

Abstract
Objective
Mechanisms by which tumor necrosis factor-α (TNF) contributes to atherosclerosis remain largely obscure. We therefore sought to determine the role of the arterial wall TNF receptor-1 (TNFR1) in atherogenesis.
Methods and Results
Carotid artery-to-carotid artery interposition grafting was performed with tnfr1−/− and congenic (C57Bl/6) wild-type (WT) mice as graft donors, and congenic chow-fed apolipoprotein E-deficient mice as recipients. Advanced atherosclerotic graft lesions developed within 8 weeks, and had 2-fold greater area in WT than in tnfr1−/− grafts. While the prevalence of specific atheroma cells was equivalent in WT and tnfr1−/− grafts, the overall abundance of cells was substantially greater in WT grafts. WT grafts demonstrated greater MCP-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression at both early and late time points, and proliferating cell nuclear antigen expression at early time points. Aortic atherosclerosis was also reduced in 14-month-old apoe−/−/tnfr1−/− mice, as compared with cognate apoe−/− mice. In coculture with activated macrophages, smooth muscle cells expressing the TNFR1 demonstrated enhanced migration and reduced scavenger receptor activity.
Conclusions
TNFR1 signaling, just in arterial wall cells, contributes to the pathogenesis of atherosclerosis by enhancing arterial wall chemokine and adhesion molecule expression, as well as by augmenting medial smooth muscle cell proliferation and migration.
Keywords: atherosclerosis, inflammation, mouse models, smooth muscle cells, tumor necrosis factor
Asubstantial body of evidence supports a proatherogenic role for the proinflammatory cytokine tumor necrosis factor-α (TNF), which exerts its cellular effects by activating the plasma membrane receptors TNF receptor-1 (TNFR1) and TNF receptor-2 (TNFR2). Most cogently, mice deficient in both apolipoprotein E (apoE) and TNF demonstrate early aortic atherosclerosis that is 50% less than that seen in apoe−/− controls—an observation that appears largely attributable to leukocyte-produced TNF.1 Moreover, systemic administration of a soluble TNFR1 reduces aortic atherosclerosis by an amount comparable to that seen with genetic TNF deficiency.1 In humans, plasma TNF concentrations predict recurrent myocardial infarction,2 and TNF is an important inducer of hepatic C-reactive protein,3 a powerful predictor of incident myocardial infarction.4 Mechanisms by which TNF may contribute to atherogenesis include myriad effects on monocyte/macrophages, as well as the induction of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) expression by endothelial cells5 and vascular smooth muscle cells (SMCs),6 endothelial cell apoptosis,7 and SMC migration and proliferation.8
Signaling mechanisms responsible for proatherogenic TNF activity remain to be clarified, however. TNFR1-deficient mice demonstrate ≈2-fold more diet-induced aortic sinus atherosclerosis than wild-type (WT) mice—perhaps because of enhanced macrophage scavenger receptor expression and cholesteryl ester accumulation.9 Furthermore, apoe−/−/tnfr1−/− mice develop innominate artery atherosclerosis comparable to that observed in apoe−/− controls.10 Together, these results suggest the possibility that the TNFR1 in macrophages may limit atherogenesis, whereas the TNFR1 in the arterial wall (SMCs and/or endothelial cells) may augment atherogenesis. To test the role of vascular wall TNFR1 in atherosclerosis, we created interposition carotid artery grafts from tnfr1−/− or WT mice in congenic apoe−/− mice.
Materials and Methods
For supplementary Methods, please see http://atvb.ahajournals.org.
Carotid Artery Interposition Grafting
WT C57Bl/6J and congenic apoe−/−, tnfr1−/−, and green fluorescent protein (GFP)-transgenic mice were obtained from the Jackson Laboratory (Bar Harbor, ME). GFP-transgenic apoe−/− and tnfr1−/− mice were derived by cross-breeding. All animal experiments followed institutional guidelines.
The surgical procedure was analogous to that reported for interposition vein grafting.11 Right common carotid arteries (≈8mm long) of WT or tnfr1−/− donor mice were anastomosed end-to-side into the right common carotid artery of recipient apoe−/− mice using 11−0 nylon suture (US Surgical). Subsequently, the intervening recipient mouse carotid was ligated and cut. Graft recipient and donor mice were 20±3 weeks old at the time of operation, and were matched for age and gender. All mice were fed low-fat Purina Rodent Chow 5058, and the graft failure rate (from anastomotic occlusion) was <2%.
To ensure that carotid interposition grafting did not promote systemic inflammation, we measured serum amyloid A by enzyme-linked immunosorbent assay (Biosource International, Inc) and serum TNF by bioassay (see online Methods) at 1 and 4 weeks postoperatively. These inflammatory markers were indistinguishable among operated and unoperated age-matched and gender-matched mice. Serum amyloid A levels were 0.6±0.3 μg/mL, and serum TNF was <5 pmol/L (n=3/group).
At harvest, carotid grafts were flushed, perfusion-fixed, and paraffin-embedded as described previously for vein grafts.11 GFP and Sudan IV-stained graft specimens were flushed with phosphate-buffered saline and embedded in OCT before sectioning.
Data Analysis
Data are presented as mean±SD in the text, and ±SE in the figures. Morphometry, protein expression, and migration data were collected by observers blinded to specimen genotype, and analyzed by 1-way ANOVA with Tukey post-hoc test for multiple comparisons.
Results
To study whether TNFR1 expression in arterial wall cells contributes to atherosclerosis, we transplanted congenic carotid arteries, WT and TNFR1-deficient, into the right common carotid artery of congenic apoe−/− mice as interposition grafts (Figure 1). These grafts developed atherosclerosis more rapidly than the contralateral native carotid of the apoe−/− mouse (Figure 1A and data not shown), but this accelerated graft atherosclerosis nonetheless demonstrated a sequence of cellular infiltration that typifies native-vessel atherosclerosis.12 In 4-week-old grafts, type II lesions13 comprising only intimal macrophages prevailed; SMCs were restricted to the tunica media, and T cells constituted only ≈1% to 2% of cells (Figure 1F and data not shown). In 8-week-old grafts, by contrast, more complex lesions comprised not only intimal macrophage foam cells but also intimal SMC-like cells, fibrous caps, extracellular lipid, necrotic core, and plaque hemorrhage (Figure 1E to 1G; supplemental Figures I and II, available online at http://atvb.ahajournals.org). Moreover, medial replacement and adventitial inflammation manifested (Figure 1; supplemental Figures I and II), characteristic of advanced atherosclerotic lesions in the apoe−/− mouse.14 The contribution of carotid graft-intrinsic cells to 8-week-old atheromata is evinced by staining for apoE, which entirely overlaps with staining for SMC actin (Figure 1G and data not shown). Thus, at both 4 and 8 weeks postoperatively, atherosclerosis in the carotid grafts demonstrated many of the characteristic morphological features that obtain in most arteries.
Figure 1.
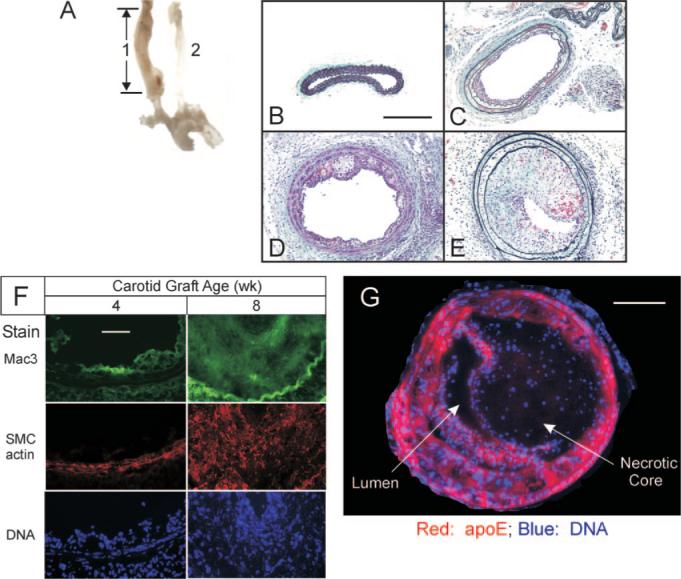
Atherosclerosis progresses rapidly in congenic carotid grafts implanted in apoe−/− mice. A, The 8-mm carotid interposition graft (1), thickened by atherosclerosis, was harvested 8 weeks postoperatively, along with the aortic arch and contralateral carotid (2). Cross-sections of carotid grafts are shown (original magnification ×220) from specimens harvested 0 (B), 4 (C), 6 (D), and 8 (E) weeks postoperatively, and stained with a modified connective tissue stain. Scale bar=200 μm. F, Carotid grafts harvested 4 or 8 weeks postoperatively were frozen, and serial sections were immunostained for either macrophages (Mac3) or SMC α-actin, and counterstained with Hoechst 33342 (DNA). The luminal surfaces are oriented upward in each panel. Images of single specimens are representative of 3 grafts of each postoperative age. Scale bar=50 μm (original magnification ×440). G, The 8-week-old carotid grafts were stained for DNA and apoE, the staining pattern for which corresponds to SMC actin and SMC myosin heavy chain (supplemental Figure IIA and data not shown, n=3). Scale bar=100 μm.
So that we could compare the extent of atherosclerosis between WT and tnfr1−/− grafts, we surveyed atherosclerotic plaque cross-sectional area along the length of the grafts. Although 8-week-old graft atheromata were often asymmetrical (Figure 1E; supplemental Figure IA and IB), the plaque cross-sectional area overall was quite consistent along the length of each graft (supplemental Figure IC to IE), therefore facilitated comparisons between grafts of distinct genotypes.
TNFR1 Expressed in Arterial Wall Cells Contributes to Atherosclerosis
WT carotid grafts demonstrated more atherosclerosis than TNFR1-deficient carotid grafts, at both 6 and 8 weeks postoperatively (Figure 2). Eight weeks postoperatively, arterial wall TNFR1 expression was associated with increases in neointimal and medial area of 98% and 68%, respectively, and a 31% increase in vessel diameter (reflecting outward remodeling; Figure 2B). Moreover, expression of TNFR1 by arterial wall cells not only increased atheroma area (and volume) but also decreased lumen diameter by 41% (Figure 2B). Thus, even when restricted to just the cells of the arterial wall, TNFR1 expression contributes to atherosclerosis.
Figure 2.
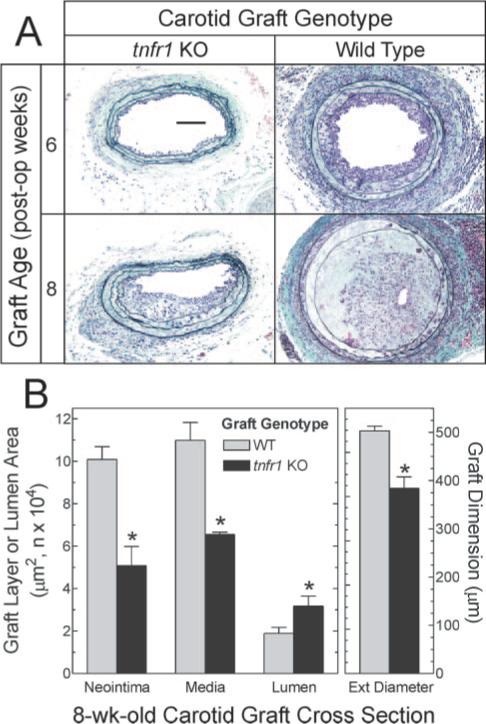
Arterial wall expression of TNFR1 contributes to atherosclerosis. Carotid arteries from WT or tnfr1−/− (tnfr1 KO) mice were grafted as in Figure 1 into apoe−/− mice, and harvested at the indicated times. A, Cross-sections from the middle of each graft were stained with a modified connective tissue stain. Specimens shown represent ≥3 obtained at each time point. B, Computerized morphometry was performed on 2 sections from the middle of each 8-week-old carotid graft specimen prepared as in (A); means±SE of 6 specimens/group are displayed. Average external diameter (ext diameter) was calculated from the perimeter of the external elastic lamina. Scale bar=100 μm (original magnification ×220). Compared with WT: *P<0.05.
To elucidate mechanisms by which the TNFR1 expressed in arterial wall cells contributes to atherosclerosis, we first assayed the grafts for cell types and cytokines typical of atheromata. Because arterial wall TNFR1 augmented atherosclerosis, we expected that TNF itself should be manifest in the atherosclerotic arterial wall. We tested this expectation in carotid grafts by immunofluorescence (supplemental Figure IIC). Whereas native carotid arteries demonstrated no TNF expression, atherosclerotic grafts did. Moreover, the ratio of TNF immunofluorescence to DNA fluorescence in WT and tnfr1−/− grafts was equivalent (not shown).
Abundant macrophages and SMC-like cells could be identified by immunofluorescence in atherosclerotic carotid grafts (supplemental Figure IIA). The prevalence of these cell types was equivalent in WT and tnfr1−/− grafts, as assessed by the ratio of DNA to SMC actin or Mac3 fluorescence (supplemental Figure IIA). Staining for CD3 and DNA demonstrated that (relatively rare) T lymphocytes were equivalently prevalent in WT and tnfr1−/− graft atherosclerotic lesions, as well (data not shown). Thus, although atherosclerotic lesions of WT carotid grafts contained a greater number of cells than those of tnfr1−/− grafts, the cell types constituting atheromata in WT and tnfr1−/− grafts were indistinguishable in type and relative abundance.
Because atherosclerotic lesion cells derive from circulating monocyte-lineage cells as well as SMCs that proliferate and migrate in from the tunica media, we assessed whether the prevalence of these 2 cell types—graft-extrinsic and graft-intrinsic—differed between WT and tnfr1−/− carotid grafts. To do so, we used GFP-expressing mice as either graft recipients (apoe−/−) or graft donors (WT or tnfr1−/−), and tracked the origin of graft atheroma cells by their expression of GFP (Figure 3). Although WT graft atheromata contained quantitatively more cells than TNFR1-deficient grafts, WT and TNFR1-deficient grafts demonstrated equivalent ratios of graft-intrinsic cells to all graft cells, assessed as the ratio of GFP to DNA fluorescence (Figure 3). Similarly, WT and TNFR1-deficient grafts demonstrated equivalent ratios of graft-extrinsic cells to all graft cells (Figure 3B). Thus, TNFR1 expression in arterial wall cells appeared to enhance both graft-intrinsic cell proliferation/migration as well as graft-extrinsic cell recruitment to the atherosclerotic grafts.
Figure 3.
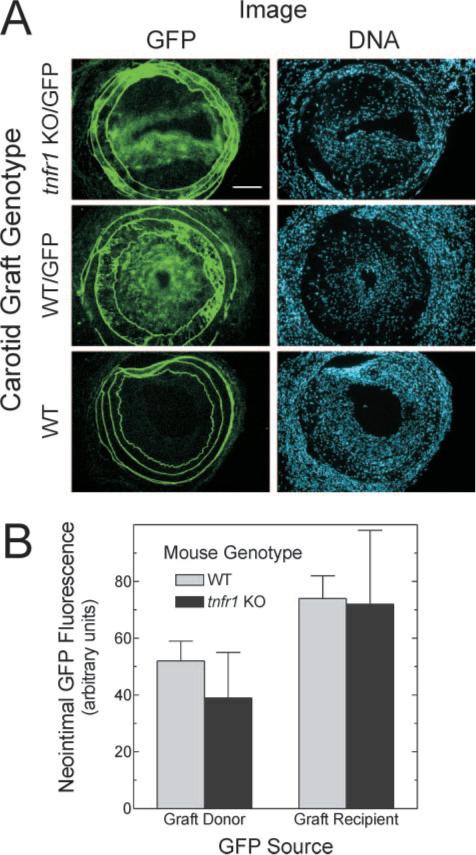
Arterial wall expression of TNFR1 does not affect the prevalence of atherosclerotic lesion cells derived from the arterial wall or circulating progenitors. Carotid grafting was performed as in Figure 2, except that either donor or recipient mice were transgenic for ubiquitous GFP expression (/GFP). A, Frozen sections of grafts were stained with Hoechst 33342 and imaged sequentially for endogenous GFP fluorescence (green) and DNA fluorescence. Images are representative of ≥4 grafts of each type (original magnification ×220); scale bar=100 μm. B, Neointimal green fluorescence in non-GFP specimens (nonspecific) was subtracted from that in GFP specimens (total fluorescence) to obtain specific GFP fluorescence. This value was divided by the cognate value for neointimal DNA fluorescence to obtain GFP/DNA (arbitrary units). These neointimal GFP/DNA values were averaged among grafts of each indicated type, and the mean±SE of ≥4 independent graft specimens is presented.
Arterial Wall TNFR1 Augments Adhesion Molecule and Chemokine Expression
The equivalence of graft-intrinsic and graft-extrinsic cell prevalence in WT and tnfr1−/− carotid grafts supported the possibilities that TNFR1 signaling in arterial wall cells enhances medial SMC proliferation and monocyte recruitment. To test these possibilities directly, we quantitated expression levels of proliferating cell nuclear antigen, the chemokine MCP-1, and the adhesion molecules ICAM-1 and VCAM-1 in 2-week-old carotid grafts, which have only isolated areas of neointimal inflammatory cells (Figure 4A and data not shown), as well as in 8-week-old grafts with advanced lesions (Figure 4B). TNFR1 expression augmented medial SMC proliferating cell nuclear antigen expression in 2-week-old grafts by 46±10% (P<0.05; Figure 4A)—suggesting that TNFR1 activity promotes SMC proliferation in the context of inflammatory stimuli associated with the preatherosclerotic arterial wall, much as it does in response to TNF in vitro. 8,15 In the advanced atheroma, however, we found no evidence for TNFR1-promoted cell proliferation (Figure 4D), as the high prevalence of graft-extrinsic cells (Figure 3B) might predict. In contrast to its time-dependent effect on arterial proliferating cell nuclear antigen expression, TNFR1 activity approximately doubled expression of MCP-1, VCAM-1, and ICAM-1 in both the preatherosclerotic tunica media (Figure 4A) and the advanced atheroma (Figure 4B). Thus, arterial wall TNFR1 signaling promotes both SMC proliferation and monocyte recruitment in early atherogenesis, and predominantly atheroma cell recruitment in late atherogenesis.
Figure 4.
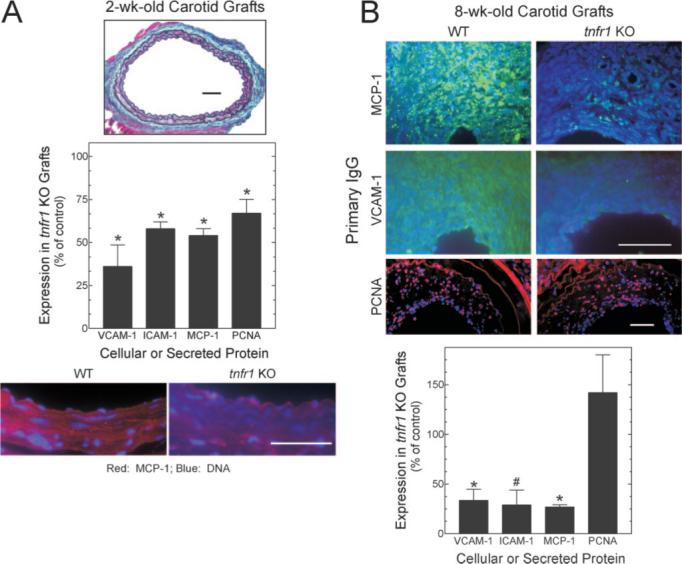
TNFR1 expression by arterial wall cells promotes chemokine and adhesion molecule expression. Carotid grafts harvested postoperatively at 2 weeks (A, with only minimal neointimal inflammatory cells) or 8 weeks (B) were stained with a connective tissue stain (A, top, ×220), or with antibodies against the indicated proteins and Hoechst 33342 (original magnification ×440, proliferating cell nuclear antigen) or ×1100; scale bars=50 μm). Immunofluorescence was quantitated as in Methods, and normalized to that obtained in WT grafts to obtain % of control; fluorescence in serial sections stained with nonimmune IgG (not shown) was subtracted from each specimen. Plotted are the means±SE from ≥3 specimens of each genotype. Compared with control: *P<0.02; #P<0.05.
To understand why our carotid grafts developed atherosclerosis more quickly than the contralateral carotid artery, we examined atherosclerosis in apoe−/− carotid grafts. Surprisingly, 8-week apoe−/− grafts demonstrated minimal atherosclerosis, predominantly at graft anastomoses (Figure 5). The contrast in atherosclerosis between these apoe−/− grafts and congenic WT grafts (Figure 2) persisted even when apoe−/− and WT mice were re-derived from matings between heterozygous parents. Thus, we reasoned that secretion of apoE by SMCs16 in the WT and tnfr1−/− grafts may elicit a mild immune response in apoe−/− graft recipients, and that this mild immune response accelerated atherosclerosis. To test this hypothesis, we first confirmed that WT and tnfr1−/− (but not apoe−/−) carotids express apoE at equivalent levels (Figure 5B). Next, we placed WT grafts in tnfr1−/− hosts to ascertain that mild immune-mediated injury (evoked by a single non-MHC protein) elicits minimal medial expansion and minimal neointimal hyperplasia in 8-week-old carotid grafts, a pattern distinctly different from that obtaining in carotid isografts (which look like unoperated carotids) or our atherosclerotic grafts (Figure 5C).
Figure 5.
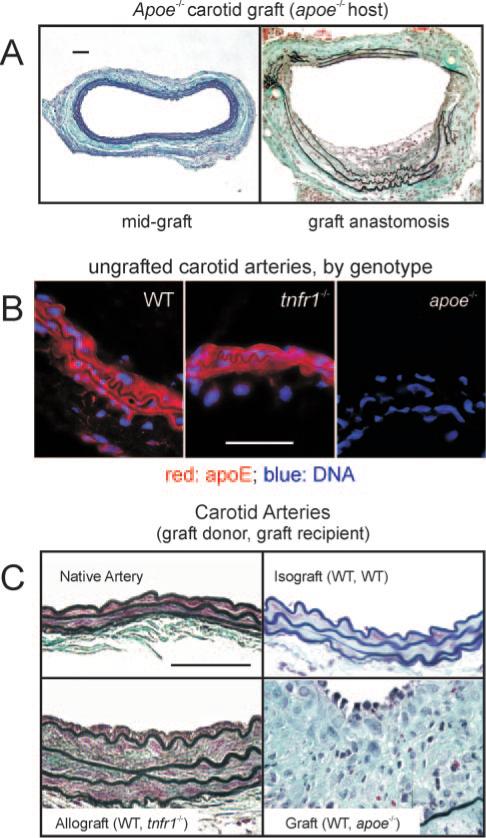
Equivalent apoE expression by arterial wall cells in WT and tnfr1−/− carotid arteries accelerates carotid graft atherosclerosis in apoe−/− mice. A, Carotid grafting with harvest at 8 weeks was performed in apoe−/− mice as in Figure 2, except that graft donors were apoe−/−. Depicted are sections from mid-graft (left) and the anastomosis (right), representative of 5 grafts harvested (original magnification ×220). B, ApoE immunofluorescence (with Hoechst 33342 counterstain) was performed on frozen sections from normal carotids of 20-week-old female mice of the indicated genotype. Results are representative of 3 carotid arteries of each genotype (original magnification ×1100). C, Carotid grafts with the indicated donor and recipient mice were harvested 8 weeks postoperatively, sectioned at mid-graft, and stained as in Figure 2. Native carotid arteries were prepared equivalently. Images are representative of results from ≥3 specimens of each type (original magnification ×1100). Scale bars=50 μm.
Effects of SMC TNFR1 on Macrophage-Evoked SMC Activity
Macrophage-evoked SMC migration from the arterial media contributes significantly to atherogenesis.12 To test the role of TNFR1 in this process, we compared the migration of WT and tnfr1−/− SMCs in response to macrophages that were activated by loading with free cholesterol.17 Whereas TNF promoted SMC migration only in WT SMCs, platelet-derived growth factor promoted SMC migration equivalently in WT and tnfr1−/− SMCs (Figure 6B). Expression of TNFR1, however, increased SMC migration toward cocultured activated macrophages by 62% (Figure 6B; P<0.05). Thus, by enhancing macrophage-promoted SMC migration, the SMC TNFR1 could quite plausibly augment atherogenesis.
Figure 6.
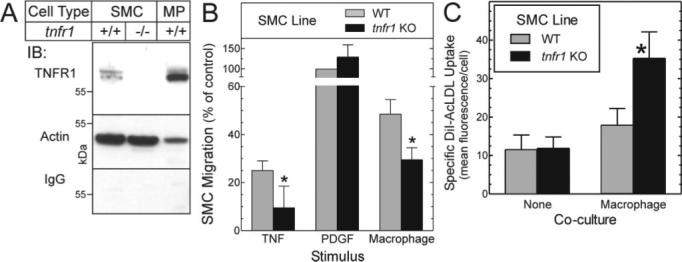
Macrophage-induced SMC migration and scavenger receptor activity: effects of SMC TNFR1 expression. A, TNFR1 expression in SMCs (WT[+/+] and tnfr1−/−) and macrophages is demonstrated by SDS-PAGE/immunoblots (IB), with 30 μg of membrane protein from each cell type. Parallel blots were probed serially with TNFR1-specific or nonimmune IgG (IgG), and then anti-actin IgG. Shown are results from a single experiment, representative of ≥4 performed. B, Migration of aortic SMCs from congenic WT and tnfr1−/− (KO) mice was assessed in response to murine TNF, human PDGF-BB (each 10 ng/mL), or cholesterol-loaded murine macrophages as described in Methods. Within each experiment (n=4), the number of SMCs migrated was normalized to the number of WT SMCs that migrated in response to PDGF (17±5 fold/basal), to obtain % of control. Basal migration values for WT and tnfr1−/− SMCs were (OD562) 0.06±0.03 and 0.06±0.04, respectively. Compared with WT: *P<0.05 (repeated measures ANOVA). For a list of cytokines secreted by these macrophages, please see http://atvb.ahajournals.org. C, WT and tnfr1−/− SMCs cultured in the absence (none)or presence of activated murine macrophages were incubated with fluorescently labeled acetylated low-density lipoprotein, and subjected to flow cytometry. Specific uptake of fluorescently labeled acetylated low-density lipoprotein is plotted (total – nonspecific) as the means±SE of 4 experiments performed in duplicate. Relative to WT SMCs: *P<0.05.
In addition to promoting SMC migration, activated macrophages are believed to secrete cytokines capable of inducing SMC scavenger receptor activity, which in turn may engender SMC foam cell formation.12 To determine whether the SMC TNFR1 affects SMC scavenger receptor activity evoked by macrophages, we subjected WT and tnfr1−/− SMCs to coculture with macrophages, and assayed scavenger receptor activity by cellular uptake of fluorescently labeled acetylated low-density lipoprotein. Stimulated SMC scavenger receptor activity was only ≈20% of that observed in macrophages (data not shown). However, as it does in macrophages,9 TNFR1 activity in SMCs reduced scavenger receptor activity by 50% (Figure 6C; P<0.05). Thus, unlike the ability of TNFR1 to promote macrophage-evoked SMC migration, the ability of the TNFR1 to reduce SMC foam cell formation would be expected to mitigate, rather than aggravate, atherogenesis.
Effects of Systemic TNFR1 Expression on Aortic Atherosclerosis
The finding that the arterial wall TNFR1 contributes to atherogenesis, in our carotid graft system, contrasts somewhat with the null effect of global TNFR1 deficiency on advanced innominate artery atherosclerosis, seen in studies of aged apoe−/− and tnfr1−/−/apoe−/− mice.10 To distinguish between TNFR1 atherogenic effects engendered by the arterial wall (in our carotid grafts) and those engendered by a combination of the arterial wall and the immune system, we compared the extent of atherosclerosis in thoracic aortas from 60-week-old female apoe−/− and tnfr1−/−/apoe−/− mice. Atherosclerosis in the thoracic aorta is most reproducible among apoe−/− mice,18 but progresses differently from that in the innominate artery,14 and not uncommonly yields comparative data distinct from those obtained from cross sections of the innominate artery.19 Serum cholesterol values were indistinguishable between apoe−/− and tnfr1−/−/apoe−/− mice (490±90 and 490±100 mg/dL, respectively). However, by en face analysis, the extent of thoracic aorta atherosclerosis was 12% less in TNFR1-deficient mice (43±1% versus 38±1% of thoracic aortic area; P<0.05; supplemental Figure III). Thus, evaluated in the context of whole-body expression, TNFR1 does contribute to advanced aortic atherosclerosis, but to an extent that appears less substantial than that observed in the context of rapidly developing atherosclerosis in our carotid grafts, in which TNFR1 expression is limited to the arterial wall. That thoracic aortas and carotid grafts yielded different magnitudes for TNFR1-dependent atherosclerosis may be attributed, in part, to cell compartment-specific TNFR1 expression (in the arterial wall versus leukocytes), and/or to fundamental differences in analytic approaches applied to these atherosclerotic vessels (lesion surface area versus lesion cross sectional area).
Discussion
With a novel arterial grafting model, this study provides the first evidence that TNFR1 expression, in just the arterial wall, contributes substantially to both early-stage and late-stage atherosclerosis. Mechanistically, TNFR1 expression in arterial wall cells enhanced adhesion molecule and chemokine expression as well as SMC proliferation, even before atherosclerotic neointimal hyperplasia supervened. Although it had no effect on the prevalence of atheroma cellular constituents, expression of TNFR1 in arterial wall cells clearly augmented the overall abundance of atheroma cells and enhanced SMC migration evoked by activated macrophages.
Although upregulation of cellular adhesion molecules and chemokines is known to be mediated through TNFR1 in response to TNF,20 we have now found that TNFR1 is a critical mediator for upregulation of cellular adhesion molecules and chemokines—even in the context of the myriad cytokines involved in atherosclerosis. Interestingly, this finding mirrors our earlier observations with TNFR1 in vein graft arterialization,13 an inflammatory process that does not involve hyperlipidemia. By demonstrating marked protein expression upregulation of MCP-1, ICAM-1, and VCAM-1 in carotid grafts from WT, as compared with tnfr1−/− mice, our work significantly extends that of Ohta et al,21 who found upregulation of mRNA encoding these molecules in aortas from 12-week-old apoe−/−, as compared with apoe−/−/tnf−/− mice. MCP-1, ICAM-1, and VCAM-1 each make significant independent contributions to atherogenesis. 22,23 Because expression of these proteins depends on the transcription factor NF-κB, and because innumerable cytokines/growth factors trigger NF-κB activation,24 it is quite remarkable that the preponderance of arterial cell ICAM-1, VCAM-1, and MCP-1 expression appears to require TNFR1 expression. The ability to target all 3 of these proatherogenic proteins through the TNFR1 pathway, or even through currently available anti-TNF therapies,1 signals novel therapeutic possibilities for atherosclerosis.
The strongly proatherogenic role for TNFR1 delineated by our studies may appear, at first, to differ from that found by Blessing et al,10 who studied advanced brachiocephalic artery atherosclerosis in 64-week-old apoe−/− and apoe−/−/tnfr1−/− mice and found no effect of systemic TNFR1 expression. However, a harmonious picture of the role of TNFR1 in atherosclerosis may be wrought by consideration of study-specific experimental approaches. Both our carotid graft and the brachiocephalic atherosclerotic lesions produce substantial luminal narrowing, outward remodeling (Figure 2B), plaque hemorrhage (Figure 1E), and variable fibrous cap formation (Figures 1 and 2; supplemental Figure II).14 It may be quite significant, however, that our carotid graft system focused exclusively on arterial wall TNFR1 expression, whereas the brachiocephalic system tested TNFR1 expression in both the arterial wall and leukocyte compartments. Moreover, whereas the carotid graft lesions develop over only 8 weeks, the brachiocephalic lesions develop over 64 weeks. Thus, not only the advanced degree but also the chronicity of the atherosclerosis in the brachiocephalic system may obscure the role of TNFR1 in atherogenesis, as Blessing et al suggest.10 It is conceivable that TNFR1, particularly in the arterial wall cells, affects early phases, or rapidly developing, more than later phases of plaque growth. This concept may also help to explain why our aortic analyses in aged apoe−/−/tnfr1−/− mice showed what appeared to be a smaller proatherogenic contribution of TNFR1 than our carotid graft analyses.
Three independent studies of TNF-deficient atherogenic mice have demonstrated that TNF promotes atherosclerosis.1,21,25 The largest effects of TNF in these studies were observed, however, in en face aortic analyses of early-stage atherosclerosis—an observation that supports a more substantial role for TNFR1 in early-stage, rather than late-stage, atherosclerosis. It should be noted, too, that these studies of tnf−/− mice may be confounded by their derivation from embryonic stem cells genetically distinct from C57Bl/6. Because MHC genes are tightly linked to the tnf gene on mouse chromosome 17, it is likely that some embryonic stem cell–derived MHC genes persist in the tnf−/− mouse.26 In light of these genetic considerations, the ability of soluble TNFR1 therapy to reduce early atherosclerosis in apoe−/− mice1 provides necessary corroboration for the proatherogenic role of TNF. Our study also substantiates a proatherogenic role for TNF and, furthermore, demonstrates the importance of the arterial wall cell TNFR1 as a proatherogenic target of TNF, the atherogenic source of which appears to be from bone marrow-derived cells.1
Study Limitations
Although our studies have not confirmed this possibility, it appears that atherosclerosis in our carotid grafts is accelerated by a mild immune reaction elicited by apoE secreted by graft SMCs (Figure 5). Additional evidence consistent with this concept derives from early graft lesions, in which neointimal macrophages are distributed somewhat more diffusely than one might expect from atherogenesis promoted by turbulent flow alone (Figure 1C, 1D).13 Does this putative alloimmune acceleration of atherogenesis compromise our ability to generalize our results to those we might expect in native-artery atherosclerosis? While extrapolating from any model system to native atherosclerosis requires caution, at least 3 aspects of our data make such extrapolation plausible. Our carotid graft atherosclerosis closely resembles native artery atherosclerosis by overall morphology and cell composition (including its relative paucity of T cells), and with regard to the sequence with which cell types infiltrate the growing atheroma. Moreover, the relative contribution of medial SMCs to the advanced atheroma in our grafts also mirrors that believed to obtain in native atherosclerosis.12 By triggering and accelerating graft atherosclerosis, extracellular apoE in our carotid grafts may serve a function similar to that intended for perivascular collars27 and endothelial denudation,28 2 techniques commonly used to accelerate carotid artery atherosclerosis.
The importance of arterial wall cell gene expression to atherogenesis has been supported largely by histological observation and a limited number of inhibitor and cell type-specific knockout studies.29 The carotid graft system presented here adds another novel approach to the study of atherogenesis mediated not just by one cell type but by all cell types constituting the arterial wall.
Sources of Funding
This work was supported by grants HL73005 and AG25462 (N.J.F.), as well as HL72842, and a grant with the Pennsylvania Department of Health (K.P.). The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Footnotes
L.Z. and K.P. contributed equally to this work and both should be considered first authors.
Disclosures
None.
Supplementary Material
References
- 1.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation. 2000;101:2149–2153. doi: 10.1161/01.cir.101.18.2149. [DOI] [PubMed] [Google Scholar]
- 3.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 4.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 5.Stannard AK, Riddell DR, Bradley NJ, Hassall DG, Graham A, Owen JS. Apolipoprotein E and regulation of cytokine-induced cell adhesion molecule expression in endothelial cells. Atherosclerosis. 1998;139:57–64. doi: 10.1016/s0021-9150(98)00052-5. [DOI] [PubMed] [Google Scholar]
- 6.Couffinhal T, Duplaa C, Labat L, Lamaziere JM, Moreau C, Printseva O, Bonnet J. Tumor necrosis factor-alpha stimulates ICAM-1 expression in human vascular smooth muscle cells. Arterioscler Thromb. 1993;13:407–414. doi: 10.1161/01.atv.13.3.407. [DOI] [PubMed] [Google Scholar]
- 7.Sato N, Goto T, Haranaka K, Satomi N, Nariuchi H, Mano-Hirano Y, Sawasaki Y. Actions of tumor necrosis factor on cultured vascular endothelial cells: morphologic modulation, growth inhibition, and cytotoxicity. J Natl Cancer Inst. 1986;76:1113–1121. [PubMed] [Google Scholar]
- 8.Peppel K, Zhang L, Orman EO, Hagen PO, Amalfitano A, Brian L, Freedman NJ. Activation of vascular smooth muscle cells by TNF and PDGF: overlapping and distinct signal transduction mechanisms. Cardiovasc Res. 2005;65:674–682. doi: 10.1016/j.cardiores.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 9.Schreyer SA, Peschon JJ, LeBoeuf RC. Accelerated atherosclerosis in mice lacking tumor necrosis factor receptor p55. J Biol Chem. 1996;271:26174–26178. doi: 10.1074/jbc.271.42.26174. [DOI] [PubMed] [Google Scholar]
- 10.Blessing E, Bea F, Kuo CC, Campbell LA, Chesebro B, Rosenfeld ME. Lesion progression and plaque composition are not altered in older apoE−/− mice lacking tumor necrosis factor-alpha receptor p55. Atherosclerosis. 2004;176:227–232. doi: 10.1016/j.atherosclerosis.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Hagen PO, Kisslo J, Peppel K, Freedman NJ. Neointimal hyperplasia rapidly reaches steady state in a novel murine vein graft model. J Vasc Surg. 2002;36:824–832. [PubMed] [Google Scholar]
- 12.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 13.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 14.Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Peppel K, Brian L, Chien L, Freedman NJ. Vein graft neointimal hyperplasia is exacerbated by tumor necrosis factor-1 signaling in graft-intrinsic cells. Arterioscler Thromb Vasc Biol. 2004;24:2277–2283. doi: 10.1161/01.ATV.0000147766.68987.0d. [DOI] [PubMed] [Google Scholar]
- 16.Hussain MM, Bucher NL, Faris B, Franzblau C, Zannis VI. Tissuespecific posttranslational modification of rat apoE. Synthesis of sialated apoE forms by neonatal rat aortic smooth muscle cells. J Lipid Res. 1988;29:915–923. [PubMed] [Google Scholar]
- 17.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterolloaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinasedependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 18.Seo HS, Lombardi DM, Polinsky P, Powell-Braxton L, Bunting S, Schwartz SM, Rosenfeld ME. Peripheral vascular stenosis in apolipoprotein E-deficient mice. Potential roles of lipid deposition, medial atrophy, and adventitial inflammation. Arterioscler Thromb Vasc Biol. 1997;17:3593–3601. doi: 10.1161/01.atv.17.12.3593. [DOI] [PubMed] [Google Scholar]
- 19.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 20.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor alpha (TNF-α)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med. 1993;177:1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, Saito K, Sekikawa K, Seishima M. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Knowles JW, Maeda N. Genetic modifiers of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2000;20:2336–2345. doi: 10.1161/01.atv.20.11.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis. J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boesten LS, Zadelaar AS, van Nieuwkoop A, Gijbels MJ, de Winther MP, Havekes LM, van Vlijmen BJ. Tumor necrosis factor-alpha promotes atherosclerotic lesion progression in APOE*3-Leiden transgenic mice. Cardiovasc Res. 2005;66:179–185. doi: 10.1016/j.cardiores.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Muller U, Jongeneel CV, Nedospasov SA, Lindahl KF, Steinmetz M. Tumour necrosis factor and lymphotoxin genes map close to H-2D in the mouse major histocompatibility complex. Nature. 1987;325:265–267. doi: 10.1038/325265a0. [DOI] [PubMed] [Google Scholar]
- 27.von Der Thusen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein e-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–1170. doi: 10.1161/01.cir.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 28.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. Rantes deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- 29.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.