

Abstract
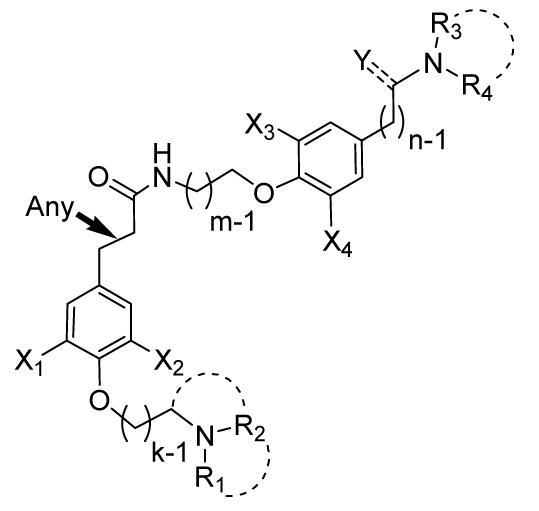
A 28-member focused library, based on the pseudosymmetric template of the marine alkaloids psammaplysenes, was prepared from combinations of components that were, in turn, derived from 4-iodophenol.
Synthetic libraries based on natural product templates have emerged as important sources of structurally diverse small molecule probes of biological function.1 In this spirit, we have extended our work on psammaplysenes A and B (Figure 1a), two closely related pseudosymmetric bromotyrosine-derived alkaloids, originally isolated from the marine sponge Psammaplysilla sp.2 They were initially identified in a high-throughput screen as inhibitors of FOXO1a nuclear export in cells with PTEN loss-of-function mutations.3 By interacting with a still unknown target in the PI3K/PTEN/Akt signaling pathway, they enhance the nuclear localization of the FOXO1a transcriptional regulator and, in effect, counteract inappropriate PI3-kinase-generated stimulatory signals, that arise due to the deficiency of tumor suppressor phosphatase PTEN. Because of the possible implications of modulating this pathway and the limited supply of material from sponge collections, we recently developed an efficient synthesis of these two compounds4 in a way that could readily accommodate the synthesis of analogues and probes, especially a focused library that would help define structure-activity relationships.
Figure 1.
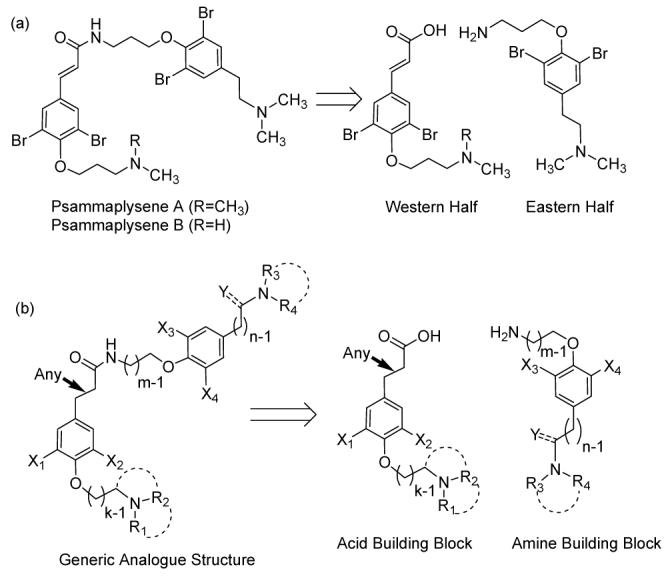
Main retrosynthetic disconnection for the original psammaplysenes and each library member.
As was the case for psammaplysenes A and B (Figure 1a), we set off to prepare a pool of acid and amine building blocks, that could be combined to provide a number of analogues (Figure 1b). Variations of the original synthetic pathway, that led from a common starting material, 4-iodophenol, to the two non-identical halves of these pseudosymmetric molecules, were used to assemble a 28-member collection of psammaplysene analogues, described herein. The compounds we elected to prepare bear a single modification compared to the most potent of the original structures, psammaplysene A (IC50=5 μM).
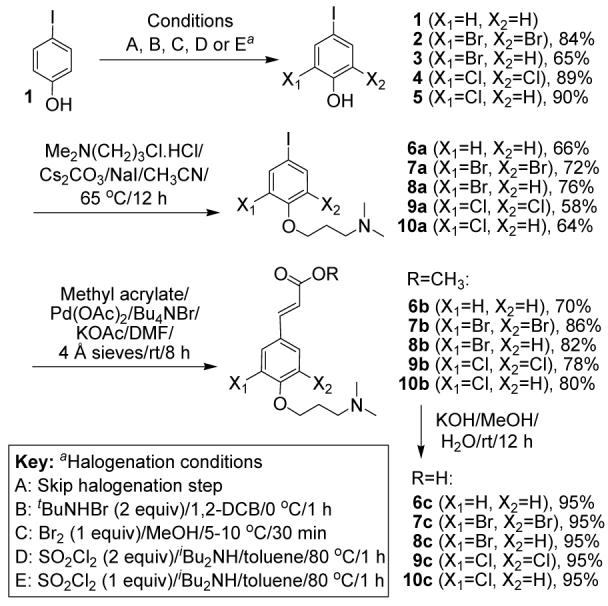
The first set of building blocks comprises acids with alternative halogen ring substituents. A variety of aromatic electrophilic substitution conditions on 4-iodophenol (1) led to compounds 2-5 (Scheme 1). Dibrominated phenol 2, a precursor to the natural products, was prepared using 2 equiv of N-bromo-tertbutylamine in 1,2-dichlorobenzene.5,6 Monobrominated phenol 3 was obtained using 1 equiv of Br2 in MeOH. The dichlorinated and monochlorinated compounds, 4 and 5, were formed using 1 and 2 equiv of SO2Cl2 in toluene respectively, in the presence of catalytic di-iso-butylamine.7 The unsubstituted counterpart, 4-iodophenol, was used directly in the next step. O-alkylation of 1-5 with 1-chloro-3-dimethylamino-propane hydrochloride in CH3CN, in the presence of Cs2CO3 and catalytic NaI, led to intermediate iodides 6a-10a respectively. These were subsequently converted to the corresponding methyl esters 6b-10b via a Heck reaction with methyl acrylate in DMF, by means of a Pd(OAc)2/Bu4NBr/KOAc system.8 The esters were hydrolyzed with KOH in MeOH/H2O to give zwitterionic acids 6c-10c.9
Scheme 1.
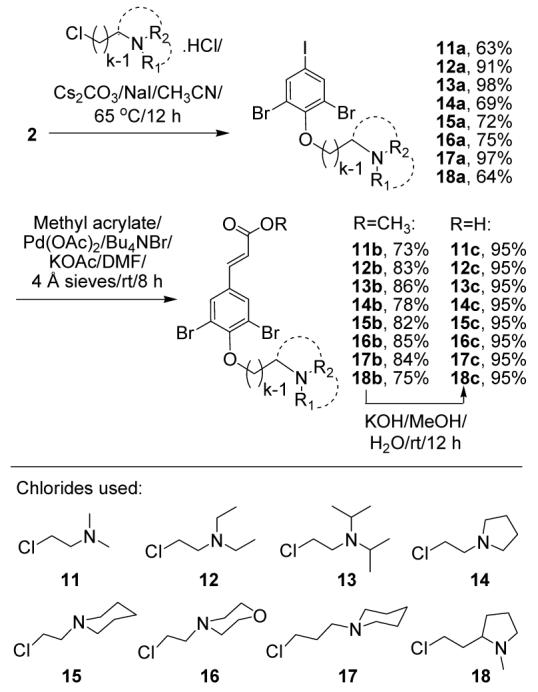
Another set of acid building blocks was chosen to have the same ring substitution as the natural product, but variable chain lengths and different tertiary amine heads (Scheme 2). The four-fold difference in IC50 values between psammaplysenes A and B,3 indicate some sensitivity at this site. Precursor 2 was alkylated with chlorides of chain lengths k=2-3 and different alkyl substituents on the nitrogen, including rings, to yield compounds 11a-18a. In most cases where bulky substituents increased the steric requirement, a shorter chain was used to offset this effect. A few large substituents were used together with a longer chain to give a sense of the steric limits. Heck reaction, followed by ester hydrolysis gave acids 11c-18c.
Scheme 2.
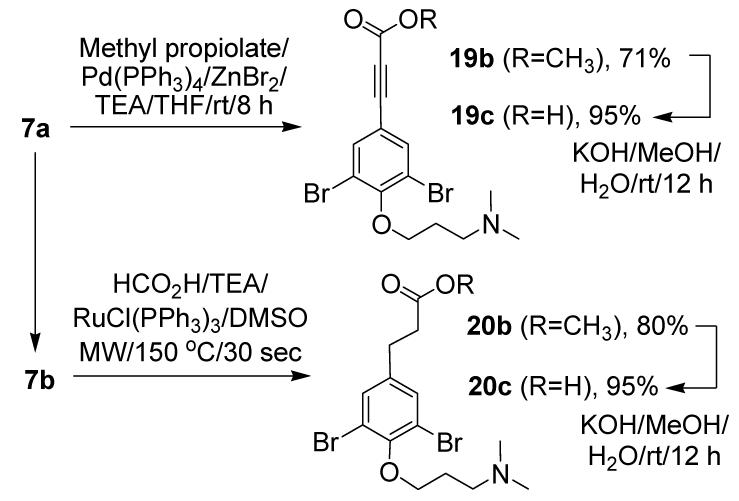
The trans double bond in psammaplysenes constitutes a distinguishing feature compared to other alkaloids of the same family. Since its significance is unknown, building blocks were prepared in which it was replaced by a triple or a single bond (Scheme 3). By carrying out a zinc-mediated Negishi cross-coupling10 with methyl propiolate and Pd(PPh3)4 in TEA on 7a instead of a Heck reaction with methyl acrylate, we obtained triple bond-containing ester 19b, which by nature is more linear than its analogue, 7b. To obtain a compound with higher flexibility, 7b was reduced under microwave-assisted conditions with formic acid/TEA/Wilkinson’s catalyst in DMSO.11 The fully saturated counterpart, 20b, was obtained in good yield after 30 sec at 150 °C. Both 19b and 20b gave the corresponding acids after ester hydrolysis.
Scheme 3.
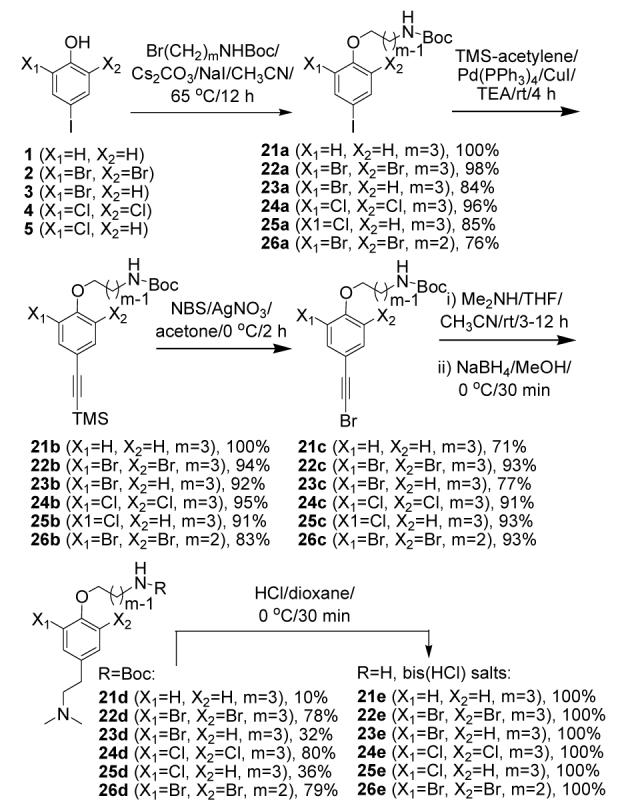
The pseudosymmetric psammaplysene skeleton allows the use of the same key precursors, 1-5, as the starting point for forming primary amine building blocks. Amines bearing various halogen substitution patterns were first prepared (Scheme 4). O-Alkylation of 1-5 with N-Boc-3-bromopropylamine afforded intermediates 21a-25a. By treating only 2 with N-Boc-2-bromoethylamine, we prepared one single building block with shorter central linker (26a, m=2). Iodides 21a-26a were submitted to standard Sonogashira12 cross-coupling with alkynyltrimethylsilane to form TMS-protected alkynes 21b-26b in excellent yields. A highly efficient silver-catalyzed desilylative bromination with NBS in acetone13 converted the latter to bromoacetylides 21c-26c. Aminolysis with dimethylamine in THF/CH3CN, followed by reduction with NaBH4 in MeOH, led to amines 21d-26d. In the aminolysis reaction the dihalogenated precursors appeared considerably more reactive than their monohalogenated and non-halogenated counterparts,14 suggesting the reaction is sensitive to stereoelectronic factors. Primary amines 21e-26e were obtained as their dihydrochloride salts via Boc deprotection with HCl in dioxane.15
Scheme 4.
The net reductive amination of aryl bromoacetylides to saturated phenethylamine systems provided the means for preparing amine building blocks with various amine heads, other than dimethyl (Scheme 5). Four examples were tried. Interestingly the reaction proceeds not only for acyclic secondary amines but also for strained cyclic secondary and for primary amines, even though in the last case, not surprisingly, the yield is considerably lower. In all cases, LC-MS analysis suggested that the intermediates were ynamines or reversible bis-amino adducts, in accord with our previous observations.4
Scheme 5.
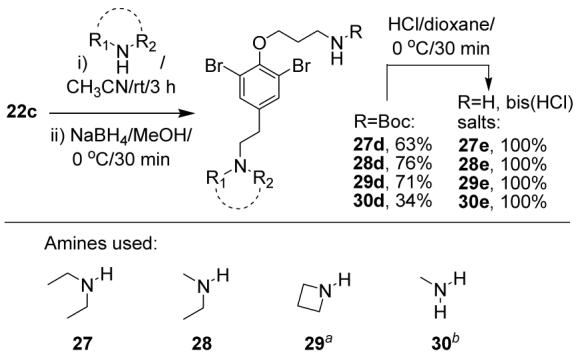
Key: aThis amine was used as the HCl salt and equimolar amount of TEA was added to the reaction mixture. bA mixed THF/CH3CN solvent was used in this case.
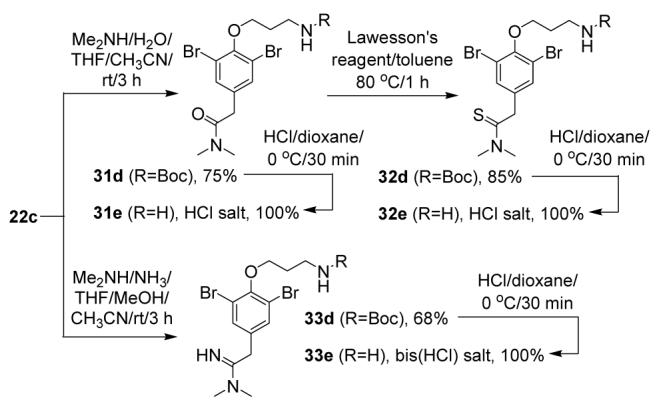
The scope of the amine addition reaction was further studied to include examples where the amine was added in mixtures with other nucleophiles (Scheme 6). If dimethylamine was added to 22c as a 1:1 mixture with H2O, an adduct was formed that tautomerized to amide 31d, in accord with a previous report.16 This amide was successfully converted to the corresponding thioamide (32d) with Lawesson’s reagent in toluene.17 Upon addition of a 1:1 dimethylamine/NH3 mixture to 22c, mixed aryl acetimidamide 33d was obtained. Compounds 31d-33d were deprotected to the free amines 31e-33e without any observed degradation.
Scheme 6.
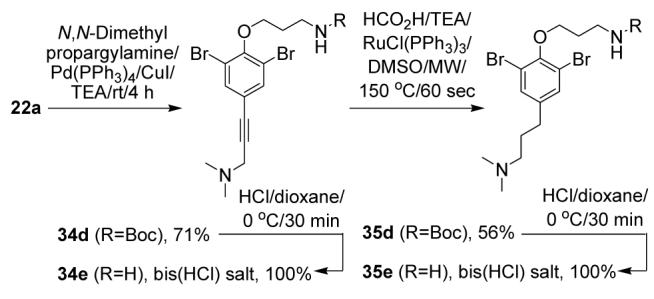
Finally, amine building blocks with a longer terminal linker were prepared (Scheme 7). A Sonogashira coupling of iodide 22a to propargyl dimethylamine afforded 34d. This was reduced under microwave-assisted conditions developed above to give fully saturated 35d (n=3). Both compounds were converted to the free amines after Boc removal.
Scheme 7.
Amide coupling, using diethyl phosphocyanidate with TEA in THF, generated 28 new compounds, resulting from combining all different acid building blocks with the primary amine eastern half of psammaplysene A, 22e, or combining all different amine building blocks with the acid western half of psammaplysene A, 7c (Scheme 8).
Scheme 8a.
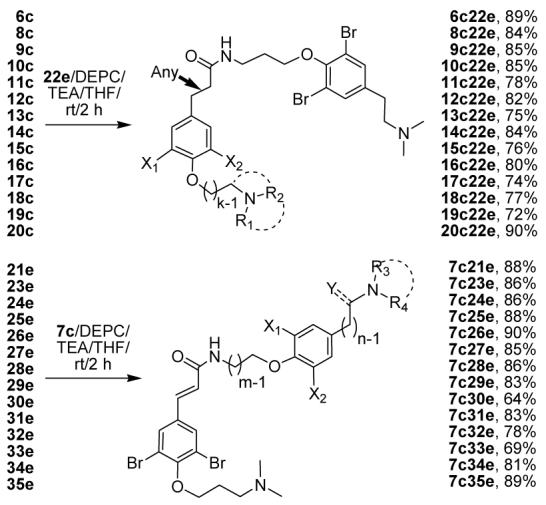
Key: aYields for final products were determined by LC-MS prior to purification. (Yields for all intermediates as shown in previous schemes are isolated yields).
In conclusion, a focused library of psammaplysene-like molecules has been prepared in solution by combining building blocks that derive via divergent pathways from a common precursor, 4-iodophenol. This library will be screened in diverse biological assays.
Supplementary Material
Acknowledgment
We gratefully acknowledge the NIH for funding of this project (CA24487 to J.C.). We thank Dr. Masaki Fujita of HMS for assistance with purifications of final products, Dr. Greg Heffron of the HMS NMR Facility for assistance with 2D-NMR and Dr. Ralph Mazitschek of the Broad Institute for inspiring discussions.
Footnotes
Supporting Information Available. Experimental procedures and spectroscopic data. This material is available free of charge via the Internet athttp://pubs.acs.org.
References
- 1.Breinbauer R, Vetter IR, Waldmann H. Angew. Chem., Int. Ed. 2002;41:2879. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Schroeder FC, Kau TR, Silver PA, Clardy J. J. Nat. Prod. 2005;68:574. doi: 10.1021/np049624z. [DOI] [PubMed] [Google Scholar]
- 3.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. Cancer Cell. 2003;4:463. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- 4.Georgiades SN, Clardy J. Org. Lett. 2005;7:4091. doi: 10.1021/ol0513286. [DOI] [PubMed] [Google Scholar]
- 5.For preparation of tBuNHBr see: Boozer CE, Moncrief JW. J. Org. Chem. 1962;27:623.
- 6.For ortho-directed bromination of unsubstituted phenol with tBuNHBr see: 11 Nov. Jpn. Kokai Tokkyo Koho: 1997.
- 7.Gnaim JM, Sheldon RA. Tetrahedron Lett. 1995;36:3893. [Google Scholar]
- 8.Jeffery T. Tetrahedron. 1996;52:10113. [Google Scholar]
- 9.Significant shifts downfield for the peaks corresponding to the methyl and methylene protons on carbons directly attached to the nitrogens of the acid building blocks suggest that the tertiary amines are protonated. Therefore, the building blocks must be overall zwitterionic, given that they are prepared and isolated in non-acidic conditions. Such pH-dependant shifts are expected for strongly basic systems.
- 10.Anastasia L, Negishi E—I. Org. Lett. 2001;3:3111. doi: 10.1021/ol010145q. [DOI] [PubMed] [Google Scholar]
- 11.Our method was a modification of the one described in: Desai B, Danks TN. Tetrahedron Lett. 2001;42:5963.
- 12.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467. [Google Scholar]
- 13 (a).Bowles DM, Anthony JE. Org. Lett. 2000;2:85. doi: 10.1021/ol991254w. [DOI] [PubMed] [Google Scholar]; (b) Tobe Y, Nakagawa N, Kishi JY, Sonoda M, Naemura K, Wakabayashi T, Shida T, Achiba Y. Tetrahedron. 2001;57:3629. [Google Scholar]; (c) Tobe Y, Utsumi N, Nagano A, Sonoda M, Naemura K. Tetrahedron. 2001;57:8075. [Google Scholar]
- 14.Our stock of 21d was supplemented by an independent pathway, alkylation of commercially available hordenine sulfate with N-Boc-3-bromopropylamine (50% yield), to compensate for the low yield obtained in the reductive aminolysis.
- 15.Han G, Tamaki M, Hruby VJ. J. Peptide Res. 2001;58:338. doi: 10.1034/j.1399-3011.2001.00935.x. [DOI] [PubMed] [Google Scholar]
- 16.Huh DH, Jeong JS, Lee HB, Ryu H, Kim YG. Tetrahedron. 2002;58:9925. [Google Scholar]
- 17.Raucher S, Klein P. Tetrahedron Lett. 1980;21:4061. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.