

Abstract
Cyclin E protein levels and associated kinase activity rise in late G1 phase, reach a peak at the G1/S transition, and quickly decline during S phase. The cyclin E/Cdk2 complex has a well-established function in regulating two fundamental biological processes: cell cycle progression and DNA replication. However, cyclin E expression is deregulated in a wide range of tumors. Our recent reports have uncovered a critical role for cyclin E, independent of Cdk2, in the cell death of hematopoietic tumor cells exposed to genotoxic stress. An 18-kD C-terminal fragment of cyclin E, p18-cyclin E, which is generated by caspase-mediated cleavage in hematopoietic cells during genotoxic stress-induced apoptosis has a critical role in the amplification of the intrinsic apoptotic pathway. By interacting with Ku70, p18-cyclin E liberates Bax, which participates in the amplification of apoptosis by sustaining a positive feedback loop targeting mitochondria. This process is independent of p53 function and new RNA or protein synthesis. Therefore, cyclin E emerges as an arbiter of the genotoxic stress response by regulating a finite physiological balance between cell proliferation and death in hematopoietic cells.
Keywords: cyclin E, Cdk2, Ku70, Bax, caspase, apoptosis, cell cycle, genotoxic stress
CDK2 Dependent Roles of Cyclin E
Cyclins regulate cell cycle progression by binding to their catalytic interacting partners, cyclin dependent kinases (Cdks). The activity of Cdk's is positively controlled by cyclins and negatively by binding to Cdk inhibitors (CKIs).1,2 The transit between the G1 and S phases of the cell cycle is primarily regulated by the cyclin E/Cdk2 complex.3,4 Even though D-type cyclins are also involved in the G1/S transition, all phenotypic and developmental defects in mice caused by cyclin D1 deficiency can be rescued by cyclin E knock-in at the cyclin D1 locus, indicating that the function of cyclin D1 may be replaced by cyclin E.5
Cyclin E1, referred to here as cyclin E, is the best known E-type cyclin and has 395 amino acids and a molecular weight of 50 kDa.6,7 In normal cells, many gene products required for S-phase progression are under the control of the E2F family of transcription factors, which become free upon phosphorylation of the retinoblastoma protein (Rb) by cyclin E/Cdk2. Constitutive or ectopic expression of cyclin E can accelerate G1 progression4 and chromosome instability,8,9 presumably because of inappropriate S-phase entry/progression.10 The cyclin E/Cdk2 complex plays an important role in the initiation of DNA replication.4,11 In Xenopus, there is strong evidence implicating cyclin E/Cdk2 in the initiation of DNA replication and in centrosome duplication.12 A recent report in mammals indicates that cyclin E also plays a kinase-independent function in DNA replication by facilitating minichromosome maintenance (MCM) loading through physical interaction with CDT1 and MCM.13
The expression of cyclin E oscillates during cell cycle progression due to tight regulation at transcriptional as well as post-transcriptional levels. At transcriptional level, the expression of cyclin E is regulated primarily by E2F14,15 and at the post-transcriptional level by its ubiquitin-proteasome-mediated degradation. Cdk2-bound cyclin E is targeted to proteasomal degradation upon autophosphorylation or phosphorylation by GSK3, with the free cyclin E being rapidly degraded.16-18 The two forms of cyclin E, either free or Cdk2-bound, are regulated by the E3 ubiquitin ligases Cdc4/Fbw7 and Cul3.19 In addition, it has recently become evident that cyclin E can be processed proteolytically, with the resulting fragments playing important biological and pathological roles. In cyclin E-overexpressing breast tumors and tumor-derived cell lines, cyclin E is proteolytically cleaved to generate N-terminally truncated low molecular weight (LMW) forms, that confer increased Cdk2 kinase activity and are much more resistant to the CDK inhibitors (CKIs) p21Waf1 and p27Kip1.20 How these LMW fragments of Cyclin E are generated and which proteases are responsible for this conversion is still controversial (Fig. 1), although both elastase21 and calpain22,23 have been implicated as key enzymes in cyclin E proteolysis. These LMW forms of cyclin E are clearly functional and regulate tumor progression and metastasis. Their role is, however, quite distinct from that of p18-cyclin E, which is generated in hematopoietic cells as a result of proteolytic cleavage. The function of this fragment is described below.
Figure 1.
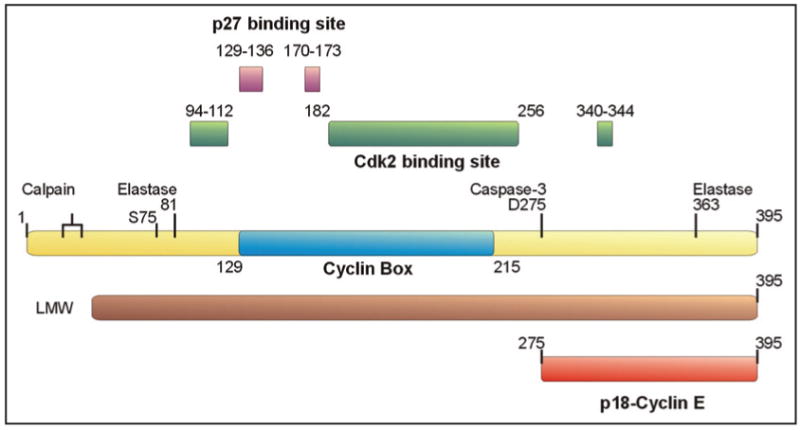
Cyclin E and its derivatives in cancer. Cyclin E has been found to be cleaved by the elastase family of proteases generating amino truncated forms, which retain Cdk2 binding capability and appear to be biochemically hyperactive (designated as LMW *).20 Interestingly, the structure of the cyclin E-Cdk2 complex was determined using a truncated form of cyclin E (81–363) following an in vitro elastase cleavage.64 Based on this structure, the interface between cyclin E and Cdk2 was defined as the 94–112, 182–256, and 340–344 amino acid domains. Through analogy with the interaction of cyclin A with p27Kip1, it was suggested that 129–136 and 170–173 are the residues responsible for the binding and inhibition of cyclin E by p27Kip1. On the other hand, calpain was also shown to be responsible for the generation of amino terminus truncated, hyperactive LMW of Cyclin E.22 The 129–215 residue of cyclin E (illustrated in blue) represents the “cyclin box” which is a conserved domain among cyclins. Recently, Ser75 has been identified to render cyclin E kinase inactive, although several other functions, such as MCM replicative helicase loading and restoration of cell-cycle reentry have not been affected.13 p18-cyclin E, on the other hand, is generated through Caspase-3 dependent cleavage at Asp275, generating a carboxy terminal cyclin E fragment, which lacks the Cdk2 or CKI binding domains, therefore, it is unable to bind and affect Cdk2 activity.27
CDK2-Independent Role of Cyclin E in Cell Death Amplification Through Generation of P18-Cyclin E
Genotoxic agents, such as ionizing radiation, induce apoptosis in many cell types, but most potently in those of hematopoietic origin.24 Apoptosis is frequently associated with proliferating cells, implying the existence of molecules in late G1 and S phase, whose activities facilitate execution of the apoptotic process. Once the cells are committed to cell death, apoptogenic factors, including cytochrome c, are released from mitochondria to initiate a caspase cascade.25 Cytochrome c acts as a cofactor to stimulate the interaction of Apaf-1 with pro-caspase 9,26 which then initiates activation of the caspase cascade. A critical downstream caspase responsible for cleavage of important cellular substrates is Caspase-3.
In addition to its role in cell cycle control,3,4 cyclin E has been shown to have a Cdk2 independent function in regulating apoptosis of hematopoietic cells.27,28 The larger N-terminal truncation of cyclin E that generates p18-cyclin E prevents its binding to either Cdk2 or CKIs p21Waf1 or p27Kip1. Therefore, unlike the LMW isoforms of cyclin E (Fig. 1), p18-cyclin E cannot bind Cdk2 and therefore, interfere with Cdk2 (bound to cyclin E1, E2, or A)-dependent kinase activity. Cleavage of cyclin E results in loss of its Cdk2 associated kinase activity, and consequently its cell cycle function.27
It has recently been discovered that the p53 tumor suppressor activates apoptosis through parallel pathways that may depend on transcription-dependent and independent events.29 Bax is one of the best-known p53 transcriptional target that may impact on apoptosis. A transcription-independent role of p53 in apoptosis has been attributed to its ability to block the anti-apoptotic function of Bcl-2 and Bcl-xL on the mitochondria.30,31 Alternatively, it has been suggested to promote, directly or indirectly, the apoptotic activities of pro-apoptotic members of the Bcl-2 family, Bax and Bak.30,32 Most tumor cells have inactivated p53, about 50% of which is achieved through mutations in its sequence and the rest by disabling key components that lie upstream or downstream of p53 in a common signaling pathway.33 To investigate the possible role of p53 in hematopoietic cells, p18-cyclin E-induced apoptosis was examined in IM-9 B-cell tumor cells in which ∼90% of p53 was depleted by stable siRNA-mediated knockdown using sipSUPER-p53 expression.34 There was no change in the extent of apoptosis mediated by HA-p18-cyclin E in these cells (Fig. 2A), even though p53 was rendered nonfunctional as evidenced by the lack of expression of its key transcriptional target p21Waf1 35 and a dramatic reduction in the extent of apoptosis following irradiation (data not shown). Importantly, there was no difference in the levels of Bax in IM-9 cells following their transfection with p18-cyclin E, regardless of the functional status of p53.
Figure 2.
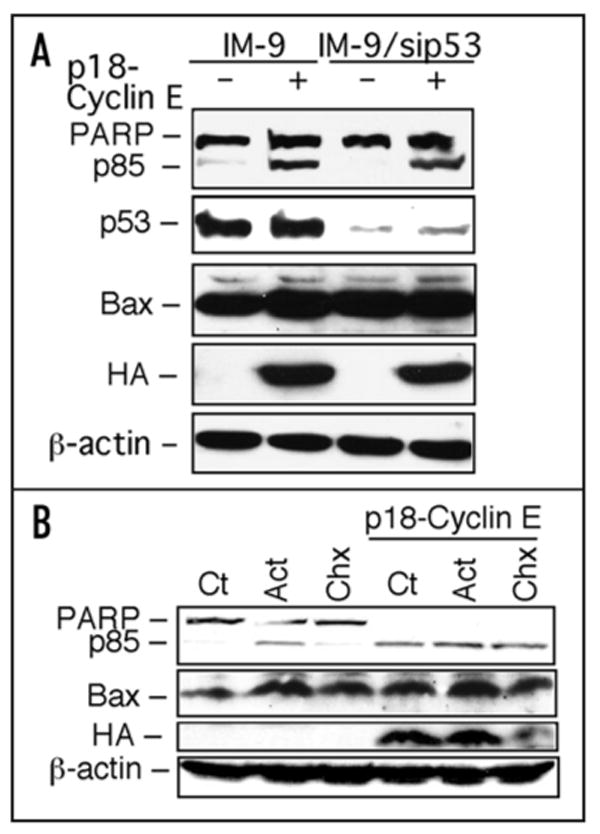
p18-cyclin E-mediated cell death is independent of p53, transcription, or translational control. (A) p18-cyclin E induces apoptosis in wild-type and p53-deficient IM-9 cells. Parental and a derivative IM-9 cell clone stably expressing sip53-SUPER (puromycin selection) were transfected with p18-cyclin E. At 24 h post transfection, cells were lysed and Western blotting was performed for PARP (full length and p85), p53, Bax, and HA-tagged p18-cyclin E. β-actin was used as a loading control. (B) p18-cyclin E induced apoptosis is independent of de novo RNA synthesis or new protein expression. NCI-H1299 cells were either untreated (Ct) or treated with actinomycin D (Act D; 10 nM) or cycloheximide (Chx; 10 μg/ml) for 24 h without or following transfection with p18-cyclin E. Western blot was used to examine PARP, HA-tagged p18-cyclin E, Bax and β-actin expression with the corresponding primary antibodies.
Next we wanted to determine whether p18-cyclin E-induced apoptosis is dependent on de novo RNA synthesis or protein expression. p18-cyclin E-induced apoptosis in NCI-H1299 cells, a cell line that has been previously used to demonstrate transcription-independent apoptosis,36 was examined in the absence or presence of either actinomycin D, a general transcription inhibitor or cycloheximide, a general protein synthesis inhibitor. In the presence of p18-cyclinE, there was no difference in the extent of apoptosis, as assayed by poly (ADP-ribose) polymerase-1 (PARP-1) cleavage, in the absence or presence of these inhibitors. These results suggest that p18-cyclin E-induced apoptosis is independent of transcriptional or translational control (Fig. 2B). As NCI-H1299 cells lack p53, this result also suggests that the function of p53 is not required for the apoptotic effect of p18-cyclin E. Taken together, these data indicate that p18-cyclin E-mediated apoptosis is independent of p53 function, as well as independent of new RNA or protein synthesis.
Bax Activation in Apoptosis
A key regulatory step in the intrinsic cell death pathway is the activation of the proapoptotic molecule Bax.37 Following its activation, Bax translocates from the cytosol to the outer mitochondrial membrane, where it oligomerizes and forms pore-like structures, leading to release of cytochrome c and other death-inducing factors.38,39 Although the mechanism of Bax activation remains unclear, a recent study indicates that Bax is kept inactive in some resistant cells by interacting with cytosolic Ku70.40,41 Overexpression of Ku70 prevents Bax-mediated apoptosis, whereas depletion of Ku70 causes cells to become more sensitive to a variety of apoptotic stimuli. These results demonstrate that Ku70 is a physiological inhibitor of Bax-induced apoptosis, a finding that expands on Ku70's previously known role in DNA repair,42 particularly through nonhomologous end-joining (NHEJ). Interestingly, cells from Ku70 knockout mice are hypersensitive to agents that induce apoptosis in the absence of DNA damage, such as staurosporine.43 This suggests that Ku70 plays a role in suppressing apoptosis that is independent of its role in DNA repair.42 Based on these findings, nuclear Ku70 is therefore considered to be mainly responsible for repair of DNA damage, whereas the cytosolic pool of Ku70 may be primarily a regulator of Bax activation.40,41
Our recent findings establish a role for Ku70 in regulating Bax-mediated apoptosis in hematopoietic cells that generate p18-cyclin E.44 By interacting with Ku70, p18-cyclin E releases Bax from Ku70 thus allowing Bax to be activated during gentoxic stress, and thereby leading to apoptosis.44 Therefore, p18-cyclin E, Bax, and Ku70 emerge as key regulators of the apoptotic pathway in hematopoietic cells. The sensitivity of these cells to genotoxic stress can be explained by their ability to generate p18-cyclin E. Therefore, Ku70 plays a dual role in NHEJ and regulation of Bax, which in turn may determine the balance between cell survival and programmed cell death in hematopoietic cells.
Ku70 may not be unique in assuring Bax anchoring/sequestration in the cytosol. Other Bax interactive proteins, such as the peptide humanin,45,46 the apoptosis repressor with caspase recruitment domain (ARC),47 or various forms of 14-3-348,49 and crystalline50 may also function in various cellular systems to sequester Bax. Moreover, other Bax-interacting proteins, such as p53,32,51 Bif-1,52 apoptosis-associated speck-like protein (ASC),53 and MOAP-154 promote Bax conformational change and mitochondrial translocation.
A schematic model depicts the key pathways for the function of cyclin E in cell cycle progression and that of p18-cyclin E in genotoxic stress-induced apoptosis. Through its dual role, cyclin E provides a physiological balance between cell proliferation and death of hematopoietic cells (Fig. 3). The model proposes an initial limited activation of Caspase-3 perhaps through a small amount of primed/activated mitochondrial Bax.37 Other factors, such as p53 may also contribute, in a transcription independent or dependent manner.29 Then, cyclin E is proteolytically cleaved by Caspase-3 to generate p18-cyclin E, which interacts with Ku70 resulting in release of Bax from the Ku70-Bax complex. This contributes to a robust activation of Bax, leading to its mitochondrial targeting and amplification of the intrinsic pathway of apoptosis. This is consistent with a feedback amplification loop model that is supported by our earlier findings that limited caspase activation is not sufficient to release enough cytochrome c to lead to significant apoptosis in hematopoietic cells. Rather, an amplification loop is required to trigger massive cytochrome c release from mitochondria resulting in abundant Caspase-3 activation leading to cell death.25 Our previous report55 and a similar model56 invoke a 2-step cytochrome c release to accomplish the apoptotic process. Moreover, assays using caspase-3- and caspase-7-deficient embryonic fibroblasts and thymocytes revealed considerable protection against different proapoptotic stimuli. These cells are also defective in several characteristic features of apoptosis. However, following ultraviolet radiation treatment, these embryonic fibroblasts show a remarkable delay in the kinetics of cytochrome c release and Bax translocation. This strongly suggests that Caspase-3 and Caspase-7 may participate in a feedback amplification loop to promote the mitochondrial apoptotic pathway.57
Figure 3.
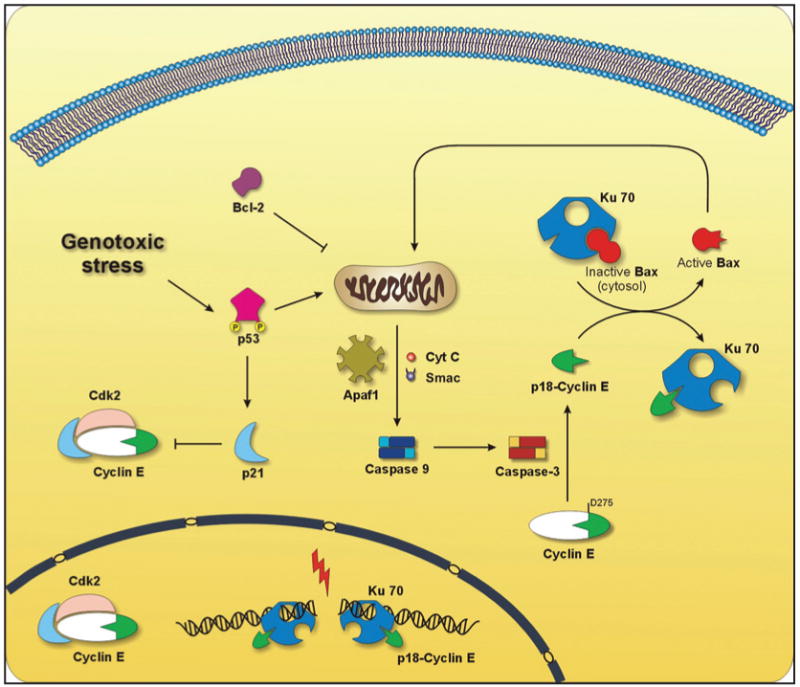
Dual role of cyclin E in apoptosis and cell cycle regulation. Genotoxic stress induced by DNA damaging agents, such as ionizing radiation, causes minimal activation of Caspase-3 which cleaves full length cyclin E. The resulting p18-cyclin E binds Ku70 in the cytosol leading to the release of inactive Bax from its complex with Ku70. Bax is translocated to the mitochondria inducing the release of several apoptogenic factors, such as Smac/Diablo and cytochrome c (cytc), which together with Apaf-1 lead to the full activation of Caspase-9, that can be prevented by Bcl-2. This in turn cleaves and activates more Caspase-3, leading to amplification of the apoptotic signal. Genotoxic stress also leads to the phosphorylation and activation of p53 causing an increase in the levels of p21Waf1. p21Waf1 binds and inhibits the cyclin E-Cdk2 complex leading to cell cycle arrest. Moreover, we have found p18-cyclin E in complex with Ku70 in the nucleus, which may also involve p18-cyclin E in regulating DNA repair, particularly nonhomologous end-joining (NHEJ).
Although caspases are classically known for their role in apoptosis, their involvement in biological processes apart from cell death has recently received increased attention. For example, they play an important role in the inflammatory signaling pathways.58,59 The non-apoptotic functions of these proteases suggest that they may become activated independently of the apoptotic cascade, thus leading to the cleavage of specific substrates, such as kinases, cytokines, transcription factors, and polymerases. Functional analysis of conditional caspase-deficient mice or derived cells confirmed the crucial roles of caspases in cell proliferation, differentiation, and inflammation. It has been reported recently that Caspase-3 is a negative regulator of B cell cycling. This study shows that while the apoptotic pathway is largely unaffected in Caspase-3 knockout mice, splenic B cell proliferation is enhanced in vivo, and in response to mitogenic stimulation in vitro, hyperproliferation is seen. Genetic and biochemical data demonstrate that Caspase-3 is essential for the regulation of B cell homeostasis.60 In contrast to Caspase-3, Caspase-8 is a positive regulator of cell cycling as B-cell proliferation since caspase-8 deficient mice fail to proliferate.61 Recently, it has been suggested that Caspase-3 and Caspase-7 together play a role in development, since double knockout mice exhibit defects in heart development, leading to death immediately after birth. In contrast, knockout of either caspase individually on the same genetic background resulted in viable animals.
Another remarkable example of the critical requirement for caspase activation is in the process of erythropoiesis, which involves nuclear condensation, followed by extrusion of the nucleus to generate enucleated reticulocytes and eventually mature erythrocytes. Red blood cell production is inhibited by caspases, which trigger the degradation of the major erythroid transcription factor GATA-1 in late erythroid progenitors and early erythroblasts, followed by reversible growth and differentiation arrest or apoptosis. Interestingly, gene knock-out studies have shown that GATA-1 is essential for erythroid cell survival and maturation. Moreover, SCL/Tal-1, a helix-loop-helix transcription factor required for blood cell development, is a key target of caspases in developing erythroblasts. Remarkably, caspase-resistant SCL/Tal-1 expression in erythroid progenitors inhibits the amplification of caspase activation.62,63
The cleavage of cyclin E as well as cell death can be prevented by Bcl-2 overexpression, indicating that Bcl-2 functions upstream of the mitochondrial apoptotic cascade.25,27 How Bax is released from its complex with Ku70 is still unknown. One mechanism may consist in a structural change in conformation of the Bax binding-domain of Ku70 upon binding of p18-cyclin E at the amino terminus. Another possibility might be the recruitment of histone acetyl transferases, which would acetylate several lysine residues at the carboxy terminus of Ku70.40,41 However, it is quite possible that other pathways and various feedback loops exist to activate Bax and thereby, to amplify the apoptotic cascade in hematopoietic and other cell types.
Conclusions
Recent developments have uncovered unexpected roles for cyclin E in cell cycle control, DNA replication, and in apoptosis. Some of these are clearly independent of the Cdk2-associated kinase activity or of Cdk2 binding altogether.13,44 Since p18-cyclin E is generated in all hematopoietic tumor cells and not in untransformed cells that we have investigated, it may become useful as a biological indicator of successful therapies. Specific targeting of cyclin E to generate p18-cyclin E or strategies that enhance the levels of p18-cyclin E could provide new clinical therapeutic approaches. In other cell types where p18-cyclin E may not be generated from cyclin E, its ectopic expression or that of its mimetics (e.g., peptide or small molecule inhibitor) may prove to be valuable therapeutically.
As cyclin E proteolysis to p18-cyclin E seems to be limited to hematopoietic cells,27,44 why epithelial cells, for example do not seem to generate p18-cyclin E is intriguing. Moreover, once cyclin E is cleaved, the resulting p18-cyclin E derivative functions quite differently. In fact, yeast two-hybrid analyses designed to identify p18-cyclin E-binding partners could not find most of the common partners of cyclin E.44 So not only does it look different, but p18-cyclin E also acts differently from cyclin E, being a killer and no longer a healer, therefore a story reminiscent of Dr. Jekyll and Mr. Hyde.
Acknowledgments
The work described in this article was supported by a research grant from the US National Institutes of Health (CA81504). We would like to thank Dr. Lisa Middleton for careful reading of the manuscript.
References
- 1.Reed SI. Ratchets and clocks: The cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4:855–64. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 2.Murray AW. Recycling the cell cycle: Cyclins revisited. Cell. 2004;116:221–34. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 3.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–24. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–9. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–77. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- 6.Lew DJ, Dulic V, Reed SI. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell. 1991;66:1197–206. doi: 10.1016/0092-8674(91)90042-w. [DOI] [PubMed] [Google Scholar]
- 7.Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, Roberts JM. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66:1217–28. doi: 10.1016/0092-8674(91)90044-y. [DOI] [PubMed] [Google Scholar]
- 8.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 9.Hubalek MM, Widschwendter A, Erdel M, Gschwendtner A, Fiegl HM, Muller HM, Goebel G, Mueller-Holzner E, Marth C, Spruck CH, Reed SI, Widschwendter M. Cyclin E dysregulation and chromosomal instability in endometrial cancer. Oncogene. 2004;23:4187–92. doi: 10.1038/sj.onc.1207560. [DOI] [PubMed] [Google Scholar]
- 10.Almasan A, Linke S, Paulson T, Huang LC, Wahl GM. Genetic instability as a consequence of inappropriate entry and progression through S-phase. Cancer Metast Rev. 1995;14:59–73. doi: 10.1007/BF00690212. [DOI] [PubMed] [Google Scholar]
- 11.Jackson PK, Chevalier S, Philippe M, Kirschner MW. Early events in DNA replication require cyclin E and are blocked by p21CIP1. J Cell Biol. 1995;130:755–69. doi: 10.1083/jcb.130.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furstenthal L, Kaiser BK, Swanson C, Jackson PK. Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J Cell Biol. 2001;152:1267–78. doi: 10.1083/jcb.152.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, Sicinski P. Kinase-independent function of cyclin E. Mol Cell. 2007;25:127–39. doi: 10.1016/j.molcel.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 14.Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004;3:1208–11. doi: 10.4161/cbt.3.12.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazumder S, Gong B, Almasan A. Cyclin E induction by genotoxic stress leads to apoptosis of hematopoietic cells. Oncogene. 2000;19:2828–35. doi: 10.1038/sj.onc.1203623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. Embo J. 1996;15:4182–93. [PMC free article] [PubMed] [Google Scholar]
- 17.Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10:1979–90. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 18.Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW. A family of mammalian F-box proteins. Curr Biol. 1999;9:1180–2. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
- 19.Reed SI. Cooperation between different Cdc4/Fbw7 isoforms may be associated with 2-step inactivation of SCF(Cdc4) targets. Cell Cycle. 2006;5:1923–4. doi: 10.4161/cc.5.17.3198. [DOI] [PubMed] [Google Scholar]
- 20.Wingate H, Zhang N, McGarhen MJ, Bedrosian I, Harper JW, Keyomarsi K. The tumor-specific hyperactive forms of cyclin E are resistant to inhibition by p21 and p27. J Biol Chem. 2005;280:15148–57. doi: 10.1074/jbc.M409789200. [DOI] [PubMed] [Google Scholar]
- 21.Porter DC, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruki S, Keyomarsi K. Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol. 2001;21:6254–69. doi: 10.1128/MCB.21.18.6254-6269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang XD, Rosales JL, Magliocco A, Gnanakumar R, Lee KY. Cyclin E in breast tumors is cleaved into its low molecular weight forms by calpain. Oncogene. 2003;22:769–74. doi: 10.1038/sj.onc.1206166. [DOI] [PubMed] [Google Scholar]
- 23.Libertini SJ, Robinson BS, Dhillon NK, Glick D, George M, Dandekar S, Gregg JP, Sawai E, Mudryj M. Cyclin E both regulates and is regulated by calpain 2, a protease associated with metastatic breast cancer phenotype. Cancer Res. 2005;65:10700–8. doi: 10.1158/0008-5472.CAN-05-1666. [DOI] [PubMed] [Google Scholar]
- 24.Gong B, Chen Q, Endlich B, Mazumder S, Almasan A. Ionizing radiation-induced, Bax-mediated cell death is dependent on activation of cysteine and serine proteases. Cell Growth Differ. 1999;10:491–502. [PubMed] [Google Scholar]
- 25.Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: Evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–33. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 27.Mazumder S, Chen Q, Gong B, Drazba JA, Buchsbaum JC, Almasan A. Proteolytic cleavage of cyclin E leads to inactivation of associated kinase activity and amplification of apoptosis in hematopoietic cells. Mol Cell Biol. 2002;22:2398–409. doi: 10.1128/MCB.22.7.2398-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazumder S, DuPree EL, Almasan A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr Cancer Drug Targets. 2004;4:65–75. doi: 10.2174/1568009043481669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuster JJ, Sanz-Gonzalez SM, Moll UM, Andres V. Classic and novel roles of p53: Prospects for anticancer therapy. Trends Mol Med. 2007;22:22. doi: 10.1016/j.molmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–50. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 31.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 32.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 33.Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage: Insights from yeast and p53. Nat Cell Biol. 2001;3:E277–86. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 34.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 35.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 36.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–81. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 37.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 39.Ray S, Almasan A. Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res. 2003;63:4713–23. [PubMed] [Google Scholar]
- 40.Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–38. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 41.Subramanian C, Opipari AW, Jr, Bian X, Castle VP, Kwok RP. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:4842–7. doi: 10.1073/pnas.0408351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Y, Lu H, Schwarz K, Lieber MR. Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: The iterative processing model. Cell Cycle. 2005;4:1193–200. doi: 10.4161/cc.4.9.1977. [DOI] [PubMed] [Google Scholar]
- 43.Chechlacz M, Vemuri MC, Naegele JR. Role of DNA-dependent protein kinase in neuronal survival. J Neurochem. 2001;78:141–54. doi: 10.1046/j.1471-4159.2001.00380.x. [DOI] [PubMed] [Google Scholar]
- 44.Mazumder S, Plesca D, Kintner M, Almasan A. Interaction of a Cyclin E fragment with Ku70 regulates Bax-mediated apoptosis in hematopoietic cells. Mol Cell Biol. 2007;27:3511–20. doi: 10.1128/MCB.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–24. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 46.Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–61. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 47.Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279:21233–8. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 48.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. Embo J. 2004;23:1889–99. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y. 14-3-3 Interacts directly with and negatively regulates pro-apoptotic Bax. J Biol Chem. 2003;278:2058–65. doi: 10.1074/jbc.M207880200. [DOI] [PubMed] [Google Scholar]
- 50.Mao YW, Liu JP, Xiang H, Li DW. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–26. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- 51.Deng Y, Wu X. Peg3/Pw1 promotes p53-mediated apoptosis by inducing Bax translocation from cytosol to mitochondria. Proc Natl Acad Sci USA. 2000;97:12050–5. doi: 10.1073/pnas.97.22.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ, Wang HG. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005;25:9369–82. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA, Lee SW. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell Biol. 2004;6:121–8. doi: 10.1038/ncb1087. [DOI] [PubMed] [Google Scholar]
- 54.Vos MD, Dallol A, Eckfeld K, Allen NP, Donninger H, Hesson LB, Calvisi D, Latif F, Clark GJ. The RASSF1A tumor suppressor activates Bax via MOAP-1. J Biol Chem. 2006;281:4557–63. doi: 10.1074/jbc.M512128200. [DOI] [PubMed] [Google Scholar]
- 55.Almasan A. Cellular commitment to radiation-induced apoptosis. Radiat Res. 2000;153:347–50. doi: 10.1667/0033-7587(2000)153[0347:cctria]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 56.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–63. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–51. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–46. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sekiyama A, Ueda H, Kashiwamura S, Sekiyama R, Takeda M, Rokutan K, Okamura H. A stress-induced, superoxide-mediated caspase-1 activation pathway causes plasma IL-18 upregulation. Immunity. 2005;22:669–77. doi: 10.1016/j.immuni.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 60.Woo M, Hakem R, Furlonger C, Hakem A, Duncan GS, Sasaki T, Bouchard D, Lu L, Wu GE, Paige CJ, Mak TW. Caspase-3 regulates cell cycle in B cells: A consequence of substrate specificity. Nat Immunol. 2003;4:1016–22. doi: 10.1038/ni976. [DOI] [PubMed] [Google Scholar]
- 61.Beisner DR, Ch'en IL, Kolla RV, Hoffmann A, Hedrick SM. Cutting edge: Innate immunity conferred by B cells is regulated by caspase-8. J Immunol. 2005;175:3469–73. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 62.Zeuner A, Eramo A, Testa U, Felli N, Pelosi E, Mariani G, Srinivasula SM, Alnemri ES, Condorelli G, Peschle C, De Maria R. Control of erythroid cell production via caspase-mediated cleavage of transcription factor SCL/Tal-1. Cell Death Differ. 2003;10:905–13. doi: 10.1038/sj.cdd.4401255. [DOI] [PubMed] [Google Scholar]
- 63.Testa U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia. 2004;18:1176–99. doi: 10.1038/sj.leu.2403383. [DOI] [PubMed] [Google Scholar]
- 64.Honda R, Lowe ED, Dubinina E, Skamnaki V, Cook A, Brown NR, Johnson LN. The structure of cyclin E1/CDK2: Implications for CDK2 activation and CDK2-independent roles. Embo J. 2005;24:452–63. doi: 10.1038/sj.emboj.7600554. [DOI] [PMC free article] [PubMed] [Google Scholar]