, 31.6, 30.2, 26.7; MS (ESI) m/z 383 (M++H), 279 (100). HRMS (ESI) m/z calcd for C22H22O6 383.1495, found 383.1499.
Methyl 2-{3-[Bis(4-hydroxyphenyl)methylene]cyclohexyl}acetate (32)
Compound 31 (3.8 g, 9.9 mmol) was dissolved with diglyme (15 mL), and the reaction mixture was heated to 160 °C for 1 h. When no bubbles were detected, the reaction mixture was cooled, and diglyme was removed under vacuum. The organic mixture was filtered through short silica gel, eluted with solvents (50% Et2OAc, 47% hexane, 2% methanol and 1% acetic acid), and the solvents were evaporated. Without further purification, the residual oil was dissolved in methanol (10 mL) under nitrogen. Thionyl chloride (1.3 g, 11 mmol) was added slowly at rt, and the mixture was stirred for 1.5 h. The reaction was quenched by saturated NaHCO3 solution (50 mL) under ice bath cooling, and H2O (100 mL) was added. Product was extracted by ethyl acetate, and organic layer was washed by brine. After drying with MgSO4, the extract was concentrated and purified by flash column chromatography (Et2OAc/hexane; 4:6). Recrystallization (Et2O and hexane) gave 32 (2.6 g, 74%) as a off-white solid: mp 157–158 °C; 1H NMR (400 MHz, methanol-d4) δ 6.87 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.674 (d, J = 8.8 Hz, 2H), 6.670 (d, J = 8.4 Hz, 2H), 3.55 (s, 3H), 2.47–2.60 (m, 2H), 2.10–2.28 (m, 2H), 1.72–1.99 (m, 4H), 1.60–1.69 (m, 1H), 1.32–1.46 (m, 1H), 1.23–1.27(m, 1H); 13C NMR (100 MHz, methanol-d4) δ 175.0, 156.77, 156.75, 136.9, 136.5, 135.9, 135.8, 131.86, 131.84, 115.5, 51.9, 42.1, 39.2, 38.0, 34.0, 32.8, 28.2; MS (EI) m/z 352 (M+), 278, 167 (100). HRMS (EI) m/z calcd for C22H24O4 352.1675, found 352.1670. Anal. (C22H24O4) C, H.
Methyl 2-{3-[Bis(4-methoxymethoxyphenyl)methylene]cyclohexyl}acetate (33b)
According to the general procedure for MOM protection with 32 (2.1 g, 5.9 mmol), methoxymethyl chloride (1.2 g, 14 mmol), sodium hydride (1.0 g, 30 mmol) and DMF (15 mL), 23b (2.5 g, 95%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 4H), 5.15 (s, 4H), 3.59 (s, 3H), 3.48 (s, 3H), 3.47 (s, 3H), 2.48–2.61 (m, 2H), 2.15–2.27 (m, 2H), 1.94–2.04 (m, 1H), 1.73–1.94 (m, 3H), 1.62–1.73 (m, 1H), 1.35–1.48 (m, 1H), 1.12–1.28 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 173.2, 155.5, 136.7, 136.55, 136.53, 134.5, 130.8, 115.5, 94.40, 94.38, 55.99, 55.95, 51.4, 41.3, 38.2, 36.5, 32.7, 31.7, 27.0; MS (EI) m/z 440 (M+, 100), 366, 167. HRMS (EI) m/z calcd for C26H32O6 440.2199, found 440.2198.
3-(2-Hydroxyethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (34b)
According to the general procedure for reduction with 33b (2.4 g, 5.5 mmol), LiAlH4 in THF solution (1 M, 5.5 mL) and THF (25 mL), 34b (2.1 g, 93%) was obtained as a white solid: mp 72–74 °C; 1H NMR (400 MHz, methanol-d4) δ 6.98 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 5.141 (s, 2H), 5.137 (s, 2H), 3.49 (td, J = 6.8, 2.0 Hz, 2H), 3.434 (s, 3H), 3.429 (s, 3H), 2.58 (bd, J = 9.2 Hz, 1H), 2.49 (bd, J = 9.2 Hz, 1H), 1.74–1.97 (m, 3H), 1.56–1.70 (m, 2H), 1.33–1.49 (m, 3H), 1.13–1.26 (m, 1H); 13C NMR (100 MHz, methanol-d4) δ 157.12, 157.09, 138.5, 138.2, 138.1, 116.72, 116.70, 95.51, 95.49, 60.7, 56.14, 56.11, 40.5, 39.8, 37.3, 34.2, 33.2, 28.4; MS (EI) m/z 412 (M+, 100), 278. HRMS (EI) m/z calcd for C25H32O5 412.2250, found 440.2251.
3-(2-Hydroxyethyl)[bis(4-hydroxyphenyl)methylene]cyclohexane (35b)
According to the general procedure for MOM deprotection with 34b (250 mg, 0.61 mmol), HCl (200 μL), methanol (7.0 mL), 35b (172 mg, 87%) was obtained as a white solid: mp 203–205 °C; 1H NMR (400 MHz, methanol-d4) δ 6.87 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H), 3.43–3.54 (m, 2H), 2.58 (bd, J = 9.6 Hz, 1H), 2.52 (bd, J = 13.6 Hz, 1H), 1.73–1.95 (m, 3H), 1.53–1.68 (m, 2H), 1.31–1.48 (m, 3H), 1.12–1.23(m, 1H); 13C NMR (100 MHz, methanol-d4) δ 156.7, 137.4, 136.20, 136.16, 136.0, 131.91, 131.86, 115.5, 60.7, 40.6, 39.8, 37.3, 34.3, 33.3, 28.5; MS (EI) m/z 324 (M+, 100), 278, 199, 149. Anal. (C21H24O3) C, H.
3-(2-Methanesulfonyloxyethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (36b)
According to the general procedure for methanesulfonylation with 35b (349 mg, 0.82 mmol), methanesulfonic anhydride (215 mg, 1.2 mmol), triethylamine (331 mg, 2.5 mmol) and CH2Cl2 (10 mL), 36b (393 mg, 98%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.002 (d, J = 8.8 Hz, 2H), 6.999 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 5.154 (s, 2H), 5.153 (s, 2H), 4.21–4.23 (m, 2H), 3.479 (s, 3H), 3.477 (s, 3H), 2.88 (s, 3H), 2.60 (bd, J = 10.0 Hz, 1H), 2.53 (bd, J = 13.6 Hz, 1H), 1.75–1.97 (m, 3H), 1.60–1.74 (m, 4H), 1.34–1.47 (m, 1H), 1.12–1.26 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 155.62, 155.58, 136.7, 136.56, 136.51, 134.5, 130.77, 130.75, 115.7, 115.6, 94.4, 68.1, 56.03, 56.02, 38.0, 37.0, 35.6, 35.5, 32.7, 32.0, 27.0; MS (EI) m/z 490 (M+, 100), 211, 141. HRMS (EI) m/z calcd for C26H34O7S 490.2025, found 490.2015.
3-(2-Fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
According to the general procedure for fluorination with 36b (350 mg, 0.71 mmol), cesium fluoride (541 mg, 3.6 mmol), H2O (20 μL), 1-butyl-3-methyl-imidazolium tetrafluoroboronate (2.0 mL) and acetonitrile (2.0 mL), 3-(2-fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (235 mg, 80%) was obtained as a slightly yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.938 (d, J = 8.8 Hz, 2H), 6.935 (d, J = 8.8 Hz, 2H), 5.161 (s, 2H), 5.156 (s, 2H), 4.42 (dm, 2JHF = 47.6 Hz, 2H), 3.49 (s, 3H), 3.48 (s, 3H), 2.60 (bd, J = 9.2 Hz, 1H), 2.54 (bd, J = 14 Hz, 1H), 1.74–1.97 (m, 3H), 1.54–1.73 (m, 4H), 1.33–1.46 (m, 1H), 1.14–1.27 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 155.57, 155.52, 137.0, 136.8, 136.7, 134.2, 130.80, 130.76, 115.56, 115.54, 94.46, 94.43, 136.7, 136.56, 136.51, 134.5, 130.77, 130.75, 115.7, 115.6, 94.46, 94.43, 81.4 (d, 1J CF = 162.4 Hz), 56.02, 55.99, 38.4, 37.0 (d, 2JCF = 18.9 Hz), 35.7 (d, 3JCF = 4.6 Hz), 32.8, 32.1, 27.2; MS (EI) m/z 414 (M+, 100). HRMS (EI) m/z calcd for C25H31O4F 414.2206, found 414.2205.
3-(2-Fluoroethyl)[bis(4-hydroxyphenyl)methylene]cyclohexane (37b)
According to the general procedure for MOM deprotection with 3-(2-fluoroethyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (215 mg, 0.52 mmol), HCl (200 μL), methanol (3.0 mL) and Et2O (1 mL), 37b (172 mg, 87%) was obtained as a white solid: mp 100–105 °C; 1H NMR (400 MHz, methanol-d4) δ 6.87 (d, J = 8.8 Hz, 4H), 6.68 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 6.935 (d, J = 8.8 Hz, 2H), 4.36 (dm, 2JHF = 47.6 Hz, 2H), 2.61 (bd, J = 9.2 Hz, 1H), 2.52 (bd, J = 13.2 Hz, 1H), 1.73–1.97 (m, 3H), 1.44–1.69 (m, 4H), 1.32–1.44 (m, 1H), 1.14–1.31 (m, 1H); 13C NMR (100 MHz, methanol-d4) δ 156. 76, 156.73, 137.1, 136.4, 136.1, 136.0, 131.9, 115.54, 115.53, 82.9 (d, 1JCF = 162.3 Hz), 39.4, 38.3 (d, 2JCF = 19.7 Hz), 37.1 (d, 3JCF = 3.8 Hz), 34.2, 33.2, 28.4; MS (EI) m/z 326 (M+, 100), 199. HRMS (EI) m/z calcd for C21H23O2F 326.1682, found 326.1672.
3-(3-Acetoxy-n-propyl)-1-[bis(4-hydroxyphenyl)methylene]cyclohexane (30c)
According to the general procedure for the McMurry coupling reaction with 4,4′-dihydroxybenzophenone (1, 0.35 g, 1.6 mmol), 15 (0.32 g, 1.6 mmol), Zn (0.79 g, 12 mmol) and titanium(IV) chloride (1.1 g, 6.0 mmol), 30c (0.43 g, 70%) was obtained as an colorless oil; 1H NMR (400 MHz, CDCl3) δ 6.91–6.96 (m, 4H), 6.738 (d, J = 8.8 Hz, 2H), 6.733 (d, J = 8.4 Hz, 2H), 4.00 (t, J = 6.8 Hz, 2H), 2.54 (bt, J = 13.6 Hz, 2H), 2.05 (s, 3H), 1.72–1.93 (m, 3H), 1.47–1.65 (m, 3H), 1.30–1.47 (m, 3H), 1.38–1.47 (m, 1H), 1.30–1-38 (m, 1H), 1.19–1.30 (m, 2H), 1.06–1.18 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 172.2, 153.89, 153.85, 137.1, 135.8, 135.6, 133.9, 131.0, 130.9, 114.73, 114.68, 65.2, 39.1, 38.5, 33.0, 32.8, 32.2, 27.3, 25.8, 21.1; MS (EI) m/z 380 (M+), 214 (100). HRMS (EI) m/z calcd for C24H28O4 380.1987, found 380.1993.
3-(3-Acetoxy-n-propyl)-1-[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (33c)
According to the general procedure for MOM protection with 30a (0.43 g, 1.1 mmol), methoxymethyl chloride (0.20 g, 2.5 mmol), sodium hydride (69 mg, 2.8 mmol) and DMF (10 mL), 33c (191 mg, 36%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.013 (d, J = 8.8 Hz, 2H), 7.008 (d, J = 9.2 Hz, 2H), 6.38 (d, J = 8.8 Hz, 2H), 6.932 (d, J = 8.8 Hz, 2H), 5.16 (s, 2H), 5.15 (s, 2H), 4.01 (t, J = 6.8 Hz, 2H), 3.48 (s, 3H), 3.47 (s, 3H), 2.50–2.63 (m, 2H), 2.04 (s, 3H), 1.72–1.95 (m, 3H), 1.30–1.68 (m, 5H), 1.20–1.30 (m, 2H), 1.09–1.20 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 171.1, 155.44, 155.41, 137.4, 136.8, 136.7, 133.8, 130.73, 130.71, 115.46, 115.43, 94.4, 94.3, 64.7, 55.90, 55.87, 39.1, 38.5, 32.86, 32.83, 32.1, 27.3, 25.8, 20.9; MS (EI) m/z 468 (M+, 100), 446, 302, 286. HRMS (EI) m/z calcd for C28H36O6 468.2512, found 468.2498.
3-(3-Hydroxypropyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (34c)
To a solution of 33c (185 mg, 0.39 mmol) in methanol (4 mL), Et2O (1 mL) and H2O (1 mL) was added K2CO3 (450 mg, 3.3 mmol). The mixture was stirred at RT for 4 h. It was then quenched by H2O and extracted with Et2OAc (50 mL). The organic layer was washed with saturated ammonium chloride solution (50 mL) and concentrated. Flash column chromatography (Et2OAc/hexane; 3:7) gave 34c (159 mg, 95%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.4 Hz, 4H), 5.150 (s, 2H), 5.147 (s, 2H), 3.56 (t, J = 6.4 Hz, 2H), 3.478 (s, 3H), 3.473 (s, 3H), 2.59 (d, J = 14.4 Hz, 1H), 2.53 (d, J = 13.2 Hz, 1H), 1.74–2.15 (m, 3H), 1.31–1.67 (m, 5H), 1.21–1.31 (m, 2H), 1.08–1.21 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 155.4, 137.7, 136.9, 136.8, 133.6, 130.77, 130.75, 115.49, 115. 46, 94.39, 94.34, 63.1, 55.95, 55.92, 39.4, 38.6, 33.0, 32.8, 32.2, 30.0, 27.4; MS (EI) m/z 426 (M+, 100). HRMS (EI) m/z calcd for C26H34O5 426.2406, found 426.2415.
3-(3-Hydroxy-n-propyl)-1-[bis(4-hydroxyphenyl)methylene]cyclohexane (35c)
To a solution of 30c (130 mg, 0.34 mmol) in methanol (7.5 mL) and H2O (2.5 mL) was added K2CO3 (500 mg, 3.6 mmol). The mixture was stirred at RT for 4 h, quenched by H2O and extracted with Et2OAc (50 mL). The organic layer was washed with saturated ammonium chloride solution (50 mL) and concentrated. Flash column chromatography (Et2OAc/hexane; 4:6) gave 35c (75 mg, 65%) as an off-white solid: mp 175–176 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.810 (d, J = 8.8 Hz, 2H), 6.807 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 4H), 3.30 (dd, J = 7.6, 6.4 Hz, 2H), 2.41 (bd, J = 13.2 Hz, 1H), 1.65–1.87 (m, 3H), 1.47–1.57 (m, 1H), 1.20–1.41 (m, 4H), 1.00–1.20 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ 155.55, 155.52, 135.7, 134.1, 133.8, 133.7, 130.4, 114.6, 61.0, 38.2, 32.9, 31.8, 31.0, 29.9, 27.1, 22.1; MS (EI) m/z 338 (M+, 100), 215, 199. HRMS (EI) m/z calcd for C22H26O3 338.1882, found 338.1878. Anal. (C22H26O3) C, H.
3-(3-Fluoropropyl)[bis(4-methoxymethoxyphenyl)methylene]cyclohexane
To a solution of 34c (190 mg, 0.45 mmol) in CH2Cl2 (5 mL) was added slowly diethylaminosulfur trifluoride (865 mg, 0.5 mmol) at −78 °C under nitrogen. The cooling was removed, and the reaction mixture was stirred for 1 h at RT. It was then placed in an ice-bath and methanol (200 μL) and H2O (200 μL) were added. The ice-bath was removed and it was allowed to stir for 30 min. Et2OAc (50 mL) was added and the organic layer was washed with NaHCO3 solution (50 mL) and ammonium chloride solution (50 mL). The organic layer was dried over sodium sulfate and concentrated. Flash column chromatography (Et2OAc/hexane; 1:9 – 3:7) gave 3-(3-fluoro-n-propyl)-1-[bis(4-methoxymethoxyphenyl)methylene]cyclohexane (71 mg, 37%) as an oil: 1H NMR (400 MHz, CDCl3) δ 6.99–7.04 (m, 4H), 6.92–6.97 (m, 4H), 5.164 (s, 2H), 5.158 (s, 2H), 4.39 (dt, 2JHF = 47.6, 6.4 Hz, 2H), 3.489 (s, 3H), 3.482 (s, 3H), 2.30–2.64 (m, 2H), 1.75–1.95 (m, 3H), 1.63–1.75 (m, 3H), 1.40–1.63 (m, 1H), 1.24–1.40 (m, 3H), 1.08–1.20 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 155.50, 155.45, 137.48, 136.9, 136.8, 133.8, 130.80, 130.75, 115.50, 115.49, 94.43, 94.38, 84.4 (d, 1J CF = 163 Hz), 55.98, 55.96, 39.2, 38.5, 32.9, 32.15, 32.12, 27.7 (d, 2J CF = 18.9 Hz), 27.3; MS (EI) m/z 428 (M+), 302 (100), 286, 165. HRMS (EI) m/z calcd for C26H33O4F 428.2363, found 428.2353.
3-(3-Fluoro-n-propyl)-1-[bis(4-hydroxyphenyl)methylene]cyclohexane (37c)
According to the general procedure for MOM deprotection with 3-(3-fluoropropyl)[bis(4-methoxymethoxyphenyl)-methylene]cyclohexane (70 mg, 0.16 mmol) and concentrated HCl (200 μL) in methanol (4.0 mL) and Et2O (1 mL), 37c (36 mg, 69%) was obtained as an off-white solid: mp 85 °C (decomposed to dark brown color); 1H NMR (400 MHz, DMSO-d6) δ 6.82 (d, J = 8.4 Hz, 4H), 6.65 (d, J = 8.4 Hz, 4H), 4.38 (dt, 2JHF = 47.6 Hz, J = 6.4 Hz, 2H), 2.36–2.52 (bm, 2H), 1.67–1.90 (m, 3H), 1.44–1.64 (m, 3H), 1.34–1.44 (m, 1H), 1.16–1.34 (m, 3H), 1.02–1,16 (m, 1H); 13C NMR (100 MHz, acetone-d6) δ 156.6, 156.5, 137.0, 135.6, 135.5, 131.6, 115.5, 115.4, 84.7 (d, 1J CF = 164 Hz), 40.1, 39.2, 33.9, 32.5 (d, 3J CF = 5.3 Hz), 32.9, 28.5 (d, 2J CF = 19.7 Hz), 28.1; MS (EI) m/z 340 (M+, 100), 199. HRMS (EI) m/z calcd for C22H25O2F 340.1839, found 340.1841.
Molecular Modeling
The experimental details of the computational method used have been reported elsewhere.55 Briefly, four of eight possible docking orientations of C4 substituted were individually examined using the FlexiDoc routine in Sybyl 7.1 (Tripos Inc.) to account for both the axial/equatorial orientation of the ring substituent and the ring flips of the cyclohexane ring. Docking of the C3 substituted cyclofenils was further complicated because they were prepared as a mixture of enantiomers, so eight of sixteen orientations were examined. We eliminated half of the docking orientations by starting from an unfavorable orientation where one pendant phenol was overlaid with the A-ring of estradiol while the other phenol was placed away from an hydrogen bonding partner (Thr347/299). The docking routine typically reoriented the cyclofenil core, placing the second phenol within hydrogen bonding distance with the Thr residue. The top ranked docking orientation in the ER binding pocket was minimized by a 3-step process that has been reported elsewhere.55
Estrogen Receptor Binding Affinity
Relative binding affinities were determined by a competitive radiometric binding assay as previously described,52 using 10 nM [3H]estradiol as tracer ([6,7-3H]estra-1,3,5,(10)-triene-3,17-β-diol, 51–53 Ci/mmol, Amersham BioSciences, Piscataway, NJ), and purified full-length human ERα and ERβ were purchased from PanVera/Invitrogen (Carlsbad, CA). Incubations were for 18–24 h at 0 °C. Hydroxyapatite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values with the RBA of estradiol set to 100%. The values given are the average ± range or SD of two to three independent determinations. Estradiol binds to ERα and uterine cytosol ER with a Kd of 0.2 nM and to ERβ‚ with a Kd of 0.5nM.
Supplementary Material
Elemental analysis data of 2-5, 18, 20, 21a-b, 24a-b, 26a-b, 27, 28, 29a-b, 30a-b, 32, and 35a-c, HPLC purity data and traces of 37b-c, and 1H and 13C NMR spectra of 2-5, 7, 10, 12, 14-16, 17a-b, 18-20, 21a-b, 22a-b, 23a-b, 24a-b, 25a-b, 26a-b, 28, 29b, 30a-c, 31, 32, 33a-c, 34a-c, 35a-c, 36b, 37b-c, and some key intermediates. This material is available free of charge via a Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by grants from the National Institutes of Health (PHS 5R37CA25836), the National Research and Development Program of MOST, Korea (2005-03184) and the Post-doctoral Fellowship Program of Korea Science & Engineering Foundation (KOSEF)
Footnotes
The following abbreviations are used: E2, 17β-estradiol; ER, estrogen receptor; F-18, fluorine-18; PET, positron emission tomography; SERM, selective estrogen receptor modulator.
References
- 1.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katzenellenbogen JA, Katzenellenbogen BS. Nuclear hormone receptors: ligand-activated regulators of transcription and diverse cell responses. Chem Biol. 1996;3:529–536. doi: 10.1016/s1074-5521(96)90143-x. [DOI] [PubMed] [Google Scholar]
- 3.Turner RT, Rickard D, Spelsberg TC, Sibonga JD. Osteoporosis. Humana Press; 2002. The basic biology of estrogen and bone; pp. 309–330. [Google Scholar]
- 4.Maran A, Turner RT. Effects of estrogen on bone. Recent Research Developments in Endocrinology. 2001:327. [Google Scholar]
- 5.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 6.Mendelsohn ME. Protective effects of estrogen on the cardiovascular system. Am J Cardiol. 2002;89:12E–17E. doi: 10.1016/s0002-9149(02)02405-0. discussion 17E–18E. [DOI] [PubMed] [Google Scholar]
- 7.Behl C. Oestrogen as a neuroprotective hormone. Nature Rev Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- 8.Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]
- 9.Katzenellenbogen BS. Estrogen receptors: bioactivities and interactions with cell signaling pathways. Biol Reprod. 1996;54:287–293. doi: 10.1095/biolreprod54.2.287. [DOI] [PubMed] [Google Scholar]
- 10.Katzenellenbogen JA, O’Malley BW, Katzenellenbogen BS. Tripartite steroid hormone receptor pharmacology: interaction with multiple effector sites as a basis for the cell- and promoter-specific action of these hormones. Mol Endocrinol. 1996;10:119–131. doi: 10.1210/mend.10.2.8825552. [DOI] [PubMed] [Google Scholar]
- 11.Weihua Z, Andersson S, Cheng G, Simpson Evan R, Warner M, Gustafsson J-Å. Update on estrogen signaling. FEBS Lett. 2003;546:17–24. doi: 10.1016/s0014-5793(03)00436-8. [DOI] [PubMed] [Google Scholar]
- 12.Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-Å. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson J-Å. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptor α and β. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 14.Register TC, Adams MR. Coronary artery and cultured aortic smooth muscle cells express mRNA for both the classical estrogen receptor and the newly described estrogen receptor beta. J Steroid Biochem Mol Biol. 1998;64:187–191. doi: 10.1016/s0960-0760(97)00155-6. [DOI] [PubMed] [Google Scholar]
- 15.Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer JM, McEwen BS, Alves SE. Immunolocalization of estrogen receptor β in the mouse brain: comparison with estrogen receptor α. Endocrinology. 2003;144:2055–2067. doi: 10.1210/en.2002-221069. [DOI] [PubMed] [Google Scholar]
- 16.Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr Opin Pharmacol. 2001;1:613–619. doi: 10.1016/s1471-4892(01)00105-9. [DOI] [PubMed] [Google Scholar]
- 17.Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional profiling of estrogen regulated gene expression via estrogen receptor (ER) α or ERβ in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 18.Kian TM, Rogatsky I, Tzagarakis-Foster C, Cvoro A, An J, Christy RJ, Yamamoto KR, Leitman DC. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors α and β. Mol Biol Cell. 2004;15:1262–1272. doi: 10.1091/mbc.E03-06-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monroe DG, Getz BJ, Johnsen SA, Riggs BL, Khosla S, Spelsberg TC. Estrogen receptor isoform-specific regulation of endogenous gene expression in human osteoblastic cell lines expressing either ERα or ERβ. J Cell Biochem. 2003;90:315–326. doi: 10.1002/jcb.10633. [DOI] [PubMed] [Google Scholar]
- 20.Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol. 2003;17:203–208. doi: 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- 21.(a) Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased expression of estrogen receptor β protein in proliferative preinvasive mammary tumors. Cancer Res. 2001;61:2537–2541. [PubMed] [Google Scholar]; (b) Palmieri C, Cheng GJ, Saji S, Zelada-Hedman M, Warri A, Weihua Z, Van Noorden S, Wahlstrom T, Coombes RC, Warner M, Gustafsson JA. Estrogen receptor beta in breast cancer. Endocrine-Related Cancer. 2002;9:1–13. doi: 10.1677/erc.0.0090001. [DOI] [PubMed] [Google Scholar]; (c) Speirs V, Carder PJ, Lansdown MRJ. Oestrogen receptor beta: how should we measure this? Br J Cancer. 2002;87:687. 688–689. doi: 10.1038/sj.bjc.6600534. author reply. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Omoto Y, Kobayashi S, Inoue S, Ogawa S, Toyama T, Yamashita H, Muramatsu M, Gustafsson JA, Iwase H. Evaluation of oestrogen receptor b wild-type and variant protein expression, and relationship with clinicopathological factors in breast cancers. Eur J Cancer. 2002;38:380–386. doi: 10.1016/s0959-8049(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 22.Mosselman S, Polman J, Dijkema R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 23.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, Ohman L, Greene GL, Gustafsson J-Å, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 24.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson J, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lonard DM, Smith CL. Molecular perspectives on selective estrogen receptor modulators (SERMs): Progress in understanding their tissue-specific agonist and antagonist actions. Steroids. 2002;67:15–24. doi: 10.1016/s0039-128x(01)00133-7. [DOI] [PubMed] [Google Scholar]
- 26.Gao H, Katzenellenbogen JA, Garg R, Hansch C. Comparative QSAR analysis of estrogen receptor ligands. Chem Rev. 1999;99:723–744. doi: 10.1021/cr980018g. [DOI] [PubMed] [Google Scholar]
- 27.Meegan MJ, Lloyd DG. Advances in the science of estrogen receptor modulation. Curr Med Chem. 2003;10:181–210. doi: 10.2174/0929867033368501. [DOI] [PubMed] [Google Scholar]
- 28.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 29.Fink BE, Mortensen DS, Stauffer SR, Aron ZD, Katzenellenbogen JA. Novel structural templates for estrogen-receptor ligands and prospects for combinatorial synthesis of estrogens. Chem Biol. 1999;6:205–219. doi: 10.1016/S1074-5521(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 30.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson KE, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: Structure-affinity/activity relationships of estrogen receptor-α-selective agonists. J Med Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 31.Mortensen DS, Rodriguez AL, Carlson KE, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Synthesis and biological evaluation of a novel series of furans: Ligands selective for estrogen receptor α. J Med Chem. 2001;44:3838–3848. doi: 10.1021/jm010211u. [DOI] [PubMed] [Google Scholar]
- 32.Meyers MJ, Sun J, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor subtype-selective ligands: asymmetric synthesis and biological evaluation of cis and trans-5,11-dialkyl-5,6,11,12-tetrahydrochrysenes. J Med Chem. 1999;42:2456–2468. doi: 10.1021/jm990101b. [DOI] [PubMed] [Google Scholar]
- 33.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 34.Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Bridged bicyclic cores containing a 1,1-diarylethylene motif are high-affinity subtype-selective ligands for the estrogen receptor. J Med Chem. 2003;46:1589–1602. doi: 10.1021/jm0204800. [DOI] [PubMed] [Google Scholar]
- 35.Malamas MS, Gunawan I, Harris HA, Keith JC, Albert LM. US Patent Application Publishers. Wyeth, John, and Brother Ltd; 2003. Preparation of naphthyl benzoxazoles and benzisoxazoles as estrogenic agents. (28 pages) [Google Scholar]
- 36.Barlaam B, Bernstein P, Dantzman C, Warwick P. PCT Int Appl WO. Astrazeneca AB; Sweden: 2002. Preparation of benzoxazoles and benzothiazoles as selective ligands for human b-estrogen receptor. [Google Scholar]
- 37.Bernstein P. PCT Int Appl WO. Astrazeneca AB; Sweden: 2003. Preparation of aminobenzothiazoles as selective ER-β ligands for treatment of Alzheimer’s disease, anxiety disorders, depressive disorders, osteoporosis, cardiovascular disease, rheumatoid arthritis or prostate cancer. [Google Scholar]
- 38.Vijaykumar D, Al-Qahtani MH, Welch MJ, Katzenellenbogen JA. Synthesis and biological evaluation of a fluorine-18 labeled estrogen receptor-alpha selective ligand: [18F] propyl pyrazole triol. Nucl Med Biol. 2003;4:397–404. doi: 10.1016/s0969-8051(02)00446-8. [DOI] [PubMed] [Google Scholar]
- 39.Yoo J, Dence CS, Sharp TL, Katzenellenbogen JA, Welch MJ. Synthesis of an estrogen receptor beta-selective radioligand: 5-[18F]fluoro-(2R3S)-2,3-bis(4-hydroxy-phenyl)pentanenitrile nd comparison of in vivo distribution with 16alpha-[18F]fluoro-17beta-estradiol. J Med Chem. 2005;48:6366–78. doi: 10.1021/jm050121f. [DOI] [PubMed] [Google Scholar]
- 40.Miques JF, Wahlstam H, Olsson K, Suxdbeck B. Synthesis of unsymmetrical diphenylalkenes. J Med Chem. 1963;6:771–780. [Google Scholar]
- 41.(a) Hospital M, Busetta B, Courseille C, Precigoux G. X-ray conformation of some estrogens and their binding to uterine receptors. J Steroid Chem. 1975;6:221–225. doi: 10.1016/0022-4731(75)90136-3. [DOI] [PubMed] [Google Scholar]; (b) Precigoux PG, Busetta B, Courseille C, Hospital M, Miquel JF. Structure de composes Oestrogenes: Structure crystalline de trios analogues du cyclofenil. Acta Cryst. 1978;B34:3300–3305. [Google Scholar]
- 42.Garg S, Bindal RE, Durani S, Kapil RS. Structure-activity relationship of estrogens: A study involving cyclofenil as the model compound. J Steroid Biochem. 1983;18:89–95. doi: 10.1016/0022-4731(83)90335-7. [DOI] [PubMed] [Google Scholar]
- 43.(a) McDonnell DP, Connor CE, Wijayaratne A, Chang CY, Norris JD. Definition of the molecular and cellular mechanisms underlying the tissue-selective agonist/antagonist activities of selective estrogen receptor modulators. Recent Prog Horm Res. 2002;57:295–316. doi: 10.1210/rp.57.1.295. [DOI] [PubMed] [Google Scholar]; (b) Silfen SL, Ciaccia AV, Bryant HU. Selective estrogen receptor modulators: tissue selectivity and differential uterine effects. Climacteric. 1999;2:268–83. doi: 10.3109/13697139909038087. [DOI] [PubMed] [Google Scholar]
- 44.Miquel JF. Molecular structure and estrogenic activities-I. Unsymmetrical diphenylethylenes and triphenylethylenes. Tetrahedron. 1960;8:205–216. [Google Scholar]
- 45.Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor β antagonist activity. J Med Chem. 2004;47:5872–5893. doi: 10.1021/jm049631k. [DOI] [PubMed] [Google Scholar]
- 46.Coe PL, Scriven CE. Cross coupling of functionalized ketones by low valent titanium (the McMurry reaction); a new stereoselective synthesis of tamoxifen. J Chem Soc Perkin Trans I. 1986;3:475–477. [Google Scholar]
- 47.Detsi A, Koufaki M, Calogeropoulou T. Synthesis of (Z)-4-hydroxytamoxifen and (Z)-2-[4-[1-(p-hydroxyphenyl)-2-phenyl]-1-butenyl]phenoxyacetic Acid. J Org Chem. 2002;67:4608–4611. doi: 10.1021/jo0255328. [DOI] [PubMed] [Google Scholar]
- 48.Suemune H, Oda K, Sakai K. Ring cleavage and reconstruction of five and six membered ring. Tetrahedron Lett. 1987;28:3373–3376. [Google Scholar]
- 49.Labaree DC, Zhang J, Harris HA, O’Connor C, Reynolds TY, Hochberg RB. Synthesis and evaluation of B-, C-, and D-ring-substituted estradiol carboxylic acid esters as locally active estrogens . J Med Chem. 2003;46:1886–1904. doi: 10.1021/jm0204340. [DOI] [PubMed] [Google Scholar]
- 50.Cahiez G, Alexakis A, Normant JF. Derives organomagnesiens ω-alcoolates: preparation et proprietes. Tetrahedron Lett. 1978;19:3013–3014. [Google Scholar]
- 51.(a) Kim DW, Song CE, Chi DY. New method of fluorination using potassium fluoride in ionic liquid: significantly enhanced reactivity of fluoride and improved selectivity. J Am Chem Soc. 2002;124:10278–10279. doi: 10.1021/ja026242b. [DOI] [PubMed] [Google Scholar]; (b) Kim DW, Song CE, Chi DY. Significantly enhanced reactivities of the nucleophilic substitution reactions in ionic liquid. J OrgChem. 2003;68:4281–4285. doi: 10.1021/jo034109b. [DOI] [PubMed] [Google Scholar]
- 52.Kim DW, Choe YS, Chi DY. A new nucleophilic fluorine-18 labeling method for aliphatic mesylates: reaction in ionic liquinds shous tolerance of water. Nucl Med Biol. 2003;30:345–350. doi: 10.1016/s0969-8051(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 53.(a) Katzenellenbogen JA, Johnson HJ, Jr, Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]; (b) Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 54.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and convenient procedure for solvent purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 55.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson KE, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α selective agonists. J Med Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data of 2-5, 18, 20, 21a-b, 24a-b, 26a-b, 27, 28, 29a-b, 30a-b, 32, and 35a-c, HPLC purity data and traces of 37b-c, and 1H and 13C NMR spectra of 2-5, 7, 10, 12, 14-16, 17a-b, 18-20, 21a-b, 22a-b, 23a-b, 24a-b, 25a-b, 26a-b, 28, 29b, 30a-c, 31, 32, 33a-c, 34a-c, 35a-c, 36b, 37b-c, and some key intermediates. This material is available free of charge via a Internet at http://pubs.acs.org.