

Abstract
Transition state theory suggests that enzymatic rate acceleration (kcat/knon) is related to the stabilization of the transition state for a given reaction. Chemically stable analogues of a transition state complex are predicted to convert catalytic energy into binding energy. Since transition state stabilization is a function of catalytic efficiency, differences in substrate specificity can be exploited in the design of tight-binding transition state analogue inhibitors. Coformycin and 2′-deoxycoformycin are natural product transition state analogue inhibitors of adenosine deaminases (ADAs). These compounds mimic the tetrahedral geometry of the ADA transition state and bind with picomolar dissociation constants to enzymes from bovine, human, and protozoan sources. The purine salvage pathway in malaria parasites is unique in that Plasmodium falciparum ADA (PfADA) catalyzes the deamination of both adenosine and 5’-methylthioadenosine. In contrast, human adenosine deaminase (HsADA) does not deaminate 5’-methylthioadenosine. 5′-Methylthio coformycin and 5’-meththio-2′-deoxycoformycin were synthesized to be specific transition state mimics of the P. falciparum enzyme. These analogues inhibited PfADA with dissociation constants of 430 and 790 pM, respectively. Remarkably, they gave no detectable inhibition of the human and bovine enzymes. Adenosine deamination is involved in the essential pathway of purine salvage in P. falciparum and prior studies have shown that inhibition of purine salvage results in parasite death. Inhibitors of HsADA are known to cause toxicity in humans and the availability of parasite-specific ADA inhibitors may prevent this side-effect. The potent and P. falciparum-specific inhibitors described here have potential for development as antimalarials without inhibition of host ADA.
Keywords: Coformycin, Deoxycoformycin, Pentostatin, transition state analogueue, ADA inhibitors, adenosine deaminase, malaria, purine salvage, isozyme specificity, Plasmodium falciparum
INTRODUCTION
Adenosine deaminase (ADA) deaminates adenosine and deoxyadenosine to form the respective inosines and thereby plays an essential role in purine metabolism.1 Genetic deficiency of ADA in humans causes severe compromised immunodeficiency disease (SCID) which results from the accumulation of dATP in B- and T-cells.2-4 Adenosine deaminase is also important in purine salvage pathways in microorganisms, and is thought to be essential for salvage pathways in purine auxotrophs like Plasmodium falciparum.5,6 The adenosine deaminase inhibitors coformycin and 2′-deoxycoformycin (also known as Pentostatin) are natural product transition state analogue inhibitors that bind with picomolar affinity to adenosine deaminases.1,7-9 Inhibition of adenosine deaminase results in the accumulation of adenosine and deoxyadenosine. Production of excess dATP from accumulated deoxyadenosine unbalances the deoxynucleotide pool, inhibits ribonucleotide reductase and initiates apoptosis in B- and T-cells.10 Deoxycoformycin has been approved by the FDA for the treatment of hairy cell leukemia since 1991.11 However, it causes renal, liver, pulmonary and central nervous system toxicities at high doses, and is therefore limited in its therapeutic profile.12
Recently, the ADA from P. falciparum (PfADA) has been characterized and found to catalyze the deamination of adenosine, as well as the conversion of 5′-methylthioadenosine (MTA) to 5′-methylthioinosine (MTI) and ammonia (Figure 1).13 5′-Methylthioinosine was not previously reported as a metabolite in any biological system. The ability of PfADA to utilize MTA is specific to the parasite and human ADA does not catalyze this reaction. Prior studies have shown that inhibition of malarial purine salvage enzymes results in parasite death,14 however potent ADA inhibitors with specificity for the enzymes from malaria parasites have not been described.
Figure 1.

The reaction catalyzed by adenosine deaminase involves nucleophilic attack on adenosine by a hydroxide ion formed by a catalytic site zinc atom. A short-lived Meisenheimer complex intermediate is protonated at the exocyclic amino group, followed by loss of NH3 to produce inosine. PfADA has substrate specificity that includes both adenosine and 5′-MeS-adenosine, an activity not shared by mammalian ADAs.
5′-Methylthiocoformycin, 5′-methylthio-2′-deoxycoformycin, and other 5′-functionalized 2′-deoxycoformycin molecules were synthesized and investigated for their ability to inhibit PfADA, as compared to the ADAs from human (HsADA) and bovine (BtADA) sources. The comparative enzymology and inhibitor specificity of malarial and human ADAs highlight the importance of examining substrate specificity in addition to transition state analysis in the design of species-specific inhibitors for use as therapeutic agents.
EXPERIMENTAL SECTION
Reagents, Bacterial Strains and Transformation
Calf spleen ADA, human erythrocyte ADA, adenosine, potassium phosphate, and EDTA were purchased from Sigma-Aldrich Chemical Company. A diastereomeric mixture of 2′-deoxy-8-(R/S)-coformycin was a gift from Dr. David C. Baker. Concentrations of inhibitors were determined spectrophotometrically using the extinction coefficient of 8250 M-1cm-1 at 282 nm. Escherichia coli cells BL21 Star-(DE3) and BL21 Star cells (Invitrogen, Carlsbad) were grown in Luria broth (LB) with 100 μg/ml Ampicillin (AMP) at 37 °C.
PfADA Gene Construction
A chemically synthesized gene encoding the adenosine deaminase from P. falciparum was synthesized, cloned into a pDONR221 vector and sequenced by DNA 2.0 (Menlo Park). The gene was synthesized using optimized codons for E. coli protein expression and the addition of an N-terminal thrombin cleavable (histidine)6 tag.
Protein Expression and Purification
The designed gene was transferred to the pDEST-14 vector with LR Clonase II. The pDEST-14-PfADA construct was transformed into BL21 Star competent cells and the protein was expressed without induction with shaking at 37 °C, overnight. The cells were ruptured by passage through a French press, the cell debris pelleted by centrifugation and the remaining supernatant was purified over a 25 mL Ni Sepharose High Performance (GE Healthcare, Piscataway) His-tag affinity column with elution by a 0 – 250 mM imidazole gradient. The enzyme was then dialyzed into 100 mM Tris-HCl, 10 μM ZnCl2, pH 8.0 and frozen in 20 μL aliquots at −80 °C. The protein was equally active with or without the (histidine)6 tag, which could be removed by combining 1 unit Thrombin (GE Healthcare, Piscataway) for every 1 mg of protein in 1 × PBS at 22 °C, incubation for one hr, followed by purification over an 8 mL Source 15Q column with elution by a 0-1 M NaCl gradient in the dialysis buffer.
Enzyme inhibition assay
Adenosine deaminase activity was determined by monitoring the conversion of adenosine to inosine via the change in absorbance at 267 nm in assay mixtures containing 20 mM potassium phosphate, pH 7.0, 100 μM adenosine and 1 μM EDTA at 30 °C.15 The kinetics for slow-onset inhibition and Ki measurement were performed using inhibitor concentrations ranging from 10 μM to 2 nM and an enzyme concentration of 1 nM. The Ki values were determined by fitting the initial rate and inhibitor concentrations to the following expression of competitive inhibition16,17:
where V′o is the initial rate in the presence of inhibitor, and Vo is the initial rate in the absence of inhibitor, [I] is the inhibitor concentration, and [S] is the substrate concentration. This expression is valid only under the condition where the inhibitor concentration is 10 times greater than the enzyme concentration. In conditions where inhibitor concentration does not exceed ten times the enzyme concentration the effective inhibitor concentration was obtained by the expression:
where I′ is the effective inhibitor concentration, V′o and Vo are the initial rate in the presence and absence of inhibitor, and Et is the total enzyme concentration.
In cases where slow onset inhibition was observed, where the inhibitor reached a tighter binding thermodynamic equilibrium with the enzyme, the equilibrium dissociation constant (Ki*) was obtained by fitting the rates to the following equation for competitive inhibition
with [I] concentrations being corrected as above.18 The KM values for the enzymes are 16 ± 2 μM for the bovine enzyme,15 29 ± 3 μM for the malarial enzyme13 and 22 ± 2 μM for the human enzyme.19
(8R)-6-(tert-Butoxycarbonyl)-8-(tert-butyldimethylsilyloxy)-3-(2’-deoxy-3’,5’-di-O-toluoyl-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine (3)
(8R)-6-(tert-Butoxycarbonyl)-8-(tert-butyldimethylsilyloxy)-3-(2’-cyanoethyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine 2a (0.76 g, 1.81 mmol) was dissolved in dry THF (15 mL) and potassium tert-butoxide (1.0 M in THF, 3.6 mL) was added at RT. The mixture turned brown immediately and was quenched by addition of glacial acetic acid (206 μL, 3.6 mmol) and coevaported with toluene (30 mL). The residue was suspended in chloroform/ethyl acetate (5 mL, 1:2 v/v) by ultrasonification and applied on a chromatography column (50 g silica, chloroform/ethyl acetate = 1:2 v/v, then ethyl acetate), which gave (8R)-6-(tert-butoxycarbonyl)-8-(tert-butyldimethylsilyloxy)-3,6,7,8-tetrahydro-imidazo[4,5-d][1,3]diazepine 2b as an amorphous yellow solid (0.51 g, 1.39 mmol, 77%).
2b was dissolved in dry acetonitrile (10 mL) and evaporated in vacuo, flushed with argon and dissolved again in dry acetonitrile (15 mL). Addition of sodium hydride (60% in mineral oil, 72 mg) gave a visible gas formation. After 30 min 2-deoxy-3,5-di-O-(p-toluoyl)-d-erythro-pentofuranosyl chloride 120 (0.70 g, 1.81 mmol) was added to the reaction mixture, a thick heterogeneous slurry was formed. After further 40 min the reaction mixture was filtered through flux calcined diatomaceous earth and rinsed thoroughly with ethyl acetate. The filtrate was evaporated to dryness in vacuo. Purification of the residue by chromatography (60 g silica, petroleum ether/ethyl acetate = 2:1 v/v) gave 3 as yellowish oil (0.85 g, 1.18 mmol, 85% calcd. from 2b). Rf = 0.31 (petrol ether/EtOAc = 3:2 v/v), [α]D20 = −17.4 (c 11.8, chloroform), 1H-NMR: (CDCl3) δ 0.05 (s, 3H, SiMe), 0.19 (s, 3H, SiMe), 0.88 (s, 9H, SitBu), 1.54 (s, 9H, OtBu), 2.40 (s, 3H, ArMe), 2.43 (s, 3H, ArMe), 2.70 (ddd, J = 2.2, 5.9, 14.2 Hz, 1H, H-2’), 2.84 (ddd, J = 6.4, 8.4, 14.2 Hz, 1H, H-2’), 3.08 (br d, J = 13.6 Hz, 1H, H-7), 4.50 (dd, J = 4.4, 13.6 Hz, 1H, H-7), 4.55–4.60 (m, 1H, H-4’), 4.61–4.65 (m, 2H, H-5’), 5.13 (br d, J = 4.4 Hz, H-8), 5.70 (ddd, J = 2.2, 2.3, 6.0 Hz, 1H, H-3’), 6.42 (dd, J = 5.9, 8.4 Hz, 1H, H-1’), 7.19–7.30 (m, 4H, Ar), 7.57 (s, 1H, H-2), 7.85–7.99 (m, 5H, 4×Ar and H-5), 13C-NMR: (CDCl3) δ −4.6 (SiMe), −4.2 (SiMe), 18.6 (SiC(CH3)3), 22.0 (ArCH3), 26.2 (SiC(CH3)3), 28.5 (OC(CH3)3), 38.9 (C-2’), 47.2 (C-7), 64.6 (C-5’), 67.6 (C-8), 75.6 (C-3’), 82.7 (C-4’), 83.7 (OC(CH3)3), 84.4 (C-1’), 127.0, 127.2, 129.6, 130.1, 130.2, 132.3 (C-2), 133.1,134.8 , 142.7 (C-5), 144.4, 144.7, 152.8, 166.3, 166.6, HRMS: (MH+) calcd. for C38H51N4O8Si+: 719.3476, found: 719.3492.
(8R)-6-(tert-Butoxycarbonyl)-8-(tert-butyldimethylsilyloxy)-3-(2’-deoxy-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine (4)
In a rubber sealed round bottom flask 3 (0.79 g, 1.10 mmol) was treated with ammonia (7 M in methanol, 75 mL) at RT. After 17 h the mixture was evaporated in vacuo, the residue was transferred into an ace pressure tube, fresh ammonia (7 M in methanol, 50 mL) was added, and the tube was sealed and warmed to 45 °C for another 4 h. After evaporation in vacuo the residue was purified by chromatography (41 g silica, chloroform/methanol = 15:1 v/v) which gave 4 as colorless oil (375 mg, 71%). Rf = 0.34 (chloroform/methanol = 10:1 v/v), [α]D20 = +10.0 (c 5.23, chloroform), 1H-NMR: (CDCl3) δ 0.06 (s, 3H, SiMe), 0.19 (s, 3H, SiMe), 0.88 (s, 9H, SitBu), 1.53 (s, 9H, OtBu), 2.25 (ddd, J = 1.1, 5.9, 14 Hz, 1H, H-2’), 2.94 (ddd, J = 5.3, 8.7, 14 Hz, 1H, H-2’), 3.08 (br d, J = 13.6 Hz, 1H, H-7), 3.74 (dd, J = 1.3, 12.2 Hz, 1H, H-5’), 3.89 (dd, J = 1.8, 12.2 Hz, 1H, H-5’), 4.10–4.15 (m, 1H, H-4’), 4.54 (dd, J = 4.7, 13.6 Hz, 1H, H-7), 4.65–4.71 (m, 1H, H-3’), 5.14 (d, J = 4.7 Hz, 1H, H-8), 6.20 (dd, J = 5.9, 8.7 Hz, 1H, H-1’), 7.49 (s, 1H, H-2), 7.85 (s, 1H, H-5), 13C-NMR: (CDCl3) δ −4.6 (SiMe), −4.3 (SiMe), 18.7 (SiC(CH3)3), 26.2 (SiC(CH3)3), 28.5 (OC(CH3)3), 41.6 (C-2’), 47.0 (C-7), 63.5 (C-5’), 67.6 (C-8), 73.3 (C-3’), 84.3 (OC(CH3)3), 87.3 (C-1’), 88.7 (C-4’), 132.7, 134.9 (C-2), 136.2, 143.3 (C-5), 152.6, HRMS: (MH+) calcd. for C22H39N4O6Si+: 483.2639, found: 483.2643.
(8R)-6-(tert-Butoxycarbonyl)-8-(tert-butyldimethylsilyloxy)-3-(2’-deoxy-5’-O-tosyl-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine (5)
4 (0.375 g, 0.777 mmol) was dissolved in dry pyridine (7 mL, 87 mmol) and tosyl chloride (0.222 g, 1.17 mmol) was added at 0 °C. After 20 min the mixture was warmed to RT and stirred overnight. The reaction mixture was diluted with chloroform (30 mL) and consecutively washed with water (40 mL), citric acid (5% w/w, 3×50 mL) and saturated NaHCO3 (60 mL), dried (MgSO4) and evaporated in vacuo. The residue was purified by chromatography (46 g silica, chloroform/methanol = 20:1 v/v) which gave the title compound as a glass (313 mg, 63%). Rf = 0.39 (chloroform/methanol = 10:1 v/v), [α]D20 = +25.8 (c 3.62, CHCl3), 1H-NMR: (CDCl3) δ 0.07 (s, 3H), 0.20 (s, 3H), 0.88 (s, 9H), 1.54 (s, 9H), 2.43 (m, 4H), 2.34–2.46 (m, 4H), 2.55–2.66 (m, 2H, 1H D2O exchangeable), 3.07 (br d, J = 13.5 Hz, 1H), 4.06–4.16 (m, 2H), 4.20–4.29 (m, 1H), 4.48 (dd, J = 4.5, 13.5 Hz, 1H), 4.60–4.70 (m, 1H), 5.12 (br d, J = 4.5 Hz, 1H), 6.30 (t, J = 6.7 Hz, 1H), 7.28–7.34 (m, 2H), 7.48 (s, 1H), 7.70–7.75 (m, 2H), 7.83 (s, 1H), 13C-NMR: (CDCl3) δ −4.6, −4.1, 18.7, 22.0, 26.2, 28.5, 40.6, 47.2, 67.6, 69.1, 72.2, 83.8, 83.9, 128.4, 130.4, 132.4, 132.6, 133.0, 134.6, 142.6, 145.6, 152.8, HRMS: (MH+) calcd. for C29H45N4O8SSi+: 637.2727, found: 637.2753.
(8R)-6-(tert-Butoxycarbonyl)-3-(2’-deoxy-5’-O-tosyl-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol (6)
5 (0.31 g, 0.49 mmol) was dissolved in dry methanol (12 mL) and ammonium fluoride (0.54 g, 14.6 mmol) was added and the mixture was heated to reflux for 6.5 h. After cooling to RT stirring was continued overnight. The next morning the mixture was refluxed for an additional 6 h. The reaction mixture was evaporated in vacuo and the residue was purified by chromatography (27 g silica, chloroform/methanol = 10:1 v/v) which gave 6 as a hard foam (147 mg, 58%). Rf = 0.23 (chloroform/methanol = 10:1 v/v), [α]D20 = +30.3 (c 2.15, chloroform), 1H-NMR: (CDCl3) δ 1.53 (s, 9H), 2.37–2.64 (m, 6H), 3.59 (br d, J = 13.5 Hz, 1H), 4.03 (dd, J = 5.9, 13.5 Hz, 1H), 4.08–4.17 (m, 3H), 4.18–4.26 (m, 1H), 4.58–4.67 (m, 1H), 5.01 (dd, J = 2.0, 5.9 Hz, 1H), 6.31 (t, J = 6.6 Hz, 1H), 7.27–7.32 (m, 2H), 7.59 (s, 1H), 7.67–7.73 (m, 2H), 7.80 (s, 1H), 13C-NMR: (CDCl3) δ 22.0, 28.4, 40.8, 46.8, 65.8, 69.4, 71.6, 84.0, 84.2, 128.3, 130.4, 132.3, 132.6, 133.0, 134.2, 142.9, 145.6, 152.9, HRMS: (MH+) calcd. for C23H31N4O8S+: 523.1863, found: 523.1848.
(8R)-3-(2’-deoxy-5’-methylthio-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol (7)
Under argon 6 (35 mg, 0.067 mmol) was dissolved in dry methanol (3 mL) and treated with sodium thiomethoxide (95% w/w, 0.30 g) at RT. After 2 h the reaction mixture was evaporated in vacuo. Purification of the residue by chromatography (12 g silica, chloroform/methanolic ammonia (7 M) = 5:1 v/v, 120 mL, 4:1 v/v 100 mL) gave 7 as a white solid (14.5 mg, 73%). Rf = 0.35 (chloroform/methanolic ammonia (7 M) = 4:1 v/v), [α]D20 = +89 (c 0.41, methanol), 1H-NMR: (D2O) δ 2.09 (s, 3H, SMe), 2.48 (ddd, J = 4.1, 6.4, 14.1 Hz, 1H, H-2’), 2.68 (ddd, J = 6.7, 7.2, 14.1 Hz, 1H, H-2’), 2.74 (dd, J = 6.6, 14.1 Hz, 1H, H-5’), 2.82 (dd, J = 5.7, 14.1 Hz, 1H, H-5’), 3.34 (br d, J = 13.5 Hz, 1H, H-7), 3.49 (dd, J = 4.4, 13.5 Hz, 1H, H-7), 4.16 (ddd, J = 3.5, 6.0, 6.2 Hz, 1H, H-4’), 4.52 (m, 1H, H-3’), 5.12 (br d, J = 3.5 Hz, 1H, H-8), 6.25 (t, J = 6.8 Hz, 1H, H-1’), 7.20 (s, 1H), 7.75 (s, 1H), 13C-NMR: (D2O) δ 15.8 (SMe), 36.7 (C-5’), 39.1 (C-2’), 47.7 (C-7), 67.2 (C-8), 73.8 (C-3’), 83.2 (C-1’), 85.8 (C-4’), 128.4, 131.4 (C-2), 135.9, 151.0 (C-5), HRMS: (MH+) calcd. for C12H19N4O3S+: 299.1178, found: 299.1187, Anal. calcd. for C12H18N4O3S·1.4 H2O: C 44.54, H 6.48, N 17.31, S 9.91; found: C 44.42, H 6.13, N 17.19, S 9.69.
(8R)-3-(2’-deoxy-5’-propylthio-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol (8)
1-Propanethiol (0.42 mL, 4.64 mmol) was diluted in dry methanol (3 mL) and sodium methoxide (30% w/w in methanol, 0.72 mL) was added. After 5 min at RT 6 (40 mg, 0.077 mmol) was added and the mixture was stirred overnight at RT. The next day the solution was evaporated in vacuo and purified by chromatography (15 g silica, chloroform/methanolic ammonia (7 M) = 5:1 v/v) which gave product as white soapy material (17 mg, 68%). Rf = 0.31 (chloroform/methanolic ammonia (7M) = 4:1 v/v). [α]D20 = +76 (c 0.85, methanol), 1H-NMR: (CD3OD) δ 0.97 (t, J = 7.3 Hz, 3H), 1.60 (sext, J = 7.3 Hz, 2H), 2.35 (ddd, J = 3.9, 6.3, 13.5 Hz, 1H, H-2’), 2.47 (dd, J = 6.9, 13.5 Hz, 1H, H-2’), 2.55 (t, J = 7.3 Hz, 2H), 2.76 (dd, J = 5.9, 13.8 Hz, 1H, H-5’), 2.81 (dd, J = 5.9, 13.8 Hz, 1H, H-5’), 3.27–3.35 (m, 1H, H-7), 3.39 (dd, J = 4.4, 13.1 Hz, 1H, H-7), 4.02 (ddd, J = 3.5, 5.9, 5.9 Hz, 1H, H-4’), 4.35–4.43 (m, 1H, H-3’), 4.98–5.05 (m, 1H, H-8), 6.27 (t, J = 6.8 Hz, 1H, H-1’), 7.08 (s, 1H), 7.66 (s, 1H), 13C-NMR: (CD3OD) δ 13.7, 24.0, 35.7, 35.9, 41.4 (C-2’), 49.1 (C-7), 68.4 (C-8), 74.5 (C-3’), 84.4 (C-1’), 87.6 (C-4’), 130.3 (C-2), 131.4, 136.8, 149.8 (C-5), HRMS: (MH+) calcd. for C14H23N4O3S+: 327.1491, found: 327.1487, Anal. calcd. for C14H22N4O3S: C, 51.51; H, 6.79; N, 17.16; found: C, 51.49; H 6.77; N, 16.94.
(8R)-3-(2’-deoxy-5’-phenylthio-β,d-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol (9)
Under argon sodium methoxide (30% w/w in methanol, 0.72 mL) was added to a solution of thiophenol (0.47 mL, 4.6 mmol) in dry methanol (3 mL). After 5 min 6 (39 mg, 0.075 mmol) was added, and the solution was stirred at RT for 24 h. Sodium thiomethoxide (95%, 0.32 g) was added to the mixture and stirred for further 3 h 20 min. The reaction mixture was concentrated in vacuo, redissolved in chloroform/methanol (4:1 v/v), silica (1 g) was added and again concentrated to dryness in vacuo and applied to a column (14 g silica, chloroform/methanol = 10:1 v/v 200 mL washed off excess thiophenol, then 4:1 v/v 100 mL eluted the product) which gave compound 9 as an oil (24mg, 90%). Rf = 0.21 (chloroform/methanol = 4:1 v/v), [α]D20 = +83 (c 0.49, methanol), 1H-NMR: (CD3OD) δ 2.35 (ddd, J = 3.4, 6.2, 13.6 Hz, 1H, H-2’), 2.51 (ddd, J = 6.2, 7.4, 13.6 Hz, 1H, H-2’), 3.19 (dd, J = 6.0, 13.8 Hz, 1H, H-5’), 3.24 (dd, J = 6.1, 13.8 Hz, 1H, H-5’), 3.27–3.34 (m, 1H, H-7), 3.39 (dd, J = 4.4, 13.2 Hz, 1H, H-7), 4.05 (ddd, J = 3.1, 6.0, 6.1 Hz, 1H, H-4’), 4.41 (ddd, J = 3.0, 3.2, 6.2 Hz, 1H, H-3’), 5.01 (dd, J = 1.4, 4.4 Hz, 1H, H-8), 6.26 (dd, J = 6.2, 7.4 Hz, 1H, H-1’), 7.07 (s, 1H, H-5), 7.14–7.22 (m, 1H), 7.23–7.32 (m, 2H), 7.36–7.43 (m, 2H), 7.59 (s, 1H, H-2), 13C-NMR: (CD3OD) δ 37.7 (C-5’), 41.3 (C-2’), 49.1 (C-7), 68.4 (C-8), 74.6 (C-3’), 84.7 (C-1’), 86.6 (C-4’), 127.3, 130.1, 130.4, 130.6, 131.5 (C-2), 136.8, 137.5, 149.8 (C-5), HRMS: (MH+) calcd. for C17H21N4O3S+: 361.1334, found: 361.1324, Anal. calcd. for C17H20N4O3S·1.7 H2O: C 52.21, H 6.03, N 14.33; found: C 52.01, H 5.75, N 14.50.
Allyl 2,3-di-O-acetyl-5-S-methyl-5-thio-α,β-d-ribofuranoside (10)
Acetyl chloride (2.0 mL, 28 mmol) was added to a stirred suspension of d-ribose (6.0 g, 40 mmol) in allyl alcohol (100 mL). After 0.5 h, pyridine (5 mL, 61.8 mmol) was added to the clear solution and it was concentrated to dryness. Chromatography afforded allyl α,β-d-ribofuranoside (7.47 g, 39.3 mmol, 98%) as a syrup. A solution of this material in dry pyridine (60 mL) was treated with 2,4,6-triisopropylbenzenesulfonyl chloride (14.2 g, 46.9 mmol) and then allowed to stand for 2 days. It was diluted with water (1L) and extracted x2 with chloroform. The combined extracts were washed with water, dil HCl and aq NaHCO3 dried and concentrated. Chromatography afforded 9.77 g of major product. A stirred solution of 8.84 g of this material in DMF (30 mL) was treated with sodium thiomethoxide (4.1 g, 58.6 mmol). After 2 h, pyridine (50 mL) and acetic anhydride (50 mL) were added and the solution was stirred for 16 h before being partitioned between toluene and water. The organic phase was washed with water, dil HCl, aq NaHCO3, dried and concentrated. Chromatography gave separately the two anomers of the title compound; first the major compound (4.16 g, 13.7 mmol) followed by the minor (0.73 g, 2.4 mmol). Total yield 40% overall. For the major anomer: 1H NMR (CDCl3) δ 5.94-5.81 (1H, m), 5.34-5.12(4H, m), 5.03 (1H, s), 4.30-4.19 (2H, m), 4.04-3.97 (1H, m), 2.77 (2H, m), 2.18 (3H, s), 2.10 (3H, s), 2.06 (3H, s); 13C NMR δ 170.1, 170.0, 133.9, 117.9, 104.7, 80.8, 75.5, 74.8, 68.9, 38.8, 20.9, 16.6. For the minor anomer: 1H NMR (CDCl3) δ 5.95-5.84 (1H, m), 5.34-5.02 (5H, m), 4.35-4.21 (2H, m), 4.12-4.05 (1H, m), 2.87 (1H, dd, J = 14.0, 4.6 Hz), 2.78 (1H, dd, J = 14.0, 5.5 Hz), 2.18 (3H, s), 2.12 and 2.11 (3H each, s); 13C NMR δ 170.8, 170.2, 134.4, 117.5, 99.8, 81.6, 72.4, 71.3, 68.9, 37.1, 21.1, 20.9, 17.4. HRMS (MH+) Calcd for C13H21O6S: 305.1059. Found: 305.1072.
8-(R)-3-(2,3-Di-O-acetyl-5-S-methyl-5-thio-β-d-ribofuranosyl)-6-tert-butoxycarbonyl-8-(tert-butyldimethylsilyloxy)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine (12)
To a solution of 10 (3.48 g, 11.5 mmol) in acetic acid (70 mL) was added tetrakis-triphenylphosphine palladium(0) (5.3 g, 4.59 mmol) and the solution was stirred under argon at 70 °C. After 1.5 h, more tetrakis-triphenylphosphine palladium(0) (1.0 g, 0.87 mmol) was added and after another 0.5 h, the solution was concentrated to dryness. Chromatography afforded de-allylated material as a syrup (1.9 g, 7.2 mmol, 63%). Trichloroacetonitrile (0.75 mL, 7.5 mmol) and then DBU (4 drops) were added to a stirred solution of this material (0.31 g, 1.17 mmol) in dichloromethane (3 mL). After 0.5 h, the solution was diluted with hexanes and chromatographed (EtOAc/Hexanes 1:3 with 0.5% Et3N) to give 0.40 g, (0.98 mmol) of syrupy trichloroacetimidate 11. A solution of this material and 2b (0.12 g, 0.328 mmol) in dry dichloromethane (5 mL) was cooled in an ice bath, trimethylsilyl trifluoromethanesulfonate (0.10mL, 0.55 mmol) was added and the solution was allowed to warm to room temperature. After 1 h, the solution was washed with aq NaHCO3, dried and concentrated. Chromatography (1% Et3N in EtOAc/CHCl3/Hexanes 1:2:4) gave syrupy 12 (0.109 g, 0.178 mmol, 54% based on the acceptor). 1H NMR (CDCl3) δ 7.88 (1H, s), 7.60 (1H, s), 6.03 (1H, d, J = 5.0 Hz), 5.80 (1H, t, J = 5.4 Hz), 5.50 (1H, t, J = 5.5 Hz), 5.13 (1H, d, J = 4.4 Hz), 4.48 (1H, dd, J = 13.5, 4.6 Hz), 4.34 (1H, dd, J = 13.5, 5.2 Hz), 3.13 (1H, d, J = 13.5 Hz), 2.87 (2H, m), 2.14, 2.10, 2.08 (3H each, s), 1.54 (9H, s), 0.88 (9H, s), 0.19, 0.05 (3H each, s); 13C NMR δ 169.9, 169.7, 152.8, 142.8, 135.0, 133.2, 86.4, 83.9, 82.0, 73.9, 72.8, 67.6, 47.2, 36.9, 28.5, 26.2, 21.0, 20.8, 18.7, 17.4, -4.2, -4.6. HRMS (MH+) Calcd for C27H45N4O8SSi: 613.2727. Found: 613.2739.
8-(R)-Hydroxy-3-(5-S-methyl-5-thio-β-d-ribofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine (13)
Tetrabutylammonium fluoride (2 mL, 1 M in THF) was added to a solution of 12 (0.25 g, 0.408 mmol) and acetic acid (0.05 mL) in THF (2 mL) and the solution was allowed to stand for 2 days. The solution was evaporated and chromatography afforded 8-(R)-3-(2,3-di-O-acetyl-5-S-methyl-5-thio-β-d-ribofuranosyl)-6-tert-butoxycarbonyl-8-hydroxy-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepine as a foam (0.143 g, 0.287 mmol, 70%). 1H NMR (CDCl3) δ 7.86 (1H, s), 7.68 (1H, s), 6.06 (1H, d, J = 5.2 Hz), 5.78 (1H, t, J = 5.4 Hz), 5.48 (1H, t, J = 5.4 Hz), 4.98 (1H, br s), 4.35 (1H, dd, J = 10.4, 5.2 Hz), 3.94-3.75 (2H, m), 2.94-2.82 (2H, m), 2.16, 2.11, 2.09 (3H each, s), 1.55 (9H, s); 13C NMR δ 170.0, 169.7, 152.7, 143.3, 133.6, 86.4, 84.4, 82.1, 73.9, 72.7, 65.7, 46.7, 36.9, 28.5, 20.9, 20.8, 17.4. Sodium thiomethoxide (0.20 g, 2.85 mmol) was added to a stirred solution of this material (0.14 g, 0.28 mmol) in methanol (3 mL). After 4 h, the solution was concentrated and chromatography of the residue gave title compound as a white solid (0.062 g, 0.197 mmol, 70%). 1H NMR (CD3OD) δ 8.00 (1H, s), 7.16 (1H, s), 5.93 (1H, d, J = 4.3 Hz), 5.01, (1H, dd, J = 4.5, 2.2 Hz), 4.43 (1H, t, J = 4.5 Hz), 4.23-4.16 (2H, m), 3.46-3.34 (2H, m), 2.94-2.79 (2H, m), 2.15 (3H, s); 13C NMR δ 150.9, 137.5, 131.5, 128.4, 90.2, 85.3, 76.4, 74.1, 67.7, 37.9, 17.1. HRMS (MH+) Calcd for C12H18N4O4S: 315.1127. Found: 315.1130.
RESULTS AND DISCUSSION
The recent determination that the adenosine deaminase from P. falciparum has catalytic activity for both adenosine and 5′-methylthioadenosine as physiological substrates,13 led to the investigation of adenosine deaminase inhibitors with 5′-methylthio and related groups. Two potent inhibitors of human adenosine deaminase, coformycin and 2′-deoxycoformycin, were targeted as parent compounds for this investigation. Both inhibitors have picomolar dissociation constants for human adenosine deaminase,7-9 and 2′-deoxycoformycin (Pentostatin) was approved by the FDA for treatment of hairy cell leukemia in 1991. The use of Pentostatin as a therapeutic agent has been limited due to the associated toxicity of the lungs, liver, kidneys and nervous system, which is related to the inhibition of ADA and also inhibitor incorporation into DNA in these tissues.12 Modification of ADA inhibitors to eliminate binding to the human enzyme and to eliminate 5’-phosphorylation for entry into DNA pathways, while retaining powerful inhibition of the malarial enzyme, would allow for therapeutic efficacy against the parasite while eliminating the toxicity of the parent compounds.
Synthesis of 5′-Functionalized Coformycins
Previous syntheses of coformycin and 2’-deoxycoformycin have focused on the preparation of 8-keto-diazepine derivatives followed by reduction of the ketone and separation of the diastereoisomeric 8-(R/S)-alcohols.21-23 The chromatographic separation of these isomers is a synthetically inefficient and difficult route. Consequently we have concentrated on methods to synthesize the desired 8-(R)-diazepine specifically. Synthesis of the chiral aglycone 2b has been reported24 and we have developed an improved synthesis of this material – details of which will be published elsewhere. The aglycone was conveniently stored as the N-protected 2a as 2b does not survive storage for even a few days.
There have been no reports of the successful glycosylation of 2b but we found that a number of 5’-substituted thio-2’-deoxycoformycin derivatives (7, 8 and 9) were accessible by using 2-deoxy-di-O-toluoylribosyl chloride 1 as glycosylating agent, which was made from 2-deoxy-d-ribose (Scheme 1). The aglycone 2a was deprotected at N-3 with potassium tert-butoxide in THF to give free amine 2b. This was deprotonated by treatment with sodium hydride followed by coupling with the α-glycosyl chloride 1 under SN2 conditions.25-27 That the isolated anomerically pure nucleoside analogue 3 was the β-anomer depicted was confirmed by NOESY NMR experiments on compounds 7, 8 and 9 which showed interactions between H-1’ ↔ H-4’, H-3’ ↔ H-2 and H-5’ ↔ H-2. This product was deacylated with ammonia in methanol to give diol 4. Selective tosylation at the primary hydroxy group afforded tosylate 5 in moderate yield (60%). Desilylation28 then gave diol 6, the treatment of which with sodium thiomethoxide simultaneously displaced the tosylate and, somewhat surprisingly, cleaved the Boc carbamate to provide methylthio ether 7 in 73% yield from compound 6. The propylthio analogue 8 was prepared in the same fashion in a similar yield (68%). However, when sodium thiophenoxide was used, the tosylate was displaced but the Boc group remained unaffected. The addition of sodium methanethiolate to this reaction mixture removed the Boc group and provided the phenylthio compound 9 which was isolated in 90% yield in a one-pot procedure.
Scheme 1. Reagents.

i, KOtBu, THF, 77%; ii, NaH, CH3CN, 85%; iii, NH3, MeOH, 71%; iv, TsCl, pyridine, 60%; v, NH4F, MeOH, reflux, 58%; vi, NaSMe, MeOH, 73%; vii, PrSH, NaOMe, MeOH, 68%; viii, PhSH, NaOMe, MeOH, then NaSMe, 90%.
The successful synthesis of the β-nucleoside derivative 3 above depends on the use of crystalline 2-deoxy-α-ribosyl chloride 1 so that essentially a single anomer of ribosyl chloride is available in solution for the SN2 reaction. There are no readily available crystalline α-ribosyl halides available, so the 5’-methylthiocoformycin derivative was prepared using a ribosyl donor with a participating group at O-2 to control the anomeric stereochemistry. We found it convenient to use a 5-methylthio-ribose donor and after some experimentation settled on a trichloroacetimidate donor as the most effective.
Thus, d-ribose was converted to the allyl 5-S-methylribofuranosides 10 (Scheme 2) in 40% overall yield. Deallylation and formation of the trichloroacetimidate donor 11 was followed directly by glycosylation of 2b in the presence of TmsOTf to give the protected coformycin derivative 12 (54%). The stereochemistry of 12 was confirmed by NMR NOESY experiments. Desilylation with Bu4NF was complicated by degradation, but when buffered with HOAc gave the desired alcohol in 70% yield. Since we have found that sodium thiomethoxide will remove the Boc carbamate from diazepines such as 12, it was convenient to use these conditions (NaSMe, MeOH) to effect deacetylation and the Boc removal to give 5’-methylthiocoformycin 13 in 70% yield.
Scheme 2. Reagents.
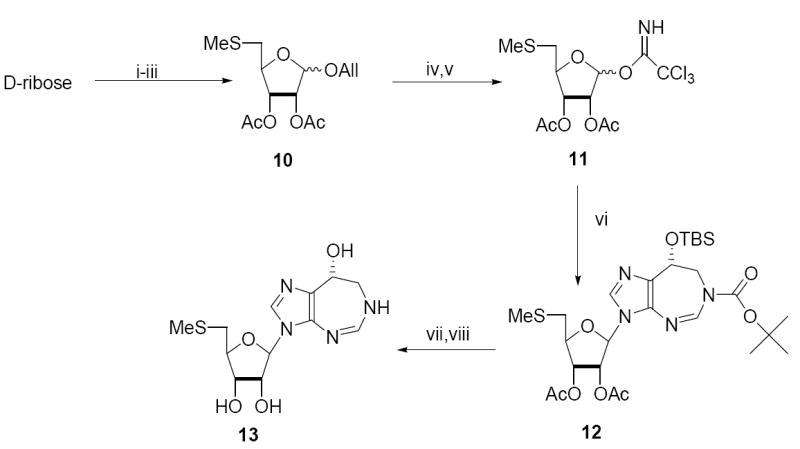
i, HCl, AllOH; ii, Triisopropylbenzenesulfonyl chloride, py; iii, NaSMe, DMF, then py/Ac2O; iv, (Ph3P)4Pd(0), HOAc; v, Cl3CN, DBU, CH2Cl2; vi, 2b, TmsOTf, CH2Cl2; vii, Bu4NF, HOAc, THF; viii, NaSMe, MeOH.
Inhibition of Adenosine Deaminase by Coformycin Analogues
The inhibition of human (HsADA), bovine (BtADA) and malarial (PfADA) adenosine deaminases was examined with the coformycins described above (Table 1, Figure 2). Both coformycin and 2′-deoxycoformycin exhibit slow onset inhibition of all three enzymes with the dissociation constants ranging from 27 to 110 pM. None of the 5′-functionalized analogues inhibited the bovine or human enzymes, whereas they all inhibited PfADA. The 5′-methylthio analogues were the most effective inhibitors of PfADA, with slow onset inhibition and dissociation constants in the range of 430 to 730 pM. The propyl- and phenylthio-analogues also inhibited PfADA, but with no detectible slow onset phase and these compounds gave dissociation constants of 12 and 61 nM, respectively.
Table 1.
Inhibition constants of transition state analogue inhibitors of adenosine deaminase
| Inhibitor | BtADA | HsADA | PfADA | |||
|---|---|---|---|---|---|---|
| Ki (nM) | Ki* (nM) | Ki (nM) | Ki* (nM) | Ki (nM) | Ki* (nM) | |
| Coformycin | 1.1 ± 0.4 | 0.06 ± 0.01 | 13.9 ± 3.4 | 0.11 ± 0.02 | 0.68 ± 0.07 | 0.08 ± 0.02 |
| 5′-MeS-Coformycin (13) | >10,000a | NDb | >10,000 | ND | 2.66 ± 0.13 | 0.43 ± 0.12 |
| 2′-deoxycoformycin | 0.39 ± 0.12 | 0.027 ± 0.004 | 0.5 ± 0.1 | 0.026 ± 0.005 | 8.2 ± 2.9 | 0.038 ± 0.009 |
| 5′-MeS-2′-deoxycoformycin (7) | >10,000 | ND | >10,000 | ND | 2.3 ± 0.8 | 0.73 ± 0.22 |
| 5′-PrS-2′-deoxycoformycin (8) | >10,000 | ND | >10,000 | ND | 12 ± 1 | ND |
| 5′-PhS-2′-deoxycoformycin (9) | >10,000 | ND | >10,000 | ND | 61 ± 11 | ND |
Ki values of >10,000 indicate that assays with 10 μM of the indicated inhibitor exhibited no inhibition under the conditions of the assay.
No slow onset phase was detected.
Figure 2.

Dissociation constants (Kd) for inhibition of ADAs from bovine, human and P. falciparum sources.
The substrate specificity of the ADA enzymes with adenosine and 5′-methylthioadenosine as reactants reveals that the mammalian enzymes, though more proficient at utilization of adenosine than PfADA, are unable to utilize 5′-methylthioadenosine (Table 2). Comparison of the amino acid sequences of PfADA, BtADA, HsADA with the structurally characterized ADAs from Mus musculus (MmADA) and Plasmodium yoelii (PyADA) reveals a high level of sequence conservation, especially of active site residues (Figure 3).29 The exception is in the amino acids that are near to the 5′-position, where residues Cys153 and Met155 in MmADA structurally and sequentially align with residues Ile171 and Asp173 in PyADA. Additionally, Tyr129 of PyADA, whose identity is maintained with respect to the mammalian homologue (corresponds to Tyr102 in MmADA), is one angstrom removed from the active site as compared to Tyr102 (corresponding to distances of 5.56 and 4.67 Å, respectively; data not shown). These differences cause an increased pocket size for the 5’-group in PfADA which is likely to be the structural change that allows for the binding and deamination of the 5′-functionalized compounds.
Table 2.
Kinetic constants for HsADA, BtADA and PfADA with adenosine and 5′-methylthioadenosine as substrates.
| Organism | adenosine | 5′-methylthioadenosine | ||||
|---|---|---|---|---|---|---|
| kcat (sec -1) | KM (μM) | kcat / KM (M-1sec-1) | kcat (sec -1) | KM (μM) | kcat / KM (M-1sec-1) | |
| B. taurus | 65 | 56 | 1.1 × 106 | <0.02 | NAa | NA |
| H. sapiens | 36 | 22 | 1.6 × 106 | <0.02 | NA | NA |
| P. falciparum | 1.8 | 29 | 6.2 × 105 | 15 | 170 | 9.6 × 104 |
Not applicable since no significant reaction rate is observed.
Figure 3.

Amino acid sequence alignment for adenosine deaminases from various sources. Residues highlighted in dark blue are completely conserved in sequence, medium blue are highly similar, and light blue are similar. Residues located in the active site (defined on the basis of structural analysis from a superposition of the mouse and P. yoelii structures, PDB ID = 1ADD and 2AMX, respectively, and being located within 10 Å of bound 1-deazaadenosine) are indicated in red. Those active site residues within 5 Å of the 5′-hydroxyl of 1-deazaadenosine, where identity is not conserved between the mammalian and Plasmodium sequences are highlighted in yellow. The insertion in the mammalian enzymes corresponding to residues 109-119 (MmADA) cause an alteration in the structural orientation of residues 101-127, relative to the malarial enzyme, which is indicated by a black bar.
CONCLUSIONS
The substrate specificity differences between mammalian and malarial ADAs have been exploited for the generation of inhibitors specific for ADA from P. falciparum. The 5′-methylthio group results in a 5-fold difference in the binding affinity of coformycin and 5′-methylthio coformycin to PfADA. However, this same change causes a >20,000-fold preference in inhibitor binding to PfADA compared to the human and bovine enzymes. The ADA isozyme specificity highlights the importance of examining substrate specificity in addition to transition state features in the design of species-specific inhibitors.
Footnotes
This work was supported by research grant AI49512 from the National Institutes of Health and contract C08X0209 from the New Zealand Foundation for Research Science and Technology.
References
- 1.Agarwal RP. Pharmacol Ther. 1982;17:399–429. doi: 10.1016/0163-7258(82)90023-7. [DOI] [PubMed] [Google Scholar]
- 2.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Lancet. 1972;2:1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal RP, Crabtree GW, Parks RE, Jr, Nelson JA, Keightley R, Parkman R, Rosen FS, Stern RC, Polmar SH. J Clin Invest. 1976;57:1025–1035. doi: 10.1172/JCI108344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ullman B, Gudas LJ, Cohen A, Martin DW., Jr Cell. 1978;14:365–375. doi: 10.1016/0092-8674(78)90122-8. [DOI] [PubMed] [Google Scholar]
- 5.Webster HK, Weismann WP, Pavia CS. Adv Exp Med Biol. 1984;165:225–229. doi: 10.1007/978-1-4684-4553-4_44. [DOI] [PubMed] [Google Scholar]
- 6.Wiesmann WP, Webster HK, Lambros C, Kelly WN, Daddona PE. Prog Clin Biol Res. 1984;165:325–342. [PubMed] [Google Scholar]
- 7.Agarwal RP, Spector T, Parks JRE. Biochem Pharmacol. 1977;26:359–367. doi: 10.1016/0006-2952(77)90192-7. [DOI] [PubMed] [Google Scholar]
- 8.Cha S, Agarwal RP, Parks JRE. Biochem Pharmacol. 1975;24:2187–2197. doi: 10.1016/0006-2952(75)90051-9. [DOI] [PubMed] [Google Scholar]
- 9.Schramm VL. Arch Biochem Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 10.Blackburn MR, Kellems RE. Adv Immunol. 2005;86:1–41. doi: 10.1016/S0065-2776(04)86001-2. [DOI] [PubMed] [Google Scholar]
- 11.Published online at: http://www.accessdata.fda.gov/scripts/cder/onctools/summary.cfm?ID=111
- 12.Johnson SA. Clin Pharmacokinet. 2000;39:5–26. doi: 10.2165/00003088-200039010-00002. [DOI] [PubMed] [Google Scholar]; Margolis J, Grever MR. Semin Oncol. 2000;27:9–14. [PubMed] [Google Scholar]
- 13.Ting LM, Shi W, Lewandowicz A, Singh V, Mwakingwe A, Birck MR, Taylor Ringia EA, Bench G, Madrid DC, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K. J Biol Chem. 2005;280:9547–9554. doi: 10.1074/jbc.M412693200. [DOI] [PubMed] [Google Scholar]
- 14.Kicska GA, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K. J Biol Chem. 2002;277:3226–3231. doi: 10.1074/jbc.M105906200. [DOI] [PubMed] [Google Scholar]
- 15.Schramm VL, Baker DC. Biochemistry. 1985;24:641–646. doi: 10.1021/bi00324a016. [DOI] [PubMed] [Google Scholar]
- 16.Merkler DJ, Brenowitz M, Schramm VL. Biochemistry. 1990;29:8358–8364. doi: 10.1021/bi00488a023. [DOI] [PubMed] [Google Scholar]
- 17.Singh V, Shi W, Evans GB, Tyler PC, Furneaux RH, Almo SC, Schramm VL. Biochemistry. 2004;43:9–18. doi: 10.1021/bi0358420. [DOI] [PubMed] [Google Scholar]
- 18.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. J Biol Chem. 2005;280:18265–18273. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 19.The human enzyme sample received from Sigma was characterized to have a kcat of 36 ± 1 sec-1 and a KM of 22 ± 2 μM.
- 20.Hoffer M. Chem Ber. 1960;93:2777–2781. [Google Scholar]
- 21.Chan E, Putt SR, Showalter HDH, Baker DC. J Org Chem. 1982;47:3457–3464. [Google Scholar]
- 22.Hawkins LD, Hanvey JC, Boyd FL, Jr, Baker DC, Showalter HDH. Nucleosides Nucleotides. 1983;2:479–494. [Google Scholar]
- 23.Jeanette TH, Riordan JM, Montgomery JA. Nucleosides Nucleotides. 1986;5:431–439. [Google Scholar]
- 24.Van Truong T, Rapoport H. J Org Chem. 1993;58:6090–6096. [Google Scholar]
- 25.Kayzimierczuk Z, Binding U, Seela F. Helv Chim Acta. 1989;72:1527–1536. [Google Scholar]
- 26.Kayzimierczuk Z, Cotton HB, Revankar GR, Robins RK. J Am Chem Soc. 1984;106:6379–6382. [Google Scholar]
- 27.Kondo KSH, Ishibashi M, Kobayashi J. Tetrahedron. 1992;48:7145–7148. [Google Scholar]
- 28.Zhang WR, M J. Tetrahedron Lett. 1992;33:1177–1180. [Google Scholar]
- 29.The PDB ID for the structurally characterized ADAs from Mus musculus and Plasmodium yoelii are 1ADD and 2AMX, respectively.