

Abstract
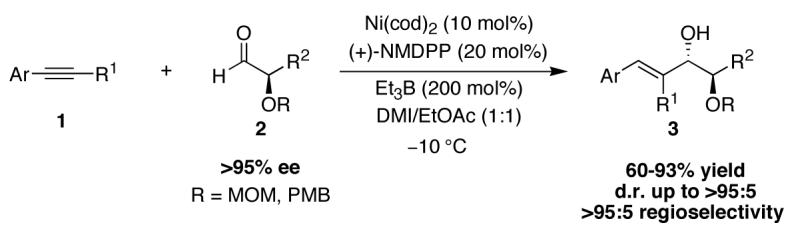
Ni-catalyzed reductive coupling of aryl alkynes (1) and enantiomerically enriched α-oxyaldehydes (2) afford differentiated anti-1,2-diols (3) with high diastereoselectivity and regioselectivity, despite the fact that the methoxymethyl (MOM) and para-methoxybenzyl (PMB) protective groups typically favor syn-1,2-diol formation in carbonyl addition reactions of this family of aldehydes.
Enantiomerically pure 1,2-diols are important and commonly occuring functional group patterns in natural products such as carbohydrates and polyketides and in chiral ligands used in asymmetric catalysis. Consequently, much effort has been invested in the development of stereoselective methods for 1,2-diol synthesis. A very powerful one for preparing syn-1,2-diols is the Sharpless asymmetric dihydroxylation of trans-disubstituted olefins.1 However, the diastereomeric anti-1,2-diols are not as easily accessed using this transformation because the corresponding dihydroxylations of cis-disubstituted olefins typically proceed with diminished enantioselectivity.1c
Auxiliary-based, anti-selective glycolate aldol addition reactions have been developed to address this limitation.2 Nevertheless, these methods are much less common than those for analogous, syn-selective addition, and this area continues to be actively investigated. Recently, MacMillan and List reported catalytic asymmetric aldol reactions that afford the anti-1,2-diol architecture.3 Aldolases,4 catalytic antibodies,5 and a heteropolymetallic catalyst6 also have been used to favor anti addition in related reactions.
A contrasting approach to the synthesis of 1,2-diols involves nucleophilic addition to aldehydes bearing protected hydroxyl groups adjacent to the carbonyl.7 Cram’s rule, after over fifty years, remains a good predictor of the stereochemical outcome of additions to these chiral α-oxyaldehydes (Fig 1).8
Figure 1.
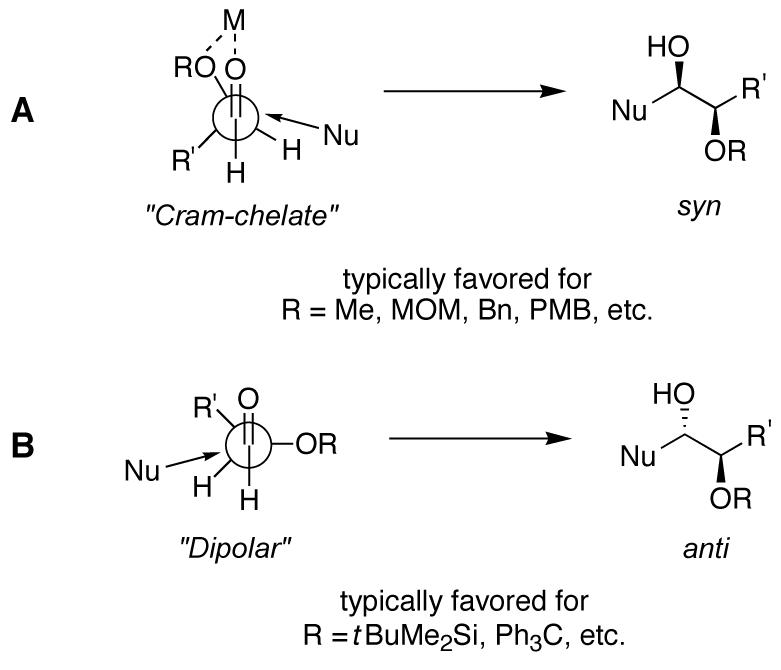
Models for stereoselective nucleophilic additions to α-oxyaldehydes. (A) The cyclic “Cram-chelate” model predicts syn-1,2-diols. (B) The “dipolar” model predicts anti-1,2-diols.
For α-alkoxy groups that have the ability to coordinate (such as MeO—, MOMO—, BnO— or PMBO—, among others), the “Cram-chelate” model typically applies, and syn-1,2-diols are favored (Fig 1, A).8b,9 When larger groups (such as tBuMe2SiO— or Ph3CO—) are employed, the “dipolar” model is invoked to account for the general preference for anti-1,2-diol products (Fig 1, B). However, due to the greater degree of flexibility in the latter process (σ—bond rotation), nucleophilic additions of this type usually proceed with moderate selectivity and therefore are not always viable means to access anti-1,2-diols.
In rare instances, α-oxyaldehydes bearing chelating groups adjacent to the carbonyl afford anti-1,2-diols with >95:5 diastereoselectivity.10 This unusual preference is particularly interesting from a mechanistic point-of-view because it suggests that even in the presence of highly coordinating groups such as MOMO— or BnO—, the nucleophilic addition occurs instead via the “dipolar” model. This phenomenon may be observed when reagents lacking the ability to chelate are utilized,10a,10c or when a reagent that imparts complete stereocontrol is employed.10d
We have previously reported that nickel-catalyzed reductive coupling reactions of aryl-substituted alkynes and aldehydes proceed with high regioselectivity and enantioselectivity when a chiral phosphine such as neomenthyldiphenylphosphine (NMDPP) is utilized (Scheme 1).11,12 We now disclose that the corresponding reaction with chiral α-oxyaldehydes preferentially affords 1,2-diol products of the anti relative configuration.
Scheme 1.
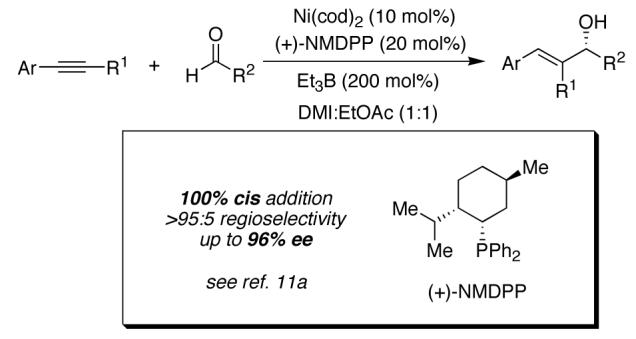
Catalytic Asymmetric Reductive Coupling of Alkynes and Aldehydes
We began our studies by investigating the role of the ligand in catalytic reductive coupling reactions of 1-phenyl-1-propyne (1a) and the known aldehyde 2a13 (eq 1).
A high level of substrate control was observed with both (+)- and (—)-NMDPP providing the coupling product 3a as predominantly the anti diastereomer (Table 1, entries 1 and 2).14 (+)-NMDPP was selected for further studies because it provided the anti product in higher yield and selectivity (Table 1, entry 1). The diastereoselectivity and yield were further improved by careful examination of reaction temperature. At —10 °C, near 9:1 diastereoselectivity was obtained without compromising the reaction yield (Table 1, entry 3). On further cooling, however, the yield diminished severely and no appreciable improvement in diastereoselectivity was observed (Table 1, entry 4).15 On the other hand, by increasing the amount of the aldehyde and extending the reaction time, a balance of yield and selectivity for the reductive coupling of 1a and 2a was achieved (Table 1, entry 6).
Table 1.
Ni/NMDPP-Catalyzed Reductive Coupling of 1-Phenyl-1-propyne (1a) and MOM-Protected α-Oxyaldehyde 2a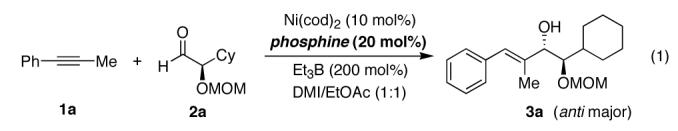
| entrya | phosphine | time(h) | temp (°C) | d.rb | yield (%)(d.r.)c |
|---|---|---|---|---|---|
| 1 | (+)-NMDPP | 6 | 0 | 84:16 | 61 90:10) |
| 1 | (-)-NMDPP | 6 | 0 | 77:23 | 32 (80:20) |
| 3 | (+)-NMDPP | 6 | -10 | 89:11 | 62 (90:10) |
| 4 | (+)-NMDPP | 6 | -20 | 90:10 | 37 (90:10) |
| 5d | (+)-NMDPP | 6 | -10 | 89:11 | 77 (90:10) |
| 6d | (+)-NMDPP | 20 | -10 | 88:12 | 87 (90:10) |
See Supporting Information for experimental procedures. 100 mol% of alkyne and 100 mol% of aldehyde were used, unless otherwise noted. All reactions proceeded with >95:5 regioselectivity.
Ratio of anti:syn determined by 1H NMR analysis of crude reaction mixtures.
Yield and d.r. of the isolated reaction product.
Conducted using 150 mol% of aldehyde.
The relative stereochemistry of 3a was determined by removal of the MOM protective group and conversion to the corresponding cyclic carbonate 4 (Scheme 2). A large nOe was observed between the carbinol protons, suggesting a cis relationship between them in 4, i.e. of the anticonfiguration in 3a. Further confirmation of the relative configuration involved conversion of 3a to ketodiol 5 whose data were consistent with those previously reported (Scheme 2).3a
Scheme 2.
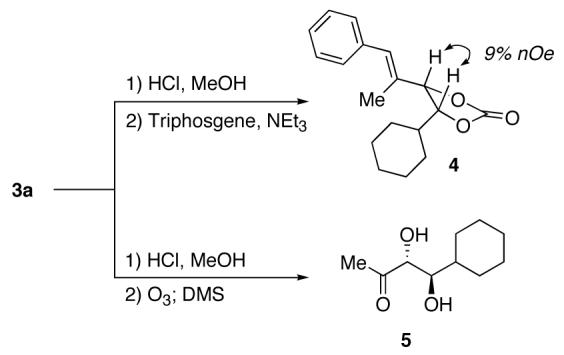
Assignment of the Relative Configuration of 3a
The scope of this novel, anti-selective reductive coupling is shown in Table 2. Placing a larger substituent on the side of the alkyne where C-C bond formation occurs (e.g. Me to Et or cyclopropyl, Table 2, entries 1, 3 and 4), dramatically improved the selectivity, albeit with depreciation in yield. Notably, heteroatom-substituted alkynes are tolerated and do not affect the anti selectivity (Table 2, entry 5). Also changing the protective group on the aldehyde to PMB improves the chemical yield while maintaining excellent diastereoselectivity (Table 2, entry 6 vs. entry 3). However, changing the cyclohexyl substituent on the aldehyde to either a phenyl group (Table 2, entry 7) or an n-hexyl group (Table 2, entry 8) resulted in lowered selectivity, likely due to a reduced conformational bias in the aldehyde.
Table 2.
Extension of the Anti-Selective Reductive Coupling to a Variety of Aryl Alkynes and α-Oxyaldehydesa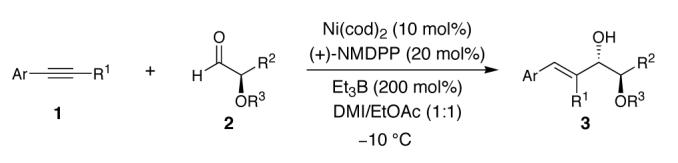
| entry | Ar | R1 | R2 | R3 | d.r.b | yield (%) (d.r.)c |
|---|---|---|---|---|---|---|
| 1 | Ph | Me | Cy | MOM | 88:12 | 87(90:10) |
| 2 | 1-Naphthyl | Me | Cy | MOM | 83:17 | 93(83:17) |
| 3 | Ph | Et | Cy | MOM | >95:5 | 54(>95:5) |
| 4 | Ph |  |
Cy | MOM | >95:5 | 62(>95:5) |
| 5 | Ph | CH2NHBoc | Cy | MOM | 90:10 | 60(95:5) |
| 6 | Ph | Et | Cy | PMB | >95:5 | 74(>95:5) |
| 7 | Ph | Et | Ph | MOM | 68:32 | 39(>95:5)d |
| 8 | Ph | Et | n-hexyl | MOM | 75:25 | 86(80:20) |
All reactions were conducted with 100 mol% of alkyne and 150 mol% of aldehyde. All reactions proceeded with >95:5 regioselectivity.
Ratio of anti:syn determined by 1H NMR analysis ofcrude reaction mixtures.
Yield and diastereoselectivity of the isolated reaction product.
The syn product was also isolated in 18% yield (>95:5 d.r.).
The simplest interpretation of the observed sense of induction is that, due to the absence of any chelating metal in the reaction, the preferred mode of addition can be rationalized by the “dipolar” model (Fig 2, B).
Figure 2.
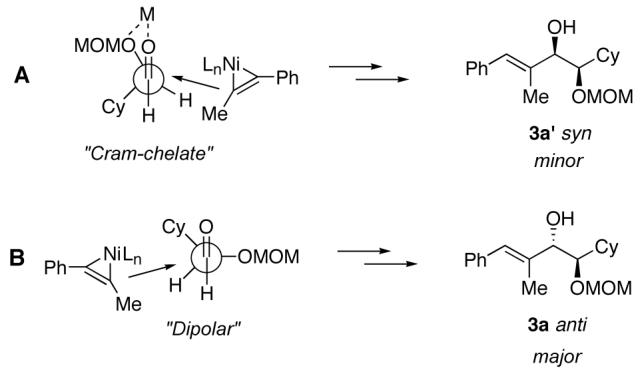
Schematic representation of divergent pathways leading to syn- and anti-1,2-diols.
In conclusion, a nickel-catalyzed reductive coupling of alkynes and easily accessible, enantiomerically enriched α-oxyaldehydes has been developed. These coupling reactions provide efficient access to a variety of differentially-protected anti-1,2-diols, despite the fact that additions to methoxymethyl- (MOM) and p-methoxybenzyl-protected (PMB) 2-hydroxyaldehydes typically display a preference for syn-1,2-diols. Currently we are investigating the utility of this novel method for preparing these useful intermediates as a catalytic, stereoselective fragment coupling reaction in target-oriented synthesis.
Supplementary Material
Acknowledgment
Support for this work was provided by the National Institute of General Medical Sciences (GM-063755). T. L. thanks the MIT Undergraduate Research Opportunities Program, and C. O. N. thanks the Pfizer Diversity in Organic Chemistry Program for fellowship support. We also thank the NIGMS (GM-072566), NSF (CAREER CHE-0134704), the Sloan Foundation, Amgen, Boehringer-Ingelheim, Bristol Myers-Squibb, GlaxoSmithKline, Johnson & Johnson, Merck Research Laboratories, Wyeth, and the Deshpande Center (MIT) for generous financial support of our program. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Footnotes
Supporting Information Available. Experimental procedures and data for all new compounds. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Jacobsen EN, Marko I, Mungall WS, Schröder G, Sharpless KB. J. Am. Chem. Soc. 1988;110:1968–1970. [Google Scholar]; (b) Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]; (c) Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2nd Ojima I, editor. VCH; New York: 2000. pp. 357–398. [Google Scholar]
- (2) (a).Mukaiyama T, Iwasawa N. Chem. Lett. 1984:753–756. [Google Scholar]; (b) Evans DA, Gage JR, Leighton JL, Kim AS. J. Org. Chem. 1992;57:1961–1963. [Google Scholar]; (c) Crimmins MT, McDougall PJ. Org. Lett. 2003;5:591–594. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]
- (3) (a).Notz W, List B. J. Am. Chem. Soc. 2000;122:7386–7387. [Google Scholar]; (b) Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew. Chem., Int. Ed. 2004;43:2152–2154. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]; (c) Northrup AB, MacMillan DWC. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- (4) (a).Bednarski MD, Simon ES, Bishofberger N, Fessner W-D, Kim M-J, Lees W, Saito T, Waldmann H, Whitesides GM. J. Am. Chem. Soc. 1989;111:627–635. [Google Scholar]; (b) Fessner W-D, Sinerius G, Schneider A, Dreyer M, Schulz GE, Badia J, Aguilar J. Angew. Chem., Int. Ed. 1991;30:555–558. [Google Scholar]
- (5) (a).List B, Shabat D, Barbas CF, III, Lerner RA. Chem. Eur. J. 1998;4:881–885. [Google Scholar]; (b) Hoffmann T, Zhong G, List B, Shabat D, Anderson J, Gramatikova S, Lerner RA, Barbas CF., III J. Am. Chem. Soc. 1998;120:2768–2779. [Google Scholar]
- (6).Yoshikawa N, Suzuki T, Shibasaki M. J. Org. Chem. 2002;67:2556–2565. doi: 10.1021/jo0162538. [DOI] [PubMed] [Google Scholar]
- (7) (a).Reviews:Reetz MT. Angew. Chem., Int. Ed. 1984;23:556–569.Reetz MT. Acc. Chem. Res. 1993;26:462–468.Eliel EL. In: Asymmetric Synthesis. Part A. Morrison JD, editor. Vol. 2. Academic; New York: 1983. pp. 125–155.
- (8) (a).Cram DJ, Abd Elhafez RA. J. Am. Chem. Soc. 1952;74:5828–5835. [Google Scholar]; (b) Gawley RE, Aube J. Principles of Asymmetric Synthesis; Tetrahedron Organic Chemistry Series. Vol. 14. Pergamon Press, Elsevier; Oxford: 1996. pp. 121–160. [Google Scholar]
- (9).Of particular interest is the NMR observation of a chelate as an intermediate in the addition of dimethylmagnesium to α-alkoxy aldehydes:Chen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1992;114:1778–1784.
- (10) (a).Banfi L, Bernardi A, Colombo L, Gennari C, Scolastico C. J. Org. Chem. 1984;49:3784–3790.Tio H, Mizobuchi T, Tsukamoto M, Tokoroyama T. Tetrahedron Lett. 1986;27:6373–6376.Marumoto S, Kogen H, Naruto S. Chem. Commun. 1998:2253–2254.An example of highly reagent-controlled nucleophilic addition to a MOMO— aldehyde:Corey EJ, Yu C-M, Kim SS. J. Am. Chem. Soc. 1989;111:5495–5496.
- (11) (a).Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y.See also: Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q.Colby EA, Jamison TF. J. Org. Chem. 2003;68:156–166. doi: 10.1021/jo0264123.
- (12) (a).Reviews of Ni-catalyzed reductive couplings:Miller KM, Molinaro C, Jamison TF. Tetrahedron: Asymmetry. 2003;14:3619–3625.Ikeda S.-i. Angew. Chem., Int. Ed. 2003;42:5120–5122. doi: 10.1002/anie.200301673.Montgomery J. Angew. Chem., Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634.
- (13).Racemic 2a:Paquette LA, Mitzel TM. J. Am. Chem. Soc. 1996;118:1931–1937.
- (14).We found no achiral phosphine that was as efficient as NMDPP, thus preventing a meaningful estimation of the “inherent selectivity” of these aldehydes in these catalytic coupling reactions:Masamune S, Choy W, Peterson JS, Sita LR. Angew Chem., Int. Ed. 1985;24:1–30.
- (15).Further cooling to —40 °C resulted in improved diastereoselectivity (>95:5) but also severely reduced conversion (<10%).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.