

Abstract
The free 2'-3' cis-diol at the 3'-terminus of tRNA provides a unique juxtaposition of functional groups that play critical roles during protein synthesis. The translation process involves universally conserved chemistry at almost every stage of this multi-step procedure, and the 2'- and 3'-OHs are in the immediate vicinity of chemistry at each step. The cis-diol contribution affects steps ranging from tRNA aminoacylation to peptide bond formation. The contributions have been studied in assays related to translation over a period that spans at least three decades. In this review we follow the 2'-and 3'-OHs through the steps of translation and examine the involvement of these critical functional groups.
Translation, the assembly of polypeptides from amino acids, is a fundamental metabolic process. The enzymes, substrates, and chemical mechanisms are universally conserved, suggesting that the basis for translation emerged very early in the evolution of terrestrial life. A large portion of cellular resources are committed to the rapid and accurate production of proteins (1, 2).
Transfer RNAs (tRNAs) are key players in this process. Often thought of as “adaptor molecules,”(3) they are responsible for bridging the information contained in messenger RNA (mRNA), which encodes a sequence of amino acids, with the chemistry of peptidyl transfer, which covalently links the chains of amino acids. tRNAs are initially recognized by their cognate tRNA synthetases, which attach the specific amino acid to the 3'-end of the cognate tRNA species. The aminoacyl-tRNA is then presented to the ribosomal A site by EF-Tu, where it acts as a peptide acceptor. The peptidyl-tRNA is translocated by EFG to the P site, where it serves as a peptide donor before finally moving to the E site and being released (4).
The A76 ribose sugar at the 3'-terminus has two free hydroxyls on adjacent carbons available for the formation of an ester linkage to an amino acid, the 2'-OH and the 3'-OH. The 3' terminal ribose of an RNA molecule is the only place anywhere in RNA or proteins where two unmodified vicinal hydroxyl groups exist, which may make the 3' terminus of the tRNA uniquely functional for many roles in translation. This 2'-3' cis-diol at the tRNA's 3'-terminus has important functions in every step of translation, providing the attachment site for the amino acid or peptide, contributing to substrate binding, and by direct involvement in the chemical transformations. Here we discuss participation of the pair of A76 hydroxyls as the tRNA progresses through the protein synthesis machinery.
Hydroxyl groups
Hydroxyl groups are common in biological molecules and their properties allow them to be employed for a variety of roles. The large difference in electronegativity between hydrogen and oxygen makes hydroxyl groups polar. They can therefore act both as hydrogen bond donors and acceptors, and a single hydroxyl can perform both roles simultaneously. This makes hydroxyls well suited for coordinating associations between molecules. Hydrogen bonding patterns at the surface of molecules are commonly used to drive molecular specificity, and hydroxyl groups are often intimately involved(5-7). Their hydrogen bonding capacity also makes hydroxyl groups useful for interactions between biomolecules and their solvents(8). They can help to increase the hydrophilic character of a molecule and can participate in positioning specifically bound water molecules or metal ions(9, 10). Hydroxyl groups also have the potential to act as proton donors or acceptors in general acid or general base catalytic mechanisms or other proton transfer events(11, 12). In most cases it is the hydroxyl groups of carboxylic acids that are involved in such roles. The pKas of other hydroxyl groups, such as those of the ribosyl cis-diol, tend to fall too far outside the physiological pH range to be good candidates for proton transfer.
The context of the ribose ring significantly affects the chemical properties of its two hydroxyl groups. The presence of the ring oxygen, an electron withdrawing group, as well as the neighboring hydroxyl group, increases the leaving group potential of each. This, in combination with the potential for intramolecular hydrogen bonds between the two hydroxyl groups, reduces their pKas from around 16 (the pKa of ethanol) to around 12.5 and increases their chemical reactivity(13, 14). The hydrolysis rate of an ester with a neighboring hydroxyl is 40-fold faster than one without(15). Simply having another reactive group in a constrained position proximal to each hydroxyl can have a strong impact on reactions of those hydroxyls. For example, nucleophilic attack by the vicinal hydroxyl accounts for the greater base lability of RNA relative to DNA(16).
The juxtaposition of hydroxyl groups also affects the sugar pucker of the ribose ring. Deoxyribose sugars of DNA generally assume a C2' endo sugar pucker, while in RNA the ribose assumes a C3' endo sugar pucker. These differences could have important implications for the placement of functional groups within the active sites of the translational enzymes.
The aminoacyl linkage
An amino acid is activated for translation by ester linkage to the 2'- or 3'-OH of the 3' terminal adenosine. These two possible regioisomeric forms are chemically similar and there is no chemical reason for one to be a better substrate than the other. However, enzymes are highly specific, so it is reasonable to expect that the ribosome, tRNA synthetases, and other translational enzymes interact exclusively with a particular isoform. Experimentation has confirmed this expectation and has demonstrated that not all translational enzymes are specific for the same regioisomer. Since sequential enzymes have different isomeric preferences, conversion between the two isoforms is required during translation.
Each isoform is readily converted to the other via a 2'/3' transacylation in aqueous solution (see figure 1). This is a base catalyzed process, and the energy difference between the two is very small. At equilibrium an aminoacyl tRNA exists as a mixture of the 3' and 2' isoforms at a ratio of about 2:1 (17). If transacylation of the amino acid or nascent peptide between the 2'- and 3'-OHs is necessary during translation, the rate must be faster than the overall rate of protein synthesis, or else it would be the rate limiting step. Either the spontaneous rate of isomerization must be fast enough or one or more of the enzymes involved must actively catalyze the isomerization.
1. Base-catalyzed isomerization of aminoacyl tRNAs.
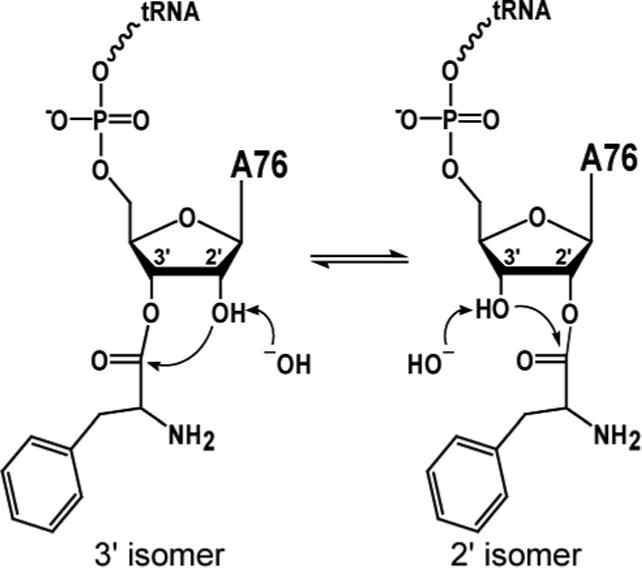
When the vicinal hydroxyl of an aminoacyl tRNA nucleophilically attacks the neighboring ester linkage, the amino acid can be transferred to the attacking hydroxyl.
The rate of spontaneous transacylation has been investigated in several contexts. An early NMR study on formyladenosine, in which a formyl group acylated on the 3'-OH of adenosine was used to mimic an amino acid, reported a transacylation rate of 4 × 103 s−1, considerably faster than the rate of amino acid incorporation during in vivo protein synthesis (∼20 s−1) (18). Based on this and similar reports, the spontaneous rate of transacylation was believed to be fast enough to occur at any point in the translation pathway, allowing each step to have its own preferred isoform, independent of the previous or subsequent steps. For the next two decades, this conclusion affected discussion of the regioisomeric state of tRNAs throughout translation. However, further investigation showed that the behavior of a formyl group may not accurately represent that of an amino acid. A series of NMR studies on aminoacyl adenosine and peptidyl adenosine demonstrated that the actual transacylation rate under physiological conditions was on the order of ∼5 s−1, slower than the rate of protein synthesis (19). Therefore, if regioisomerization of the aminoacyl or peptidyl tRNAs is required, the reaction would need to be catalyzed to keep up with protein synthesis.
Aminoacylation of tRNAs
Before the ribosome or any other translational machinery can incorporate amino acids into proteins, the amino acids must first be linked to tRNAs by aminoacyl tRNA synthetases. This step in the translation pathway accounts for much of the energy consumption as well as fidelity of protein synthesis. ATP is hydrolyzed during formation of an aminoacyl-adenylate intermediate, which is then the target of nucleophilic attack by the 2'- or 3'-OH of A76, resulting in transfer of the amino acid to the tRNA. This ATP hydrolysis is the only essential energy input for peptide bond formation, and once the aminoacyl tRNA is formed all of the energy required for peptide bond formation is contained in the ester linkage between the amino acid and the 2'- or 3'-OH.
Half of the amino acids are initially acylated on one hydroxyl, and half on the other. Aminoacyl tRNA synthetases are distributed into two evolutionarily unrelated classes, each containing 10 synthetases, based on sequence homology and three-dimensional structural similarity (20, 21). The two classes catalyze essentially the same reactions, but are significantly different in terms of sequence motifs and active site topology. Class I synthetases acylate tRNAs exclusively at the 3'-OH, while Class II synthetases, with the exception of PheRS, acylate exclusively at the 2'-OH. Prior to designation of each synthetase as either class I or class II based on sequence and structure, individual preferences for the 2'- or 3'-OHs were determined experimentally by testing their ability to aminoacylate tRNAs where A76 was substituted by 2' deoxyadenosine (2'-dA) or 3' deoxyadenosine (3'-dA) (22-24). The discovery that half of the amino acids were initially attached at one site, and half at the other clearly meant that if subsequent steps required one or the other isoform, then transacylation would be absolutely required, whether or not it was a catalyzed event.
The non-connective hydroxyl group has been present at the site of tRNA aminoacylation throughout the evolution of modern synthetase enzymes. It would be surprising if this potentially useful functional group had no role in the aminoacylation reaction. Although regioisomerization occurs readily, the synthetases' preferences for aminoacylation sites have been mostly maintained throughout evolution (25). Perhaps this reflects the conserved use of the other hydroxyl group during the aminoacylation process. Although, aminoacylation of deoxyadenosine substrates indicates that in each case the non-targeted hydroxyl group is not required for activity, the other hydroxyl group is not irrelevant. Within the two classes of synthetases a great deal of mechanistic diversity has evolved for recognition of cognate tRNAs and accurate aminoacylation, and each synthetase uses the vicinal hydroxyl to a different extent. For example, structural studies of AspRS showed that the non-acylated 2'-OH group forms hydrogen bonds with a conserved active site serine and the α-amino group of the adenylated aspartate (figure 2). These interactions facilitate transfer of the amino acid to the 3'-OH and position the nucleophile hydroxyl for attack (26). Replacing the 2'-OH with a proton results in 25-fold reduction of catalytic efficiency (27); mutation of hydrogen bonding partners results in 25−100 fold reduction of efficiency, with minimal effect on tRNA binding (28). Kinetic studies on charging of other dA76-substituted tRNAs show the range of the vicinal hydroxyls' importance for different synthetases. Phenylalanyl-tRNA synthetase charges 3'dA76 tRNAPhe only 2-fold less efficiently than the wild type substrate(24), but a 2'dA76 substitution in tRNAAla results in a greater than 100-fold reduction in efficiency relative to wild type(29).
2. X-ray crystal structure of E. coli tRNAAsp and aspartyl-adenylate bound in the active site of aspartyl tRNA synthetase [PDB entry 1C0A(26)].
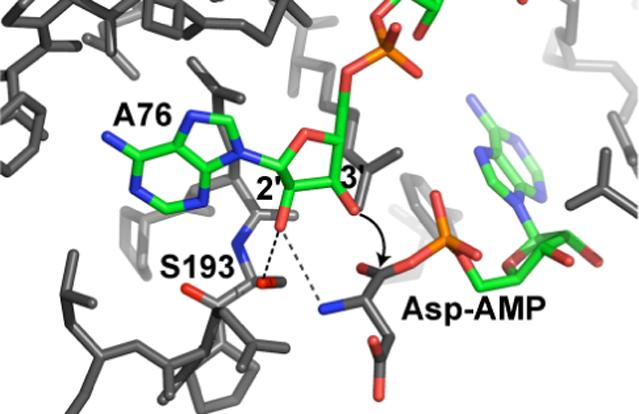
The A76 3'-OH is positioned for attack on the aspartyl-adenylate carboxyl carbon. The 2'-OH hydrogen bonds with serine 193, which is invariant in all AspRSs, and the aspartyladenylate α-amino group. These interactions contribute to the organization of the active site prior to aminoacylation.
The A76 hydroxyls can also participate in the aminoacyl adenylate formation, the amino acid activation step that precedes tRNA charging. In three aminoacyl tRNA synthetases, GlnRS, GluRS, and ArgRS, this reaction requires the presence of the cognate tRNA. The tRNAs are predicted to be critical components of the enzymes' active sites. Subsequent studies of GlnRS have demonstrated that this is at least partially due to the involvement of the 2'-OH. GlnRS, a class I synthetase, aminoacylates at the 2'-OH; a 2'-deoxy modification at A76 completely abolishes aminoacylation (30). However, the 2'-deoxy substitution also results in a 104-fold reduction in catalytic efficiency of the tRNA dependent adenylation of glutamine, implicating this functional group for an important role in adenylate formation (31). Structural studies of the GlnRS/tRNA/ATP complex demonstrate that the 2'-OH interacts with the α-phosphate of ATP, but that removal of this functional group does not result in significant conformational changes in the active site. Together these data suggest that the 2'-OH promotes the glutamine adenylation reaction through positioning, and possibly activating, the α-phosphate of the ATP for attack by the incoming amino acid (31-33).
Proofreading
tRNA synthetases are responsible for efficient and accurate attachment of the correct amino acid to its cognate tRNA. Synthetases employ different proofreading mechanisms to achieve a high level of fidelity. Non-cognate aminoacyl adenylates can be specifically hydrolyzed by synthetases, or these enzymes can use a kinetic proofreading mechanism wherein dissociation of a non-cognate aminoacyl adenylate occurs more rapidly than transfer to the tRNA. Some synthetases also specifically hydrolyze misacylated aminoacyl-tRNAs. Different synthetases (even the same synthetases from different species) utilize different discrimination strategies to varying degrees, but all synthetases achieve a high level of accuracy (34-36). Because modifications such as 2'-dA and 3'-dA and 2' or 3' aminoadenosine can render a tRNA non-functional for aminoacylation or particular proofreading steps, these substrate analogues have allowed individual steps to be isolated and studied in detail.
Post-transfer editing, in which mischarged tRNAs are specifically hydrolyzed, can require the vicinal hydroxyl for efficient reaction. Early studies indicated that the non- accepting hydroxyl group of A76 is essential for the enzymatic hydrolysis of amino acids from the tRNA by several synthetases, a reaction assumed to be related to proofreading (37-40). Some synthetases that utilize a post-transfer editing strategy require acylation at a specific hydroxyl, and these requirements differ between synthetases. This is clearly demonstrated by the three closely related class I synthetases LeuRS, IleRS, and ValRS, all of which specifically acylate the 2'-OH (41), and have a separate active site for editing, distinct from the synthetic site (42, 43). Based on activity assays of modified tRNAs as well as binding of small analogue inhibitors, IleRS deacylates only from the 3'-OH, and therefore requires transacylation of the amino acid from the 2' to the 3' position prior to hydrolysis (35, 44). In contrast, LeuRS and ValRS hydrolyze mischarged tRNAs specifically from the 2'-OH, which is the original site of acylation (44, 45). Alanine scanning mutagenesis of the LeuRS editing site has failed to identify catalytic residues for hydrolysis within the active site (44). Structural studies also do not provide any obvious candidates for direct involvement in catalysis, but rather suggest that the editing site acts by rigidly positioning substrates for nucleophilic attack by water. Based on an X-ray crystal structure of LeuRS bound to an analog of a mischarged tRNA, the 3'-OH hydrogen bonds with two conserved threonines as well as a specific water molecule that sits close to the 2' ester bond and may be directly involved in hydrolysis (45) (figure 3). The importance of these interactions was further demonstrated by studies of a mutant LeuRS that is compromised for discrimination against the cognate amino acid but retained wild-type editing kinetics. In the context of this mutant active site, further mutagenesis of one of the conserved threonines eliminates editing activity(46).
3. X-ray crystal structure of the post-transfer editing substrate analog 2_-(L-norvalyl)amino-2_-deoxyadenosine bound to the editing site of Thermus thermophilus Leucyl tRNA synthetase [PDB entry 1OBC(45)].
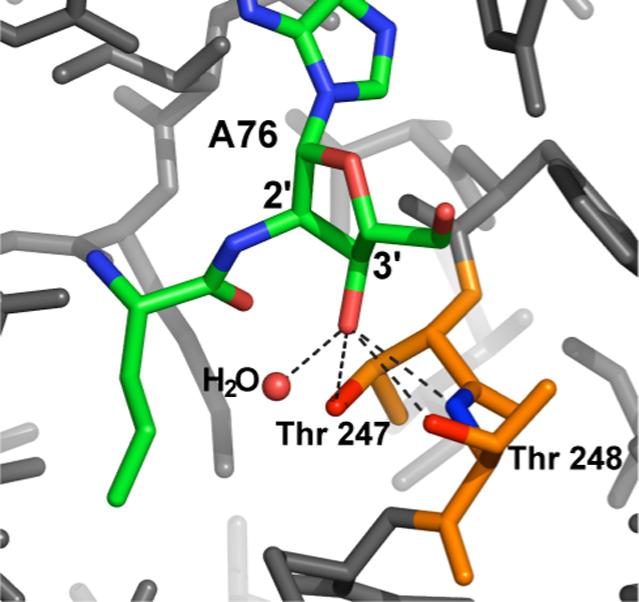
The A76 3'-OH is positioned to hydrogen bond with the _-OHs of threonines 247 and 248, and the backbone amide of threonine 248. The 3'-OH also coordinates a specifically bound water molecule that may be directly involved in hydrolysis of the editing substrate.
EF-Tu and accommodation
Following aminoacylation, aminoacyl tRNAs bind to elongation factor Tu (EF-Tu), which catalyzes the delivery of tRNAs to the ribosomal A site. In this process the tRNAs are matched with codons on the mRNA and positioned for acceptance of a growing peptide. EF-Tu must specifically bind aminoacyl tRNAs, independent of the amino acid, and discriminate against uncharged tRNAs. The 2'/3' ester linkage to the amino acid differentiates aa-tRNAs from uncharged tRNAs and is likely to be instrumental in recognition of aa-tRNAs by EF-Tu.
EF-Tu binding to aminoacyl-tRNAs stabilizes the 3' ester prior to arrival in the ribosomal A site. Several early studies examined binding to EF-Tu and the associated GTP hydrolysis by short oligonucleotide analogues of the 3' end of aa-tRNAs as well as full aa-tRNAs. Results from these studies indicated that both isomers of aa-tRNAs bind to EF-Tu, but the 2' isomer binds more efficiently and is better able to promote the hydrolysis of GTP than the 3' isomer (47-51). These inferences, coupled with the fact that only the 3' isomer is active as a peptide acceptor in the A site (see below) lead to the hypothesis of a transacylation event in the A site following EF-Tu binding and delivery of the the 2' isomer. However, when wild-type Phe-tRNAPhe was bound to EF-Tu and frozen in its isomeric state by treatment with phenol and acetic anhydride, the 3' isomer was predominantly seen, in contradiction with the earlier conclusions (52). An NMR study on 13C labeled tRNAs bound to EF-Tu in solution suggested that EF-Tu stabilizes an orthoester intermediate between the two normally stable forms (53). X-ray crystal structures of EF-Tu bound to aminoacyl tRNAs showed binding in the 3' configuration (54-56). The structures showed numerous interactions stabilizing the 3' ester, including tetrahedral coordination of the α-amino group, hydrogen bonding between an EF-Tu arginine and the carbonyl group of the ester, and acceptance of a hydrogen bond from the 2'-OH by an EF-Tu glutamate. This hydrogen bond would be interrupted by a 2' ester and therefore energetically favors the 3' isomer. The structural data seem to indicate that EFTu is designed to force aa-tRNAs into the 3' configuration in preparation for delivery to the A site, where the 3' ester is required (see below). These data also discount the stabilization of an orthoester, and imply that while both isomeric forms may bind to EFTu, the 3' ester is the lowest energy form. A recent revisitation of the binding experiments attempted a more quantitative analysis of the relative binding of 2'dA and 3'dA substrates. This study found that neither modification has a substantial effect on overall binding of the aa-tRNA to EF-Tu, but postulates that the hydrogen bonding interaction between the 2'-OH and EF-Tu stimulates the GTPase activity that is necessary for delivery of the aa-tRNA to the ribosome (57).
Acceptor activity in the A Site
Soon after the discovery in 1957 (58, 59) of tRNA as Francis Crick's postulated “adaptor molecule”(3), the field became focused on how tRNAs functioned as “adaptors” in protein synthesis. Much early biochemical work used ribosomal assays to examine tRNAs and tRNA fragments with ribose modifications of A76. A consensus emerged from these studies that the A-site substrate must have a 3'-linkage to the amino acid. The initial evidence was the observation that puromycin, in which a methyl-tyrosine is stably linked to the 3' carbon of dimethyl adenosine via an amide bond, functions as an A-site substrate, while the 2' linked version does not (60). A number of other biochemical studies demonstrated that for a variety of ester-linked substrates, both 2' and 3' linkages allow A-site binding, but only 3' linkages allow acceptor activity (51, 61-63). The crystal structure of the Haloarcula marismortui 50S ribosomal subunit in complex with the “Yarus inhibitor”, an analog of the peptidyl transfer reaction's tetrahedral intermediate with CCdA in the P site and puromycin in the A site, suggests a hydrogen bond between the A-site 2'-OH and a conserved active site uracil (64). This interaction is maintained in the more recent structure of individual substrates bound simultaneously(65) and probably contributes to substrate binding and positioning, and perhaps assists in maintaining the ester at the 3'-position.
The absolute requirement for a 3' ester linkage between the tRNA and the amino acid for acceptor activity has implications about the function of EF-Tu and the transition from EF-Tu bound tRNA to the active form in the A site. If the transition from the synthetases to the A site (via EF-Tu) is more rapid than the rate of transacylation in solution, then enzymatic transacylase activity would be required for the half of the tRNAs that are acylated at the 2' position. If aa-tRNAs exist in equilibrium prior to A-site binding, then the fraction that is in the 2' configuration would need to assume the 3' configuration in order to act as acceptors. In either case, some tRNAs will be in the incorrect configuration prior to EF-Tu binding. Based on NMR and EF-Tu structural data, the aatRNAs are forced into the 3' configuration when bound. The active site of the ribosome lacks space for extra waters. It is conceivable that once the end of an aa-tRNA enters the active site, the ribosome does not provide an environment conducive to transacylation. If so, to achieve the protein synthesis rates seen in vivo, it would be imperative that the incoming tRNAs are properly configured for activity prior to entering, and thus transacylation by EF-Tu is critical, regardless of transacylation rates in solution.
Translocation and donor activity in the P Site
The most fundamental question regarding peptidyl transferase is: “how does the ribosome catalyze the reaction?” This question illuminates the central purpose of the ribosome and has implications for the architecture and complexity of the full, modern translational machinery, as well as the ancient roots and evolutionary development of this critical reaction. Possibly the best opportunity for the vicinal hydroxyl to get involved is in the P site. The ester linkage to the tRNA is the object of nucleophilic attack during formation of the new peptide bond. During this step, the non-acetylated hydroxyl is directly adjacent to the center of chemistry, and is ideally situated to promote the reaction. 40 years ago participation by this hydroxyl group in peptidyl transfer was proposed based on model aminolysis reactions and theoretical grounds (66). Recent biochemical and structural results strongly support this conclusion.
The X-ray crystal structure of the Haloarcula marsimortui 50S ribosomal subunit afforded the first opportunity to develop mechanistic models for ribosome function based on the relative locations of functional groups within the active site (64, 67). This initially lead to a model where A2451 of the ribosomal RNA catalyzes the peptide bond formation by acting as a general base to extract a proton from the attacking amino group and then stabilizing the oxyanion of the tetrahedral intermediate. However, studies of mutant ribosomes purified from mixed populations have demonstrated that none of the rRNA residues in the active site, even those that are universally conserved including A2451, eliminates ribosome activity when mutated (68-71). This leaves few functional groups proximal to the reaction center that remain candidates for essential catalytic roles. The A76 2'-OH is one such group.
Two early studies regarding the role of the A76 ribose hydroxyls resulted in conflicting conclusions regarding the requirement for these groups in the P site tRNA. The first study tested tRNAPhe with a 2'-dA or 3'-dA substitution at A76. While the 2'-dA substituted tRNA was active as an A-site substrate (as mentioned above), neither modified tRNA was active as a donor, suggesting that both hydroxyls are required in the P site. The second study found that dA76 substituted tRNALys cannot support multiple turnover poly(A)-mRNA-dependent synthesis of poly(Lys), but is active as a donor when nonenzymatically loaded directly into the P-site and puromycin is used as the A-site substrate (51). In a subsequent report, the same research group found that during translation of a poly-A mRNA, 2'dA76 tRNALys cannot act as a P-site substrate following acceptance of a peptide in the A site (72). They also found that N-acetylated Lys-tRNALys, which has no free α-amino group for A-site activity, can act as the initial P-site substrate for multiple turnover polymerization, but N-acetylated Lys-2'dA76 tRNALys is inactive in this assay. These data, in combination with their previous observation that a 2'-dA tRNA is active in the P site, lead them to the conclusion that the 2'-dA substitution prevents proper translocation from the A site to the P site following acceptor activity, as well as tRNA release from the P site following donor activity. These studies utilized poorly defined minimal assays, and the conclusions were mainly based on reaction endpoints rather than kinetics. Two other studies, one 25 years ago and one more recently, examined small tRNA fragments with modified A76 ribose moieties as P-site substrates. In each of these studies neither dA substrate acted as a donor, suggesting that both hydroxyl groups were essential for the activity of a single residue P-site substrate (73, 74).
In a recent report from our group, 2'-dA and 2'-deoxy-2'-fluoroadenosine (2'-FA) substituted tRNALys were tested using a translation system, which proceeds at a physiologically relevant rate with rate limiting chemistry. Both 2' modified tRNAs were active as A-site substrates, but were completely inactive as P-site substrates, resulting in at least a 106-fold rate reduction. A toe-printing assay demonstrated that both substrates were translocated from the A site to the P site in an EF-G dependant manner (75). Based on this report and the collective evidence from the other related studies, it is now clear that neither A76 hydroxyl group is dispensable for donor activity. The question remains: what is the role of the vicinal hydroxyl group in the peptidyl transfer reaction? How does the ribosome catalyze peptide bond formation?
It has long been assumed that the 3'-linked regioisomer is the active peptidyl-tRNA form in the P site. This assumption followed from the deduction that the 2'-dA substrate was active in the P site, and the fact that the A-site substrate must be 3' acylated. Since neither deoxy substrate is active the biochemical data cannot rule out a transacylation event during the reaction. However, the crystal structure of the ribosome bound to CCA-pcb, a P-site substrate in which the peptidyl moiety should be free to isomerize between the 2' and 3' positions, shows the 3' isomer bound in the P site (76). Furthermore, a tetrahedral intermediate analog in which the phosphate is connected to the 3' carbon binds in the active site, while the 2'-linked version does not(77). These data support the assumption that the P-site substrate is acylated at the 3'-position and argue that the peptide is not transacylated to the 2'-position in the course of the reaction. Thus, once the 3'-aminoacylated tRNA is presented to the A-site, it is not required to undergo isomerization at any subsequent step of translation.
If the 2'-OH does not participate in a direct linkage to the nascent peptide, why does substitution of this functional group eliminate peptidyl transferase activity? In our report demonstrating the importance of the 2'-OH we suggested that the 106 rate effect of removing the 2'-OH implies a substrate-assisted catalytic role (75), but the exact nature of this contribution has not yet been defined. One possibility is that the 2'-OH does not play an active role in the reaction, but is essential for the active site to assume an active conformation or for proper positioning of the substrates for the reaction because of important hydrogen bonding and steric interactions. The 2'-OH might be involved in proton transfer, acting either as a general base to extract a proton from the amine or as a general acid to provide a proton to the 3'-OH leaving group. Additionally, the 2'-OH could participate in a critical interaction with a catalytic metal ion. It is likely that it is not any single one of these possibilities, but a combination of several that gives the 2'-OH its importance.
X-ray crystal structures of the ribosome in complex with different substrates and intermediate mimics have provided clues about the peptidyl transferase mechanism and the involvement of the P-site vicinal hydroxyl. A recent study presented crystal structures of A-site and P-site substrates bound simultaneously (figure 4A), as well as an analog of the tetrahedral intermediate that includes the A76 2'-OH (figure 4C), providing the most complete picture of the active site to date (65, 78). According to these structures, the 2'-OH hydrogen bonds with the α-amino group of the A-site substrate and is furthermore the only atom within hydrogen bonding distance of the α-amino group in the substrate structure. The 2'-OH is most certainly involved in aligning the nucleophile prior to reaction, however it is also noted that the 2'-OH is the only functional group positioned to act as a general base in the intermediate structure. These results agree closely with models of the active site containing both substrates or the tetrahedral intermediate that were developed earlier by in silico combination of ribosome structures with A-site and P-site substrates bound separately (76). The structural studies support a model of the intermediate/transition state that has S-chirality, and a 2'-OH situated immediately between the α-amino nucleophile and the O3' leaving group, poised to interact with both.
4. X-ray crystal structure of Saccharomyces cerevisiae Phe-tRNAPhe bound to Thermus aquaticus EF-Tu [PDB entry 1TTT(55)].
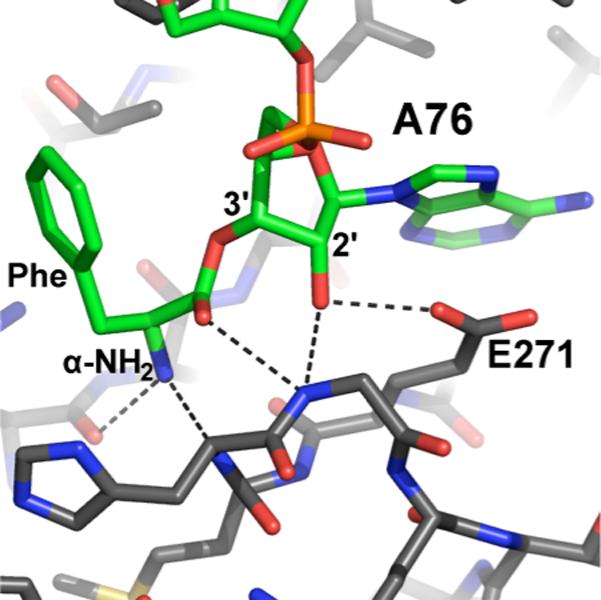
Numerous hydrogen bonds between EF-Tu and the aminoacyl tRNA stabilize the 3' configuration.
Crystal structures of the active site show that the 2'-OH is positioned for a role in proton transfer, but cannot determine if it actually participates in such a mechanism. They can, however, indicate whether the hydroxyl's local environment has other features to promote activity as a general base or general acid, by shifting its pKa downward and stabilizing the required negatively charged state. In the intermediate analog structures, a solvent molecule sitting adjacent to the A76 ribose coordinates both the O2' and the O3' as well as several ribosomal functional groups. Attempts to associate this electron density with a metal ion were unsuccessful, so it is most likely a specifically bound water molecule (78). Coordination by a water molecule is unlikely to provide enough stabilization for the 2'-OH to assume a full negative charge at physiological pH, so the structural data argue against a role as a traditional general acid or general base. However, kinetic isotope effect and Bronsted coefficient measurements on other model reactions occurring in organic solvent suggest that in the absence of an aqueous environment the 2'-OH behaves as a general base (79).
Comparison of the temperature dependence of the ribosomal reaction and uncatalyzed model reactions indicated that the ribosome drives peptide bond formation solely by increasing the change in entropy (ΔS) of activation (80). This led to the conclusion that ribosomal catalysis is based only on the juxtaposition of substrates and exclusion of water from the active site, rather than any direct chemical mechanism. Molecular dynamic and free energy perturbation simulations of the ribosomal and aqueous reactions similarly suggest that an increase in ΔS is the primary thermodynamic motivation for catalysis (81). This computational study attributes the entropy change mainly to ordered water molecules surrounding the transition state in solution that are eliminated in the pre-ordered active site of the ribosome, rather than positioning of the substrates.
In the crystal structures of the pre-reaction complex and the intermediate, the α-amino group is completely solvent inaccessible (78), yet it must release a proton for the reaction to proceed. The 2'-OH is the only nearby functional group capable of accepting a proton. Similarly, except for the bound water molecule, the O3' leaving group is only minimally exposed, but must receive a proton during the reaction. The computational and crystallographic reports both invoke a model where the 2'-OH acts as a “proton shuttle” by simultaneously accepting a proton from the nucleophile while donating a proton to the leaving group(78, 81). This mechanism differs from traditional acid/base catalysis in that it does not require the 2'-OH to assume a net positive or negative charge, alleviating the need for a significantly shifted pKa. Furthermore, since these proton transfer functions can be assumed by readily available water molecules in the solution reaction, the proton shuttling may not have a significant contribution to the change in the enthalpic change (ΔΔH) of activation. However, since the proton transfers are required for the reaction, the 2'-OH becomes essential in the context of the ribosomal active site, and the absence of the 2'-OH results in a large ΔΔH penalty.
Translocation
Following the peptidyl transfer reaction the deacylated P-site tRNA must exit the ribosome in preparation for the next round of peptide bond formation. The 3' end of the deacylated tRNA spontaneously translocates to the E site following peptide bond formation. This is required for EF-G catalyzed translocation of the full tRNAs (82, 83). To allow movement from one site to the next, the free energy of binding must be progressively lower for each binding site (A, P, and E), and each site must have a specific affinity for the substrates that bind there. Therefore, the ribosomal tRNA binding sites must necessarily recognize the 3' ends of the tRNAs, the site at which they differ (82). Studies of E-site binding and translocation kinetics have shown that removal of either ribose hydroxyl group from A76 results in 10−40 fold reductions in both E-site binding and translocation rates (84-86). The magnitudes of these effects are consistent with a direct interaction, such as hydrogen bonds, between the E site and the A76 ribose hydroxyls. However, the 2'-OH is not absolutely essential for translocation and tRNA release, as was concluded earlier(72). Lill et al. also found that aminoacylation of the tRNA, or inclusion of any other additional bulk at the 3' end reduces the affinity of the tRNA for the E site by at least 100-fold. The 2'- and 3'-OHs themselves apparently play only a minor role in promoting translocation, but must be deacylated to allow E-site binding. A structural study of tRNA mimics bound to the E site supports these conclusions. The 3'-OH of A76 hydrogen bonds with the 2'-OH of C75, stabilizing the unusual extended conformation of A76, C75, and C74 that allows significant stacking interactions between these bases and ribosomal bases of the E site. The 2'-OH of A76 hydrogen bonds to the N3 of universally conserved ribosomal base C2394 in the E site, a biochemically predicted interaction (87). Any additional atoms on the 2'-OH would interrupt this hydrogen bond and result in steric clash with C2394. Acylation of the 3'-OH would interrupt this interaction and inhibit the E-site bound conformation.
Conclusions
The 3' end of tRNAs are at the site of chemistry at every step of translation, and no part of the tRNA is more closely associated with the reaction centers than the 3' terminal ribose. In addition to being the site of covalent attachment of the amino acids and peptides, the A76 cis-diol plays numerous roles, including an essential catalytic function in the ribosomal P site.
For the most part, tRNAs are considered to be simply the substrates for the translational reactions, carrier molecules that are subjected to the catalytic activity of the real translational machinery: tRNA synthetases, initiation and elongation factors, and of course the ribosome. Less thought has been put into considering the tRNAs themselves as active parts of the mechanism, essential components whose involvement goes back to the most ancient peptide producing assemblies. In a provocative essay, Woese condemns naïve acceptance of tRNAs as passive adaptors and calls for a reassessment of their roles, both in the modern mechanism as well as the evolution of translation(88). Such a reevaluation seems to be underway, and important, proactive roles for tRNAs have been suggested in decoding, storage of mechanical energy, and movement through the ribosome (75, 89, 90). The cis-diol of the 3' terminal ribose is important, even essential, at a number of steps in the translation pathway. This direct involvement should shape the way we fit tRNAs into our view of translation.
Another important consideration is the development of the ribosomal active site. RNA, while capable of catalyzing biological reactions, has a much more limited chemical repertoire than proteins, which in many cases can achieve more robust catalysis than their RNA counterparts. For this reason, most modern biological catalysis is done in protein active sites. The ribosome, with its all-RNA active site, is a notable exception. Why? The peptidyl transferase center is constrained to the available RNA repertoire, which includes phosphates, functional groups on the bases, 2'-OHs and the rare 3' terminal cis-diol. The A76 2'-OH is near the reaction center and has a specific geometrical orientation relative to the labile ester. This configuration has likely been consistent since the early days of peptidyl transfer evolution. If an RNA based peptidyl transfer center was not capable of providing adequately efficient catalysis, then evolution would have replaced the ribosome with a protein-based enzyme that could. This has not yet occurred, and the active site remains exclusively RNA, including the necessary 2'-OH.
5. The P-site A76 2'-OH is positioned for a critical role in proton transfer.
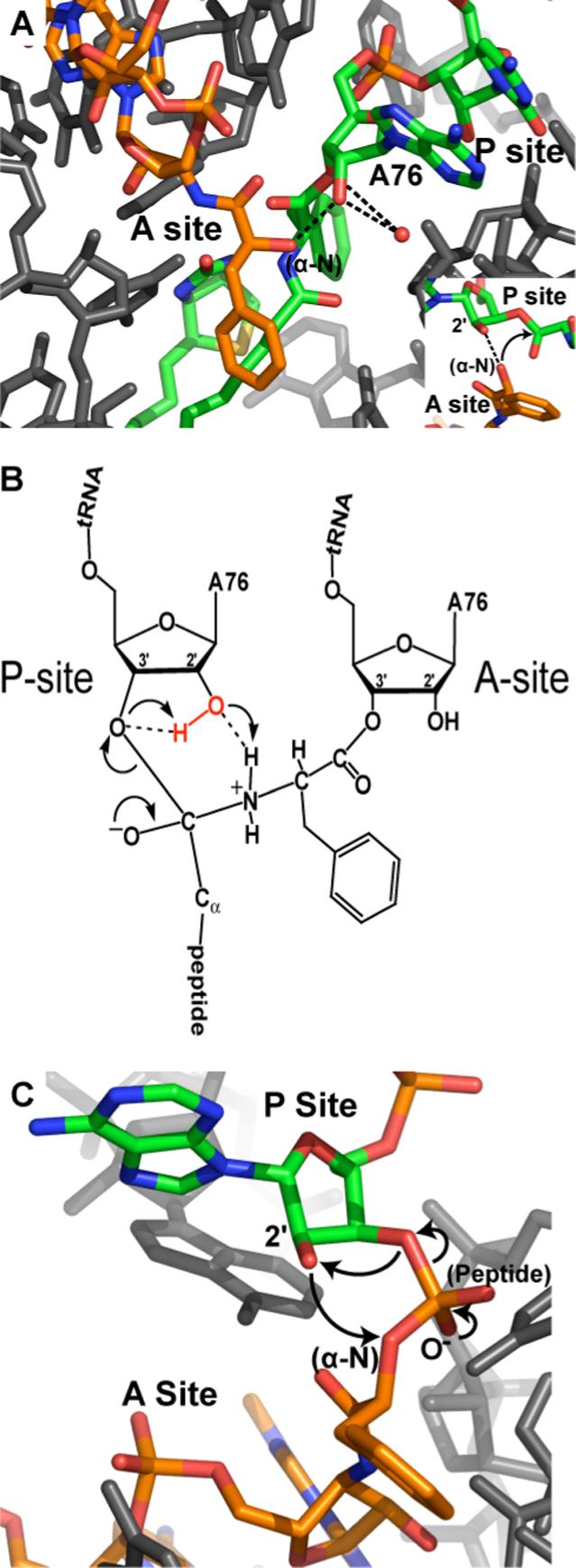
(A) X-ray crystal structure of ribosomal substrates bound in the active site [PDB entry 1VQN (78)]. The α-NH3 group of the A-site substrate has been replaced with a hydroxyl group to inhibit the reaction. The A76 2'-OH hydrogen bonds with the α-NH3, and the 2'- and 3'-OHs coordinate a solvent molecule on their far side. (B) Schematic of the peptidyl transfer intermediate and the proposed mechanism for resolution into products. The 2'-OH acts as a proton shuttle by simultaneously donating a proton to the 3'-OH and accepting a proton from the α-NH3. (C) X-ray crystal structure of the transition state mimic CCA-phosphate-puromycinPhe-CC [PDB entry 1VQP (78)]. The non-bridging phosphate oxygens mimic the peptide and the oxyanion. The proposed proton transfers are indicated with arrows.
Acknowledgements
We thank Dieter Söll and Jesse Cochrane for helpful discussions. This review was prepared with support from NIH grant GM54839.
References
- 1.Hill WE, Dahlberg A, Garrett RA, Moore PB, Schlessinger D, Warner JR. The Ribosome: Structure, Function and Evolution. American Society for Microbiology; Washington, D.C.: 1990. [Google Scholar]
- 2.Nierhaus KH, Franceschi F, Subramanian AR, Erdmann VA, Wittmann-Liebold B. The Translational Apparatus: Structure, Function, Regulation, Evolution. Plenum Press; New York: 1994. [Google Scholar]
- 3.Crick FH. On protein synthesis. Symp. Soc. Exp. Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- 4.Lewin B. Genes VI. Oxford University Press; New York: 1997. pp. 151–241. [Google Scholar]
- 5.Westhof E, Fritsch V. RNA folding: beyond Watson-Crick pairs. Structure Fold. Des. 2000;8:R55–65. doi: 10.1016/s0969-2126(00)00112-x. [DOI] [PubMed] [Google Scholar]
- 6.Sinha N, Smith-Gill SJ. Electrostatics in protein binding and function. Curr. Protein Pept. Sci. 2002;3:601–614. doi: 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- 7.Creighton TE. Stability of folded conformations. Curr. Opin. Struct. Biol. 1991;1:5–16. [Google Scholar]
- 8.Saenger W. Structure and dynamics of water surrounding biomolecules. Annu. Rev. Biophys. Biophys. Chem. 1987;16:93–114. doi: 10.1146/annurev.bb.16.060187.000521. [DOI] [PubMed] [Google Scholar]
- 9.DeRose VJ. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol. 2003;13:317–324. doi: 10.1016/s0959-440x(03)00077-0. [DOI] [PubMed] [Google Scholar]
- 10.Pyle AM. Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem. 2002;7:679–690. doi: 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- 11.Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 12.Schowen KB, Limbach HH, Denisov GS, Schowen RL. Hydrogen bonds and proton transfer in general-catalytic transition-state stabilization in enzyme catalysis. Biochim. Biophys. Acta. 2000;1458:43–62. doi: 10.1016/s0005-2728(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 13.Izatt RM, Rytting JH, Hansen LD, Christensen JJ. Thermodynamics of proton dissociation in dilute aqueous solution. V. An entropy titration study of adenosine, pentoses, hexoses, and related compounds. J. Am. Chem. Soc. 1966;88:2641–2645. doi: 10.1021/ja00964a003. [DOI] [PubMed] [Google Scholar]
- 14.Velikyan I, Acharya S, Trifonova A, Foldesi A, Chattopadhyaya J. The pKa's of 2'-hydroxyl group in nucleosides and nucleotides. J. Am. Chem. Soc. 2001;123:2893–2894. doi: 10.1021/ja0036312. [DOI] [PubMed] [Google Scholar]
- 15.Bruice TC, Fife TH. Hydroxyl Group Catalysis .3. Nature of Neighboring Hydroxyl Group Assistance in Alkaline Hydrolysis of Ester Bond. J. Am. Chem. Soc. 1962;84:1973–1979. [Google Scholar]
- 16.Oivanen M, Kuusela S, Lonnberg H. Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by Bronsted acids and bases. Chem. Rev. 1998;98:961–990. doi: 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- 17.Reese CB, Trentham DR. Acyl Migration in Ribonucleoside Derivatives. Tetrahedron Lett. 1965:2467–2472. doi: 10.1016/s0040-4039(01)84008-9. [DOI] [PubMed] [Google Scholar]
- 18.Griffin BE, Jarman M, Reese CB, Sulston JE, Trentham DR. Some Observations Relating to Acyl Mobility in Aminoacyl Soluble Ribonucleic Acids. Biochemistry. 1966;5:3638–3649. [Google Scholar]
- 19.Taiji M, Yokoyama S, Miyazawa T. Transacylation Rates of (Aminoacyl)Adenosine Moiety at the 3'-Terminus of Aminoacyl Transfer Ribonucleic-Acid. Biochemistry. 1983;22:3220–3225. doi: 10.1021/bi00282a028. [DOI] [PubMed] [Google Scholar]
- 20.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 21.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of Transfer-RNA Synthetases into 2 Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 22.Hecht SM. Utilization of Isomeric Aminoacyl Transfer-Ribonucleic Acids in Peptide-Bond Formation. Accounts. Chem. Res. 1977;10:239–245. [Google Scholar]
- 23.Sprinzl M, Cramer F. Accepting Site for Aminoacylation of Transfer-RNAPhe from Yeast. Nature-New Biol. 1973;245:3–5. doi: 10.1038/newbio245003a0. [DOI] [PubMed] [Google Scholar]
- 24.Sprinzl M, Cramer F. Site of Aminoacylation of Transfer-RNAs from Escherichia-Coli with Respect to 2'-Hydroxyl Group or 3'-Hydroxyl Group of Terminal Adenosine. Proc. Natl. Acad. Sci. U.S.A. 1975;72:3049–3053. doi: 10.1073/pnas.72.8.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woese CR, Olsen GJ, Ibba M, Soll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eiler S, Dock-Bregeon A, Moulinier L, Thierry JC, Moras D. Synthesis of aspartyl-tRNAAsp in Escherichia coli--a snapshot of the second step. Embo J. 1999;18:6532–6541. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vortler CS, Fedorova O, Persson T, Kutzke U, Eckstein F. Determination of 2'-hydroxyl and phosphate groups important for aminoacylation of Escherichia coli tRNAAsp: a nucleotide analogue interference study. RNA. 1998;4:1444–1454. doi: 10.1017/s1355838298980967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavarelli J, Eriani G, Rees B, Ruff M, Boeglin M, Mitschler A, Martin F, Gangloff J, Thierry JC, Moras D. The active site of yeast aspartyl-tRNA synthetase: structural and functional aspects of the aminoacylation reaction. Embo. J. 1994;13:327–337. doi: 10.1002/j.1460-2075.1994.tb06265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pleiss JA, Wolfson AD, Uhlenbeck OC. Mapping contacts between Escherichia coli alanyl tRNA synthetase and 2' hydroxyls using a complete tRNA molecule. Biochemistry. 2000;39:8250–8258. doi: 10.1021/bi0001022. [DOI] [PubMed] [Google Scholar]
- 30.Englisch-Peters S, Conley J, Plumbridge J, Leptak C, Soll D, Rogers MJ. Mutant enzymes and tRNAs as probes of the glutaminyl-tRNA synthetase: tRNAGln interaction. Biochimie. 1991;73:1501–1508. doi: 10.1016/0300-9084(91)90184-3. [DOI] [PubMed] [Google Scholar]
- 31.Gruic-Sovulj I, Uter N, Bullock T, Perona JJ. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J. Biol. Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. [DOI] [PubMed] [Google Scholar]
- 32.Rath VL, Silvian LF, Beijer B, Sproat BS, Steitz TA. How glutaminyl-tRNA synthetase selects glutamine. Structure. 1998;6:439–449. doi: 10.1016/s0969-2126(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 33.Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. doi: 10.1021/bi00085a006. [DOI] [PubMed] [Google Scholar]
- 34.Freist W. Mechanisms of aminoacyl-tRNA synthetases: a critical consideration of recent results. Biochemistry. 1989;28:6787–6795. doi: 10.1021/bi00443a001. [DOI] [PubMed] [Google Scholar]
- 35.Freist W, Cramer F. Isoleucyl-tRNA synthetase from Baker's yeast. Catalytic mechanism, 2',3'-specificity and fidelity in aminoacylation of tRNAIle with isoleucine and valine investigated with initial-rate kinetics using analogs of tRNA, ATP and amino acids. Eur. J. Biochem. 1983;131:65–80. doi: 10.1111/j.1432-1033.1983.tb07232.x. [DOI] [PubMed] [Google Scholar]
- 36.Gabius HJ, von der Haar F, Cramer F. Evolutionary aspects of accuracy of phenylalanyl-tRNA synthetase. A comparative study with enzymes from Escherichia coli, Saccharomyces cerevisiae, Neurospora crassa, and turkey liver using phenylalanine analogues. Biochemistry. 1983;22:2331–2339. doi: 10.1021/bi00279a005. [DOI] [PubMed] [Google Scholar]
- 37.Baldwin AN, Berg P. Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J. Biol. Chem. 1966;241:839–845. [PubMed] [Google Scholar]
- 38.Eldred EW, Schimmel PR. Rapid deacylation by isoleucyl transfer ribonucleic acid synthetase of isoleucine-specific transfer ribonucleic acid aminoacylated with valine. J. Biol. Chem. 1972;247:2961–2964. [PubMed] [Google Scholar]
- 39.von der Haar F, Cramer F. Isoleucyl-tRNA synthetase from baker's yeast: the 3'-hydroxyl group of the 3'-terminal ribose is essential for preventing misacylation of tRNAIle-C-C-A with misactivated valine. FEBS Lett. 1975;56:215–217. doi: 10.1016/0014-5793(75)81094-5. [DOI] [PubMed] [Google Scholar]
- 40.von der Haar F, Cramer F. Hydrolytic action of aminoacyl-tRNA synthetases from baker's yeast: “chemical proofreading” preventing acylation of tRNAIle with misactivated valine. Biochemistry. 1976;15:4131–4138. doi: 10.1021/bi00663a034. [DOI] [PubMed] [Google Scholar]
- 41.Cramer F, Faulhammer H, von der Haar F, Sprinzl M, Sternbach H. Aminoacyl-tRNA synthetases from baker's yeast: reacting site of enzymatic aminoacylation is not uniform for all tRNAs. FEBS Lett. 1975;56:212–214. doi: 10.1016/0014-5793(75)81093-3. [DOI] [PubMed] [Google Scholar]
- 42.Lin L, Hale SP, Schimmel P. Aminoacylation error correction. Nature. 1996;384:33–34. doi: 10.1038/384033b0. [DOI] [PubMed] [Google Scholar]
- 43.Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakama T, Fukai S, Konno M, Hendrickson TL, Schimmel P, Yokoyama S. Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science. 1998;280:578–582. doi: 10.1126/science.280.5363.578. [DOI] [PubMed] [Google Scholar]
- 44.Nordin BE, Schimmel P. Plasticity of recognition of the 3'-end of mischarged tRNA by class I aminoacyl-tRNA synthetases. J. Biol. Chem. 2002;277:20510–20517. doi: 10.1074/jbc.M202023200. [DOI] [PubMed] [Google Scholar]
- 45.Lincecum TL, Jr., Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grotli M, Martinis SA, Cusack S. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol. Cell. 2003;11:951–963. doi: 10.1016/s1097-2765(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 46.Mursinna RS, Lee KW, Briggs JM, Martinis SA. Molecular dissection of a critical specificity determinant within the amino acid editing domain of leucyl-tRNA synthetase. Biochemistry. 2004;43:155–165. doi: 10.1021/bi034919h. [DOI] [PubMed] [Google Scholar]
- 47.Sprinzl M, Kucharzewski M, Hobbs JB, Cramer F. Specificity of elongation factor Tu from Escherichia coli with respect to attachment to the amino acid to the 2' or 3'-hydroxyl group of the terminal adenosine of tRNA. Eur. J. Biochem. 1977;78:55–61. doi: 10.1111/j.1432-1033.1977.tb11713.x. [DOI] [PubMed] [Google Scholar]
- 48.Ringer D, Chladek S. Interaction of elongation factor Tu with 2'(3')-O-aminoacyloligonucleotides derived from the 3' terminus of aminoacyl-tRNA. Proc. Natl. Acad. Sci. U.S.A. 1975;72:2950–2954. doi: 10.1073/pnas.72.8.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ringer D, Chladek S. Enzymatic binding of aminoacyl transfer ribonucleic acid to ribosomes: the study of binding sites of 2' and 3' isomers of aminoacyl transfer ribonucleic acid. Biochemistry. 1976;15:2759–2765. doi: 10.1021/bi00658a008. [DOI] [PubMed] [Google Scholar]
- 50.Chinali G, Sprinzl M, Parmeggiani A, Cramer F. Participation in protein biosynthesis of transfer ribonucleic acids bearing altered 3'-terminal ribosyl residues. Biochemistry. 1974;13:3001–3010. doi: 10.1021/bi00712a001. [DOI] [PubMed] [Google Scholar]
- 51.Wagner T, Cramer F, Sprinzl M. Activity of the 2' and 3' isomers of aminoacyl transfer ribonucleic acid in the in vitro peptide elongation on Escherichia coli ribosomes. Biochemistry. 1982;21:1521–1529. doi: 10.1021/bi00536a009. [DOI] [PubMed] [Google Scholar]
- 52.Taiji M, Yokoyama S, Miyazawa T. Slow transacylation of peptidyladenosine allows analysis of the 2'/3'-isomer specificity of peptidyltransferase. Biochemistry. 1985;24:5776–5780. doi: 10.1021/bi00342a013. [DOI] [PubMed] [Google Scholar]
- 53.Forster C, Limmer S, Zeidler W, Sprinzl M. Effector region of the translation elongation factor EF-Tu.GTP complex stabilizes an orthoester acid intermediate structure of aminoacyl-tRNA in a ternary complex. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4254–4257. doi: 10.1073/pnas.91.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nissen P, Kjeldgaard M, Thirup S, Clark BF, Nyborg J. The ternary complex of aminoacylated tRNA and EF-Tu-GTP. Recognition of a bond and a fold. Biochimie. 1996;78:921–933. doi: 10.1016/s0300-9084(97)86714-4. [DOI] [PubMed] [Google Scholar]
- 55.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 56.Nissen P, Thirup S, Kjeldgaard M, Nyborg J. The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure Fold. Des. 1999;7:143–156. doi: 10.1016/s0969-2126(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 57.Pleiss JA, Uhlenbeck OC. Identification of thermodynamically relevant interactions between EF-Tu and backbone elements of tRNA. J. Mol. Biol. 2001;308:895–905. doi: 10.1006/jmbi.2001.4612. [DOI] [PubMed] [Google Scholar]
- 58.Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958;231:241–257. [PubMed] [Google Scholar]
- 59.Hoagland MB, Zamecnik PC, Stephenson ML. Intermediate reactions in protein biosynthesis. Biochim. Biophys. Acta. 1957;24:215–216. doi: 10.1016/0006-3002(57)90175-0. [DOI] [PubMed] [Google Scholar]
- 60.Nathans D, Neidle A. Structural requirements for puromycin inhibition of protein synthesis. Nature. 1963;197:1076–1077. doi: 10.1038/1971076a0. [DOI] [PubMed] [Google Scholar]
- 61.Ringer D, Chladek S. Inhibition of the peptidyl transferase A-site function by 2'-O-aminoacyloligonucleotides. Biochem. Biophys. Res. Commun. 1974;56:760–766. doi: 10.1016/0006-291x(74)90670-6. [DOI] [PubMed] [Google Scholar]
- 62.Bhuta A, Quiggle K, Ott T, Ringer D, Chladek S. Aminoacyl Derivatives of Nucleosides, Nucleotides and Polynucleotides .33. Stereochemical Control of Ribosomal Peptidyltransferase Reaction - Role of Amino-Acid Side-Chain Orientation of Acceptor Substrate. Biochemistry. 1981;20:8–15. doi: 10.1021/bi00504a002. [DOI] [PubMed] [Google Scholar]
- 63.Hecht SM, Kozarich JW, Schmidt FJ. Isomeric phenylalanyl-tRNAs. Position of the aminoacyl moiety during protein biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1974;71:4317–4321. doi: 10.1073/pnas.71.11.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 65.Schmeing TM, Huang KS, Strobel SA, Steitz TA. An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature. 2005;438:520–524. doi: 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- 66.Griffin BE, Reese CB. Some Observations on the Mechanism of the Acylation Process in Protein Synthesis. Proc. Natl. Acad. Sci. U.S.A. 1964;51:440–444. doi: 10.1073/pnas.51.3.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 68.Thompson J, Kim DF, O'Connor M, Lieberman KR, Bayfield MA, Gregory ST, Green R, Noller HF, Dahlberg AE. Analysis of mutations at residues A2451 and G2447 of 23S rRNA in the peptidyltransferase active site of the 50S ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9002–9007. doi: 10.1073/pnas.151257098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polacek N, Gaynor M, Yassin A, Mankin AS. Ribosomal peptidyl transferase can withstand mutations at the putative catalytic nucleotide. Nature. 2001;411:498–501. doi: 10.1038/35078113. [DOI] [PubMed] [Google Scholar]
- 70.Youngman EM, Brunelle JL, Kochaniak AB, Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- 71.Beringer M, Adio S, Wintermeyer W, Rodnina M. The G2447A mutation does not affect ionization of a ribosomal group taking part in peptide bond formation. RNA. 2003;9:919–922. doi: 10.1261/rna.5600503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wagner T, Sprinzl M. Inhibition of ribosomal translocation by peptidyl transfer ribonucleic acid analogues. Biochemistry. 1983;22:94–98. doi: 10.1021/bi00270a013. [DOI] [PubMed] [Google Scholar]
- 73.Quiggle K, Kumar G, Ott TW, Ryu EK, Chladek S. Donor site of ribosomal peptidyltransferase: investigation of substrate specificity using 2'(3')-O-(N-acylaminoacyl)dinucleoside phosphates as models of the 3' terminus of N-acylaminoacyl transfer ribonucleic acid. Biochemistry. 1981;20:3480–3485. doi: 10.1021/bi00515a027. [DOI] [PubMed] [Google Scholar]
- 74.Dorner S, Polacek N, Schulmeister U, Panuschka C, Barta A. Molecular aspects of the ribosomal peptidyl transferase. Biochem. Soc. Trans. 2002;30:1131–1136. doi: 10.1042/bst0301131. [DOI] [PubMed] [Google Scholar]
- 75.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat. Struct. Mol. Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 76.Hansen JL, Schmeing TM, Moore PB, Steitz TA. Structural insights into peptide bond formation. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11670–11675. doi: 10.1073/pnas.172404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang KS, Weinger JS, Butler E, Strobel SA. Regiospecificity of the P site substrate in the ribosomal peptidyl transferase reaction. J. Am. Chem. Soc. 2006 doi: 10.1021/ja0554099. (submitted) [DOI] [PubMed] [Google Scholar]
- 78.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2' hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 79.Changalov MM, Ivanova GD, Rangelov MA, Acharya P, Acharya S, Minakawa N, Foldesi A, Stoineva IB, Yomtova VM, Roussev CD, Matsuda A, Chattopadhyaya J, Petkov DD. 2'/3'-O-peptidyl adenosine as a general base catalyst of its own external peptidyl transfer: implications for the ribosome catalytic mechanism. Chembiochem. 2005;6:992–996. doi: 10.1002/cbic.200400349. [DOI] [PubMed] [Google Scholar]
- 80.Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7897–7901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trobro S, Aqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Noller HF, Yusupov MM, Yusupova GZ, Baucom A, Cate JH. Translocation of tRNA during protein synthesis. FEBS Lett. 2002;514:11–16. doi: 10.1016/s0014-5793(02)02327-x. [DOI] [PubMed] [Google Scholar]
- 83.Joseph S. After the ribosome structure: how does translocation work? RNA. 2003;9:160–164. doi: 10.1261/rna.2163103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feinberg JS, Joseph S. Identification of molecular interactions between P-site tRNA and the ribosome essential for translocation. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11120–11125. doi: 10.1073/pnas.211184098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lill R, Lepier A, Schwagele F, Sprinzl M, Vogt H, Wintermeyer W. Specific recognition of the 3'-terminal adenosine of tRNAPhe in the exit site of Escherichia coli ribosomes. J. Mol. Biol. 1988;203:699–705. doi: 10.1016/0022-2836(88)90203-3. [DOI] [PubMed] [Google Scholar]
- 86.Lill R, Robertson JM, Wintermeyer W. Binding of the 3' terminus of tRNA to 23S rRNA in the ribosomal exit site actively promotes translocation. Embo J. 1989;8:3933–3938. doi: 10.1002/j.1460-2075.1989.tb08574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bocchetta M, Xiong L, Shah S, Mankin AS. Interactions between 23S rRNA and tRNA in the ribosomal E site. RNA. 2001;7:54–63. doi: 10.1017/s1355838201001650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woese CR. Translation: in retrospect and prospect. RNA. 2001;7:1055–1067. doi: 10.1017/s1355838201010615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. Science. 2005;308:1178–1180. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frank J, Sengupta J, Gao H, Li W, Valle M, Zavialov A, Ehrenberg M. The role of tRNA as a molecular spring in decoding, accommodation, and peptidyl transfer. FEBS Lett. 2005;579:959–962. doi: 10.1016/j.febslet.2004.10.105. [DOI] [PubMed] [Google Scholar]