

Abstract
An mAb was raised to the C5 phagosomal antigen in Paramecium multimicronucleatum. To determine its function, the cDNA and genomic DNA encoding C5 were cloned. This antigen consisted of 315 amino acid residues with a predicted molecular weight of 36,594, a value similar to that determined by SDS-PAGE. Sequence comparisons uncovered a low but significant homology with a Schizosaccharomyces pombe protein and the C-terminal half of the β-fructofuranosidase protein of Zymomonas mobilis. Lacking an obvious transmembrane domain or a possible signal sequence at the N terminus, C5 was predicted to be a soluble protein, whereas immunofluorescence data showed that it was present on the membranes of vesicles and digestive vacuoles (DVs). In cells that were minimally permeabilized but with intact DVs, C5 was found to be located on the cytosolic surface of the DV membranes. Immunoblotting of proteins from the purified and KCl-washed DVs showed that C5 was tightly bound to the DV membranes. Cryoelectron microscopy also confirmed that C5 was on the cytosolic surface of the discoidal vesicles, acidosomes, and lysosomes, organelles known to fuse with the membranes of the cytopharynx, the DVs of stages I (DV-I) and II (DV-II), respectively. Although C5 was concentrated more on the mature than on the young DV membranes, the striking observation was that the cytopharyngeal membrane that is derived from the discoidal vesicles was almost devoid of C5. Approximately 80% of the C5 was lost from the discoidal vesicle-derived membrane after this membrane fused with the cytopharyngeal membrane. Microinjection of the mAb to C5 greatly inhibited the fusion of the discoidal vesicles with the cytopharyngeal membrane and thus the incorporation of the discoidal vesicle membranes into the DV membranes. Taken together, these results suggest that C5 is a membrane protein that is involved in binding and/or fusion of the discoidal vesicles with the cytopharyngeal membrane that leads to DV formation.
INTRODUCTION
Membrane transport, flow, and recycling during the digestive cycle in Paramecium multimicronucleatum have been studied extensively. A digestive cycle is initiated by the fusion of the discoidal vesicles with the cytopharyngeal membrane (Allen, 1974). As more discoidal vesicles fuse with the cytopharyngeal membrane, the nascent vacuole grows in size. Acidosomes that are responsible for phagosomal acidification in this cell dock at the cytosolic surface of a growing nascent vacuole (Allen and Fok, 1983). When a nascent vacuole becomes sufficiently large, an unknown stimulus will trigger the release of the nascent vacuole from the cytopharynx, and a young digestive vacuole (DV), designated DV-I, is formed. Fusion of the acidosomes with the young DV-I then results in a rapid drop in vacuolar pH from 7 to below 3. Concomitant retrieval of much of the discoidal vesicle membrane leads to the more condensed vacuole of stage II (DV-II) (Allen and Staehelin, 1981; Fok et al., 1982; Allen et al., 1995). The lysosomes are now able, for the first time, to recognize and bind to these acidic and condensed DVs. Lysosomal fusion brings about the digestive phase of the cycle (DV-III) (Fok et al., 1984). After digestion, lysosomal membrane is retrieved from the late DV-III (Allen and Fok, 1984), and these DVs become defecation-competent (DV-IV). Subsequent to defecation, the spent DV membrane is retrieved and transported back in the form of discoidal vesicles to the cytopharynx where the membrane is reused to form new DVs (Allen and Wolf, 1974; Allen and Fok, 1980).
Recently we showed that an H+-ATPase is located on the membrane of the acidosomes, suggesting that these proton pumps may be responsible for the phagosomal acidification process (Ishida et al., 1997). Although phagosomal acidification is a common event in mammalian cells (Metchnikoff, 1893; Geisow et al., 1981) and proton pumps have been implicated in such acidification (Mellman et al., 1986; Sturgill-Koszycki et al. 1994), an organelle similar to the acidosomes of Paramecium has yet to be found in mammalian cells. In mammalian cells, proton pumps on the phagosomal membrane are thought to be derived from the plasma membrane or from endosomal membrane as a result of phagosome–endosome fusions.
To further study the digestive system in Paramecium, we developed mAbs against proteins associated with the phagosomal system as well as with other structures in this ciliate. We obtained mAbs specific against antigens B2 and Q2, proteins associated mostly with the discoidal vesicles and DV-I, and mAbs specific against B3, D6, E9, and L1, proteins associated mostly with the acidosomes and DV-II (Allen et al. 1995). In an effort to obtain a marker for the lysosomal membrane, a highly purified lysosomal fraction was obtained through a combination of differential centrifugation, free-flow electrophoresis, and sucrose gradient centrifugation and was used as an immunogen. Several hybridoma clones reacting with the same ∼33 kDa band were obtained, and the C5 clone was selected for further study. Immunofluorescence study showed that this 33-kDa polypeptide is largely associated with the DV membranes and that there was a general increase in the percentage of DVs containing this antigen, and in the intensity of this label, as the DVs mature (Fok et al. 1996).
This report details our effort to determine the molecular structure and function of the C5 antigen. The cloning results show that C5 is composed of 315 amino acid residues and that it has limited homology to two known proteins. The gene encoding the C5 does not appear to contain either a signal peptide sequence at the N terminus, which is usually cleaved off from an endoplasmic reticulum-targeted precursor protein, or a typical transmembrane domain. Cryoelectron microscopic and immunofluorescence studies showed that C5 was specifically located on the cytosolic surface of the membranes. Immunoblotting proteins of purified and KCl-washed DVs showed that C5 was relatively tightly bound to the membrane. Thus, the lack of a transmembrane domain and the finding that <20% of the C5 antigen initially present on the discoidal vesicles remains on the cytopharyngeal or nascent DV (NDV) membrane indicate that the C5 antigen is not permanently associated with the phagosomal membrane. More importantly, microinjection of the mAb to C5 greatly inhibited the rate of DV formation and the size of the DVs that were formed, as a result of the inhibition of the fusion of discoidal vesicles with the cytopharyngeal membrane.
In summary, these results show that the C5 antigen 1) is a relatively tightly bound cytosolically located membrane protein and 2) is involved in the binding and/or fusion of the discoidal vesicles with the cytopharyngeal membranes that leads to DV formation. Finally, it may also be involved in the binding and/or fusion of the acidosomes with the DV-I and of the lysosomes with the DV-II.
MATERIALS AND METHODS
Cell Culture and Chemicals
Cells of P. multimicronucleatum, syngen 2, cultured in an axenic medium (Fok and Allen, 1979), were harvested at mid-log phase of growth. Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). [α-32P]dCTP (111 TBq/mmol) and [γ-32P]ATP (222 TBq/mmol) were purchased from ICN (Costa Mesa, CA) and New England Nuclear (Boston, MA), respectively. Other reagents for molecular biology were purchased from Wako (Osaka, Japan). Protein G Sepharose was purchased from Pharmacia (Uppsala, Sweden), and supplies for electrophoresis were obtained from Bio-Rad Laboratories (Hercules, CA). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Enzymes for Cloning
Restriction enzymes were obtained from Takara (Kyoto, Japan), Boehringer Mannheim (Penzberg, Germany), or New England Biolabs (Beverly, MA). T4 polynucleotide kinase and RNase H were purchased from Pharmacia (Uppsala, Sweden), and EcoRI methylase, Klenow enzyme, calf intestinal alkaline phosphatase, and a DNA ligation kit were obtained from Takara. MMLV reverse transcriptase, Escherichia coli DNA polymerase I, E. coli DNA ligase, and T4 DNA polymerase were obtained from Bethesda Research Laboratories (Rockville, MD). Taq DNA polymerase was from Perkin Elmer-Cetus (Norwalk, CT). A multiprime DNA labeling kit and a DNA sequencing kit were purchased from Amersham (Buckinghamshire, UK) and Toyobo (Tokyo, Japan), respectively. A λgt11 cloning kit and an in vitro packaging kit were obtained from Stratagene (La Jolla, CA), and proteinase K was from Merck (Darmstadt, Germany).
Purification of C5 for N-Terminal and Internal Sequencing
mAb antibody (clone C33-1-1) against the C5 antigen was obtained using the standard procedures for hybridoma production (Kohler and Milstein, 1975). This mAb was purified from the ascites fluid using Protein G Sepharose according to the manufacturer’s instructions. The IgG bound to the Protein G was covalently coupled to the Protein G Sepharose according to Schneider et al. (1982), which provided a solid matrix for the C5 antigen purification. The lysosomal and microsomal fractions obtained by differential centrifugation were solubilized using 1.0% Triton X-100 and then incubated with mAb-Protein G Sepharose beads. After incubation and extensive washing, the C5 antigen was eluted from the solid matrix and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto nylon filters. N-terminal sequencing was carried out by the Biotechnology Instrumentation Facility at the University of California at Riverside.
For internal sequencing, the C5 antigen was subjected to electrophoresis, and the gel was fixed and stained with Coomassie brilliant blue for internal sequencing. The protein band was digested with trypsin. Sequencing was performed by the Protein Structure Laboratory at the University of California at Davis.
Construction of a cDNA Library and Its Screening
Total RNA was prepared by the acid guanidinium thiocyanate–phenol–chloroform method (Chomczynski and Sacchi, 1987). Poly(A)+ RNA was isolated by adsorption to and elution from oligo(dT) cellulose (Aviv and Leder, 1972). A cDNA library was constructed according to the method of Young and Davis (1983), with slight modifications. The cDNA was ligated into the EcoRI restriction site of λgt11 DNA and packaged in vitro. The recombinant phages were then plated onto E. coli Y1090.
The cDNA library was screened with either the 32P end-labeled oligonucleotides or the 32P internally labeled cDNA fragment that was amplified by PCR. Based on the N-terminal amino acid sequence obtained from the C5 antigen, two oligonucleotide screening probes synthesized by the Biotechnology Facility (University of Hawaii at Manoa) (see Figure 1) were labeled with T4 polynucleotide kinase. The cDNA library adsorbed on the nylon filters was hybridized overnight at 42°C with a mixture of these two oligonucleotide probes (2 × 107 cpm/ml) in a hybridization buffer containing 50 mM HEPES at pH 7.0, 10% dextran sulfate, 15% formamide, 100 μg/ml denatured salmon sperm DNA, 10× Denhardt’s solution, 10× SSC, and 0.1% SDS. The filters were washed two times at room temperature with 6× SSC and then with 4× SSC containing 0.1% SDS. Alternatively, the same library was screened with the cDNA fragment that was amplified by PCR using a mixture of the two internal primers (see Figure 1) and a vector primer. Cycling conditions of PCR were as follows: 30 cycles, each consisting of 94°C for 1 min, 55°C for 2 min, and 72°C for 3 min. The amplified cDNA fragments were directly labeled with 32P using a multiprime labeling kit. The cDNA library immobilized on nylon filters was hybridized at 42°C overnight with the PCR product (0.5 × 107 cpm/ml) in a hybridization buffer containing 50 mM HEPES buffer at pH 7.0, 100 μg/ml denatured salmon sperm DNA, 10× Denhardt’s solution, 4× SSC, and 50% formamide. The filters were washed three times at room temperature, each with 2× SSC containing 0.1% SDS and 0.1× SSC containing 0.1% SDS. The dried filters were autoradiographed on Fuji RX-U film (Fuji Medical Systems, Stamford, CT) for 2–3 d at room temperature. The inserts were subcloned into the KpnI/SacI site of pUC19 or the EcoRI site of pBluescript SK(−).
Figure 1.

Synthetic oligonucleotides for cloning the cDNA for the C5 antigen. Oligonucleotides 1 (arrow) and 2 (arrow with broken line) were synthesized at a region corresponding to the amino acid residues at 3–21. The amino acid sequence was determined directly by protein sequencing. Codon selections (+) were made on the basis of the highest frequency in the codon usage reported for Paramecium proteins (Martindale, 1989; Dupuis, 1992; Yamauchi et al. 1992). The amino acid residue that was mismatched between the determined protein sequence and the deduced protein sequence (Figure 2B) is shown in parentheses. The residue marked XXX, which was not determined by protein sequencing, was predicted to be cysteine, and TGT was assigned as the possible codon.
Southern Blotting
Enzyme digests of the genomic DNA were electrophoresed on 1% agarose gels, transferred onto a nylon filter, and hybridized to the 32P-labeled p36-C EcoRI fragment of the cDNA (see Figure 2, 1 × 108 cpm/μg), as described (Southern, 1975). The filter was washed three times with 2× SSC containing 0.1% SDS and then once with 0.1× SSC containing 0.1% SDS at room temperature. It was autoradiographed on Kodak (Eastman Kodak, Rochester, NY) XAR-5 film for 8–16 h at −80°C.
Figure 2.

Restriction map and amino acid sequence of P. multimicronucleatum C5 antigen deduced from the nucleotide sequence. (A) The open box indicates the location of the coding region. E, EcoRI; P, PvuII. (B) The nucleotide number is marked on the left, and the amino acid number is marked on the right. The termination codon TGA is marked with *. Putative regulatory signals in the 5′ and 3′ nucleotide sequences are underlined. The portions of the N-terminal and internal amino acid sequence that were determined by direct protein sequencing are also underlined. The first in-frame TGA triplet present upstream from the tentative translational start site ATG is in boldface.
Northern Blotting
Total RNA was prepared from frozen or fresh cells by the method described above and was electrophoresed (10 μg/lane) on a 1% agarose gel containing 2.6 M formaldehyde. The separated RNA was transferred onto a nylon filter. Hybridization and washing were performed as described above (Sambrook et al., 1989). Probes used for hybridization were the p36-C EcoRI fragment of the cDNA as described above, the 0.4-kbp P. caudatum hemoglobin cDNA (Yamauchi et al. 1992), and the 0.5-kbp P. caudatum genomic DNA encoding the glyceraldehyde-3-phosphate dehydrogenase. The last probe was produced by PCR with two specific primers (O’Hara and Mikami, unpublished observations).
Cloning the Genomic Fragment that Contained the 5′ Region of the Gene Encoding the C5 Antigen
The 5′ region of the gene encoding C5 antigen was isolated by colony hybridization using the p36-C EcoRI fragment of the cDNA as a probe (Sambrook et al. 1989). Genomic DNA digested with EcoRI was electrophoresed on a 1% agarose gel and probed with p36-C. Fragments of reasonable sizes were recovered from the gel and ligated into the EcoRI site of pBluescript SK(−).
DNA Sequencing
The inserted DNA in the plasmid vectors was treated stepwise with exonucleases III (Takara) and VII (New England Biolabs). Immediately after alkali denaturation (Hattori and Sakaki, 1986), sequencing of the purified DNA was performed on a double-stranded plasmid by the dideoxynucleotide chain termination method (Chen and Seeburg, 1985). The reaction mixture was loaded onto a 6% polyacrylamide gel containing 7 M urea.
Computer Analyses
Sequence similarity and prediction of subcellular location of the C5 antigen were carried out using the programs FASTA (Pearson and Lipman, 1988) from the National Institute of Genetics (Mishima, Japan) and PSORT (Nakai and Kanehisa, 1992) from the National Institute for Basic Biology (Okazaki, Japan).
Ultracryoelectron Microscopy
For immunoelectron microscopy, cells previously fed latex beads of various sizes to time the DVs (Fok et al., 1986) were fixed in 0.25% glutaraldehyde in 50 mM phosphate buffer (pH 7.6) at room temperature for 30 min. These cells were pelleted and embedded in 4% gelatin and processed for cryosectioning and immunogold staining as described previously (Allen et al. 1990). To reduce the nonspecific staining, all sections were preincubated with 5% normal goat serum and/or bovine serum albumin before incubation with antibodies.
Determination of the Location of C5 on the Phagosomal Membranes
The plasma membrane of live Paramecium cells was permeabilized for 3 min using 0.01% Triton X-100. Half of the cells were incubated for 30 min at 4°C with the mAb against C5. As a control to insure that the DVs in the cells were intact, the other half of the cells were incubated with the mAb against L1, an antigen known to be on the luminal side of the acidosome and DV-II membranes (Allen et al. 1995). After washing, both groups of cells were fixed in 3% formaldehyde buffered in 50 mM phosphate (pH 7.6). These cells were then incubated with a secondary antibody and examined in a Zeiss light microscope (Thornwood, NY) equipped with epifluorescence illumination.
Microinjection Study
mAb 132F4G4 against the B2 antigen was chosen as the irrelevant mAb for this microinjection study. This antigen has been shown to be located on the exoplasmic surface of the cytopharyngeal membrane as well as on the luminal surface of the DV-I membrane (Allen et al., 1995) and was expected to have a minimal effect on the fusion of the discoidal vesicles with the cytopharyngeal membranes. The mAbs to B2 and C5 were both purified from their ascites fluid using Protein G Sepharose according to the manufacturer’s instructions. Each mAb was suspended in an injection buffer containing 20 mM phosphate and 60 mM NaCl at pH 7.4. Microinjection was performed as described by Ishida et al. (1993). Forty-five minutes after injection, cells were pulsed with latex beads for 3 min to induce DV formation and then fixed with 3% formaldehyde in phosphate buffer. To determine the rate of incorporation of the discoidal vesicle membrane into the cytopharyngeal membrane, the number and the diameter of the DVs formed during the pulse period were determined.
Other Procedures
Isolation and purification (Fok et al., 1996) and immunoblotting (Fok et al. 1988) of the DVs as well as electron microscopy (Fok and Allen, 1979) were described previously
RESULTS
When N-terminal sequencing was performed on the purified C5 antigen, a sequence of 25 amino acid residues was obtained. From this sequence two oligonucleotide probes were designed. The first was 39 nucleotides long and the second was 33 nucleotides long based on the amino acid residues 3–15 and 11–21, respectively (Figure 1). When these two probes were used to screen the λgt11 P. multimicronucleatum cDNA library, no positive clones were obtained. To determine whether these two probes could be used, PCR was performed using either one of these two oligonucleotides as well as either the λgt11 forward (5′-GGTGGCGACGACTCCTGGAGCCCG-3′) or the reverse (5′-TTGACACCAGACCAACTGGTAATG-3′) primer, in the presence of the recombinant phage particles (1 × 106 pfu) as templates. The resulting PCR product gave a single band of ∼1 kbp after agarose gel electrophoresis. Because this was a reasonable size for the cDNA encoding the C5 antigen, it was used as a probe to screen the cDNA library. Five positive clones were isolated, and because all five had the same cDNA insert, restriction site, and size of 0.87 kbp, they were called the p36-C fragment (Figure 2A). Sequencing revealed that all five clones lacked the 5′ portion that contained the translation start site. To obtain information about the 5′ region around this site and the promoter region of the gene encoding C5, Southern blotting of the total genomic DNA was performed using the p36-C fragment as a probe. A 20-kbp BamHI, a 1.3-kbp EcoRI, a 9-kbp HindIII, and a 12-kbp PstI fragment all containing the translation start site were obtained (Figure 3). The 1.3-kbp fragment, subsequently called the p36-G fragment (Figure 2A), was cloned by colony hybridization.
Figure 3.

Southern hybridization of P. multimicronucleatum genomic DNA with the p36-C fragment of the cDNA for the C5 antigen. Total genomic DNA (10 μg/lane) was digested with BamHI (lane 1), EcoRI (lane 2), HindIII (lane 3), or PstI (lane 4). Positions of the HindIII-digested λ DNA (kbp) are marked on the right side.
By combining the sequence information from the p36-C and p36-G fragments, the nucleotide sequence for the DNA encoding the C5 and its predicted amino acid sequence were obtained (Figure 2B). Except for five amino acid residues, the deduced amino acid sequence matched completely the N-terminal sequence and the internal sequence between amino acid residues 256 and 269 (Figures 1 and 2B).
The nucleotide sequence of the p36-C fragment had 996 bp (positions 184-1170) with an internal EcoRI site at positions 1048–1053. A putative polyadenylation signal, AAATAA, was located at 1148–1152. The p36-G fragment contained a 1.3-kbp region upstream from the nucleotide position 1053. The combined sequence of these two fragments had a large open reading frame beginning at position 172 and ending at 1116 and encoded a protein of 315 amino acid residues with a predicted molecular weight of 36,594. This value agreed well with the relative molecular mass of 33 kDa as determined by SDS-PAGE (incorrectly reported to be 31 kDa in Fok et al. 1996). The translation start site was assigned to the methionine codon ATG at positions 172–174, because it was the first ATG triplet downstream of the in-frame nonsense codon TGA at positions 139–141. The sequence flanking this ATG codon, ATTAAATGT, also partially matched the consensus sequence for the ciliate start site, TA/TAAAATGA/G (Yamauchi, 1991). Three putative TATA-like sequences were found at the 5′ upstream region, TATAA, TAATA, and TATATAA at positions 87–91, 101–105, and 113–119, respectively, suggesting that one of these three sequences might function as a promoter. These results indicate that the protein predicted from the cDNA and genomic DNA is C5 and the gene might be functional.
During its purification, C5 was found to be an abundant protein. To verify this observation and to examine the C5 expression, Northern blots of total RNA from P. multimicronucleatum (Figure 4A) and P. caudatum (Figure 4B) were performed. These blots were compared with Northern blots of two abundant proteins in P. caudatum, the 0.5-kbp genomic DNA encoding the glyceraldehyde-3-phosphate dehydrogenase (Figure 4, A and B, lane 2) and the 0.4-kbp P. caudatum hemoglobin cDNA (Figure 4, A and B, lane 3). The mRNA signal for C5 was very strong when the RNA and DNA were both from P. multimicronucleatum (Figure 4A, lane 1), suggesting that C5 is an abundant protein; however, the signal was rather weak when total RNA from P. caudatum was hybridized with the p36-C fragment from P. multimicronucleatum (Figure 4B, lane 1), suggesting that the C5 DNA sequence may be species specific.
Figure 4.

Northern hybridization of Paramecium RNA. Total RNA (10 or 15 μg/lane) was prepared from P. multimicronucleatum (A) and P. caudatum (B) and electrophoresed. Probes for hybridization were the 0.87-kbp p36-C fragment of the P. multimicronucleatum C5 antigen cDNA (lane 1), the 0.5-kbp P. caudatum genomic DNA encoding glyceraldehyde-3-phosphate dehydrogenase (lane 2), and the 0.4-kbp P. caudatum hemoglobin cDNA (lane 3).
The amino acid sequence of C5 appeared to be rather unique, because homology was found in only two other proteins when the program FASTA was used. Sequence similarity was observed in almost all regions of a S. pombe protein but was restricted to the C-terminal half of the β-fructofuranosidase protein of Z. mobilis (Figure 5). Sequence identity was only 14–15%, and sequence homology including conserved residues was only 22–25%; however, the predicted pI value of 6.49 for C5 was very similar to the 6.05 value for the yeast protein.
Figure 5.

Comparison of amino acid sequence of P. multimicronucleatum C5 antigen with those of two other proteins. The amino acid sequence of C5 antigen (Pm C5) from Figure 2B is aligned with those of a S. pombe hypothetical protein (Sp p36; accession No. S62543 in PIR protein database) and the β-fructofuranosidase protein of Z. mobilis (accession No. A37803 in PIR protein database). Identical residues and conserved residues are marked by double and single dots, respectively.
To obtain more information about these three proteins, PSORT, the computer program for predicting the intracellular location of proteins, was used (Nakai and Kanehisa, 1992). The S. pombe protein having 9–10 clusters of hydrophobic residues was predicted to be a transmembrane protein. The β-fructofuranosidase having no clear transmembrane domains was predicted to be a soluble protein. The C5 was also predicted to be a soluble protein, because it also has neither a clear transmembrane domain nor a cleavable signal sequence.
Our earlier immunofluorescence work had suggested that C5 was probably not a soluble protein because it was located on the DV membranes (Fok et al., 1996). To confirm its location on the DV membranes, immunolocalization at the electron microscopic level was used. The C5 was found to be located specifically on the membranes of the discoidal vesicles (Figure 6A), acidosomes (Figure 6B), lysosomes (Figure 6C), DV-II (Figure 6C), DV-III, and DV-IV (Figure 6D). The antigens appeared to be located more on the cytosolic than on the luminal surfaces of these vesicle and DV membranes (Figures 6A–D). Strikingly, only a few gold particles were seen on the membranes of the cytopharyngeal or NDV membranes (Figure 6A, arrow) and the DV-I membrane (Figure 6B, arrow), although these membranes are derived directly from the discoidal vesicles. More label was seen on the membranes of the DV-II (Figure 6C, arrow), and DV-III. The highest number of gold particles was seen on the DV-IV membrane (Figure 6D, arrow).
Figure 6.

Cryosections of cytopharynx and DV membranes (arrows) and associated organellar membranes. Some membranes are positive for the C5 antigen using the immunogold technique. (A) Discoidal vesicles (dsv), but not the cytopharyngeal membrane (cyx), are labeled. The label appears mostly on the cytosolic side of the vesicle membranes. (B) Acidosomes (ac), but not the DV-I membrane (I), are labeled. The vacuole contained 1.1 μm beads, indicating that it was 0–3 min old. (C) Lysosomes (ly) are aligned along the sparsely labeled 15- to 18-min-old DV-II (II). This DV contained 0.3 μm beads. (D) The DV-IV (0.1 μm beads indicate that the DV was 20–23 min old) shows the most label of all four DV stages. A labeled lysosome (arrowhead) lies near the DV-IV. Bar, 1 μm.
To provide a quantitative estimate of the relative C5 density on the various phagosomal membrane pools, the numbers of gold particles per micrometer on cross-sectioned membranes were counted. There was a 12-fold increase in C5 density on the DV-IV membranes over that on the NDV (Figure 7). The density of C5 on the DV-II membranes was slightly lower than that on the acidosome membranes, whereas the reverse was true for the lysosome and the DV-III membranes; however, the most striking observation was the difference in C5 densities between the membranes of the discoidal vesicles (3.2 gold particles/μm) and the NDV (0.6 gold particles/μm). That 80% of the C5 antigen could be removed or lost from the NDV membrane after the discoidal vesicles had fused with the cytopharynx supports our contention that C5 is not a transmembrane or integral protein.
Figure 7.

The average number of gold particles/micrometer of membrane profile in electron micrographs for discoidal vesicles, acidosomes, lysosomes, and the four stages of DVs. NDV refers to the nascent DV and includes the cytopharynx membrane as well. DCV, discoidal vesicles.
To further verify that C5 was on the cytosolic surface of the phagosomal membranes, two different light microscopic labeling procedures were used. The first method used our standard procedure for immunofluorescence. Cells were fixed, acetone permeabilized, and incubated with the mAb against C5. Labeling could be seen concentrated in the cytoplasm interior to the cell cortex, where it appeared to be on vesicles and vacuoles (Figure 8A) rather than being randomly dispersed in the background cytosol. These results indicated that C5 is most likely membrane bound and not a soluble protein.
Figure 8.

Immunofluorescence in cells labeled with mAbs against C5 and L1 antigens. (A) Cell fixed, acetone-permeabilized, and then labeled with mAb against C5 antigen. (B) The cell was briefly and weakly permeabilized with Triton X-100 first and then incubated with mAb against C5 antigen before fixation and secondary antibody incubation. The positive result shows that the mAb was able to bind to the antigenic sites on the cytosolic surfaces of the DVs. (C) This cell was similarly treated as in B but was exposed to mAb against L1, an antigen known to be on the luminal side of the DV-II membranes. The negative result shows that this mAb was not able to penetrate the DV-II membrane to get to the antigenic site on the luminal side of the DV membranes. Bar, 20 μm.
The second method involved the permeabilization of the cell’s pellicle while the DV membrane was kept intact. Half of these permeabilized cells were incubated with the mAb against C5 and were then fixed and incubated with a secondary antibody. Several DVs were positive for the C5 label (Figure 8B). To show that the DVs in these treated cells were intact, the other half of the permeabilized cells were incubated with the mAb against L1, an antigen known to be located on the luminal side of the DV-II membranes (Allen et al., 1995). No label was seen on any DV (Figure 8C), indicating that this mAb could not penetrate the intact DV membrane and therefore could not bind to the luminally exposed L1 antigen. These observations showed that the reactive epitope on C5 was indeed located on the cytosolic surface of phagosomal membranes.
To determine whether the C5 antigen is loosely or tightly bound to the DV membranes, DVs of stages II and III were purified and washed with 0.2 M KCl according to Fok et al. (1996). Proteins from these DV preparations were electrophoresed and blotted with the mAb against C5. A very strong reaction was observed in the immunoblot of DV-II (Figure 9) and DV-III, suggesting that C5 was bound rather tightly to the phagosomal membranes.
Figure 9.
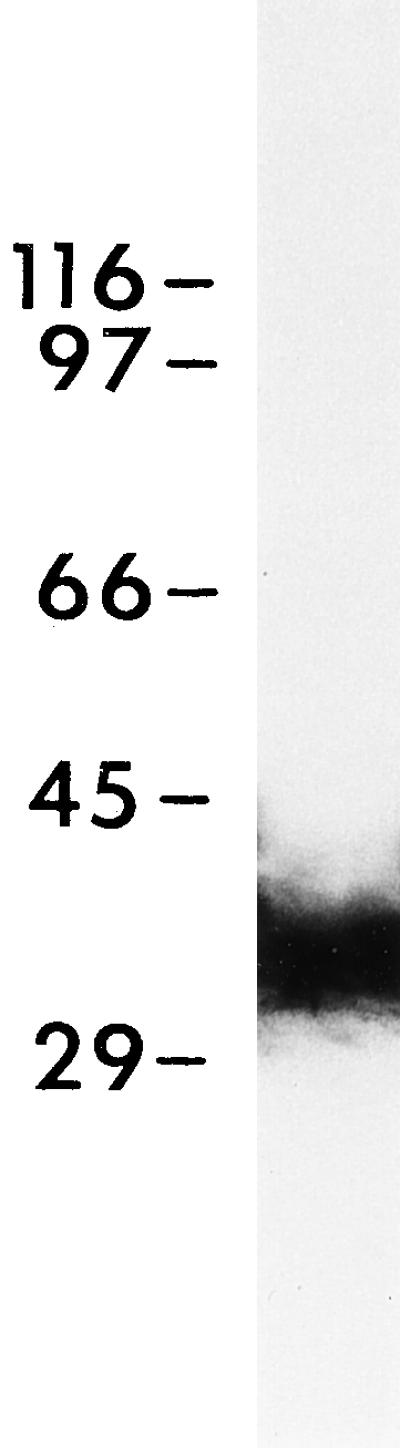
Digestive vacuoles of stage II were purified from cells that had been pulse-fed magnetic beads and chased as described in Fok et al. (1996) and processed for SDS-PAGE and immunoblotting according to Fok et al. (1988). DV-II gave a very strong result.
We were unable to obtain more information on the function of the C5 antigen from the amino acid sequence, so microinjection studies were performed to determine whether C5 played a role in the discoidal vesicle–cytopharyngeal membrane docking or fusion. Cells were injected with the purified mAbs to either C5 or B2 and allowed to recover for 45 min. After this recovery period, cells were pulsed with latex beads to induce DV formation, and the total amount of discoidal vesicle membrane incorporated was calculated from the rate of DV formation and the size of the DVs formed.
When cells were injected with 10.7 pg/cell of the mAb to B2, DV formation, i.e., discoidal membrane fusion, was not inhibited, although at twice the concentration, a 33% inhibition was observed (Figure 10). In contrast, in cells injected with as little as 1.34 pg/cell of the mAb to C5, membrane fusion was inhibited by 10%. Inhibition was linear from 1.34 to 10.7 pg/cell. At 10.7 pg/cell, the inhibition was close to 86%, whereas at 21.4 pg/cell, the inhibition was almost complete (Figure 10). Electron microscopy (Figure 11) showed a normal accumulation of the discoidal vesicles lying next to the cytopharynx in the cell injected with the mAb to C5. These results showed that 1) the inhibition of membrane fusion was not due to an interference in the formation or retrieval of the discoidal vesicles, the DV membrane precursors, and 2) C5 played a role in some step of binding or fusion of the discoidal vesicles with the cytopharyngeal membrane.
Figure 10.

The effects of microinjected mAbs on the incorporation of discoidal vesicle membrane. ▴ indicates mAb to B2 (140 kDa, on the luminal surface of the DV-I); ▪ indicates mAb to C5. Discoidal vesicle membrane incorporation was determined from the rate of DV formation and the size of the DVs formed in the injected cells. For each time point, data were obtained from 12–17 injected cells.
Figure 11.

A grazing section through the lip of the cytopharynx where the cytopharyngeal membrane (CYX) is continuous with the nascent digestive vacuole (NDV) membrane in a cell injected with 10.7 pg of the mAb to the C5 antigen. There has been no decrease in the number of discoidal vesicles (dsv) along the cytopharyngeal microtubular ribbons (mt). These discoidal vesicles are waiting to fuse with the cytopharyngeal membrane. ac, acidosome; c, cilia. Bar, 1 μm.
DISCUSSION
This report represents the first cloning attempt in P. multimicronucleatum in which a phagosomal membrane antigen known as C5 was purified, N-terminal and internal sequences were obtained, and finally the gene encoding the antigen was cloned. The amino acid sequence of C5 appears to be rather unique, because only two other proteins share some homology with it. Sequence identity between C5 and a S. pombe protein or with the β-fructofuranosidase protein of Z. mobilis is only 14–15%, and sequence homology, including conserved residues, is only 22–25% (Figure 5). Of these two proteins, C5 resembles more closely the bacterial β-fructofuranosidase in that both proteins lack any obvious transmembrane domains. The yeast protein is predicted to be a transmembrane protein located in the membranes of the endoplasmic reticulum or the plasma membrane, and its sequence has half as many charged amino acids and a lower content of histidine, arginine, and aspartic acid than C5. On the other hand, C5 and the yeast protein have very similar pI values. Until proteins with known functions that have much more homology to C5 have been sequenced, it will not be possible to determine the function of C5 based on sequence data alone.
The C5 antigen is known to be an abundant protein as shown by immunoprecipitation studies. This is verified in Northern blots, because the message for C5 was as high as those for hemoglobin and glyceraldehyde-3-phosphate dehydrogenase, which are known to be abundant proteins, in P. caudatum (Figure 4). On the other hand, the signal was rather weak when the p36-C fragment was used to hybridize with the total RNA from P. caudatum, indicating that the C5 DNA sequence is probably species specific. This latter finding is confirmed by our observation that the mAb against C5 also did not cross-react with DVs in P. tetraurelia. It is our experience that many mAbs raised against P. multimicronucleatum membrane proteins do not cross-react with proteins of other Paramecium species.
From the amino acid sequence, it is predicted that C5 would be a soluble protein because it lacks a transmembrane domain or a cleavable signal sequence. Also it could not be a myristate-anchored membrane protein because it does not have a glycine residue at the N terminus, nor can it be a farnesyl-anchored membrane protein because it does not have a CAAX box at its C terminus; however, cryoelectron microscopy (Figure 6) and immunofluorescence (Figure 8) show that C5 is most likely a membrane-associated protein, because it is specifically located on the cytosolic surface of the phagosomal membranes. That much of the C5 is not transferred to the NDV membrane after discoidal vesicles fuse with the NDV membrane indicates that C5 can be removed quickly from these membranes (Figure 7). Therefore C5 is probably not a transmembrane protein, although it does seem to be relatively tightly bound to the phagosomal membrane surface (Figure 9). On the basis of these findings, we conclude that C5 could be a relatively tightly bound peripheral protein found on the cytosolic surface of most phagosomal membranes.
The 25 residues at the N-terminal end of this antigen are quite hydrophobic, giving this region the property shared by signal peptides of some mammalian proteins. Thus, it is not surprising that the computer program PSORT recognized this region as a signal peptide; however, because these amino acid residues are on the mature protein, this region cannot serve as a signal peptide unless it is an uncleaved signal. This hydrophobic region would be a good candidate for a membrane-binding site for the phagosomal membrane, but that remains to be determined.
It is now known that many proteins must be recruited to the site of vesicular membrane contact with an acceptor membrane before fusion with the acceptor membrane can be achieved. The most extensively studied example is that of neurosecretion as synaptic vesicles fuse with the presynaptic membrane in neurons (see review by Martin, 1997). Proteins recruited to the site of fusion include those involved in anchoring, docking, priming, and fusion. The observations that a normal accumulation of discoidal vesicles remains in the vicinity of the cytopharynx in cells microinjected with mAb to C5 and that the microinjected mAb inhibited the discoidal vesicle–cytopharyngeal membrane fusion in a dose-dependent manner suggest that C5 does play a role in one of the steps leading to membrane fusion between the discoidal vesicles and the cytopharyngeal membrane. Because C5 is also present on the acidosomes and lysosomes, it is reasonable to assume that this antigen may also be involved in binding and/or fusion of these two vesicular pools with DV-I and DV-II, respectively. Exactly what role C5 plays will require further experimentation.
This study lays the foundation for future cloning of other phagosomal proteins for which we have the mAbs. In particular, it will be interesting to clone the genes encoding the antigen Q2, which is also found on the surface of discoidal vesicles (Allen et al., 1995). In contrast to C5, this antigen remains with the DV-I after the discoidal vesicles have fused with the cytopharyngeal membrane. Information obtained for these antigens can eventually be compared with proteins in other ciliates, making it possible to begin to follow the evolutionary divergence of the different Paramecium species.
ACKNOWLEDGMENTS
We thank Professor K. Mikami (Research Institute for Science Education, Miyagi University of Education, Sendai, Japan) for supplying the oligonucleotides specific for glyceraldehyde-3-phosphate dehydrogenase for PCR. We thank T. Toyozato for help with running the sequencing gels, and K. Mochizuki, Y. Sato, and N. Nishiyama for technical assistance. This research was supported by National Science Foundation (NSF) grants. The Biological Electron Microscope Facility is supported in part by National Institutes of Health grant RR-03061 and by NSF instrumentation grants.
REFERENCES
- Allen RD. Food vacuole membrane growth with microtubule-associated membrane transport in Paramecium. J Cell Biol. 1974;63:904–922. doi: 10.1083/jcb.63.3.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RD, Bala NP, Ali RF, Nishida DM, Aihara MS, Ishida M, Fok AK. Rapid bulk replacement of acceptor membrane by donor membrane during phagosome to phagoacidosome transformation in Paramecium. J Cell Sci. 1995;108:1263–1274. doi: 10.1242/jcs.108.3.1263. [DOI] [PubMed] [Google Scholar]
- Allen RD, Fok AK. Membrane recycling and endocytosis in Paramecium confirmed by horseradish peroxidase pulse-chase studies. J Cell Sci. 1980;45:131–145. doi: 10.1242/jcs.45.1.131. [DOI] [PubMed] [Google Scholar]
- Allen RD, Fok AK. Nonlysosomal vesicles (acidosomes) are involved in phagosome acidification in Paramecium. J Cell Biol. 1983;97:566–570. doi: 10.1083/jcb.97.2.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RD, Fok AK. Retrieval of lysosomal membrane and acid phosphatase from phagolysosomes of Paramecium caudatum. J Cell Biol. 1984;99:1955–1959. doi: 10.1083/jcb.99.6.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RD, Staehelin LA. Digestive system membranes: freeze-fracture evidence for differentiation and flow in Paramecium. J Cell Biol. 1981;89:9–20. doi: 10.1083/jcb.89.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RD, Ueno MS, Pollard LW, Fok A K. Monoclonal antibody study of the decorated spongiome of contractile vacuole complexes of Paramecium. J Cell Sci. 1990;108:1263–1274. doi: 10.1242/jcs.96.3.469. [DOI] [PubMed] [Google Scholar]
- Allen RD, Wolf RW. The cytoproct of Paramecium caudatum, structure and function, microtubules, and fate of food vacuole membranes. J Cell Sci. 1974;14:611–631. doi: 10.1242/jcs.14.3.611. [DOI] [PubMed] [Google Scholar]
- Aviv H, Leder P. Purification of biologically active globin mRNA by chromatography of oligothymidynic acid-cellulose. Proc Natl Acad Sci USA. 1972;69:1408–1412. doi: 10.1073/pnas.69.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Seeburg PH. Laboratory methods. Supercoil sequencing, a fast and simple method for sequencing plasmid DNA. DNA. 1985;4:165–170. doi: 10.1089/dna.1985.4.165. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dupuis P. The β-tubulin genes of Paramecium are interrupted by two 27 bp intron. EMBO J. 1992;11:3713–3719. doi: 10.1002/j.1460-2075.1992.tb05456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok AK, Allen RD. Axenic Paramecium caudatum. I. Mass culture and structure. J Protozool. 1979;26:463–470. doi: 10.1111/j.1550-7408.1979.tb04654.x. [DOI] [PubMed] [Google Scholar]
- Fok AK, Lee Y, Allen RD. The correlation of digestive vacuole pH and size with the digestive cycle in Paramecium caudatum. J Protozool. 1982;29:409–414. [Google Scholar]
- Fok AK, Ma L, Aihara MS, Allen RD. High resolution view of the true cytosolic membrane surface of phagosomes of known ages purified from Paramecium. Eur J Cell Biol. 1996;71:259–269. [PubMed] [Google Scholar]
- Fok AK, Muraoka JH, Allen RD. Acid phosphatase in the digestive vacuoles and lysosomes of Paramecium caudatum, a timed study. J Protozool. 1984;31:216–220. [Google Scholar]
- Fok AK, Olegario RP, Ueno MS, Allen RD. Characterization of monoclonal antibodies to trichocyst antigens in Paramecium. J Cell Sci. 1988;91:191–199. doi: 10.1242/jcs.91.2.191. [DOI] [PubMed] [Google Scholar]
- Fok AK, Ueno MS, Allen RD. Differentiation of Paramecium membrane and stages using monoclonal antibodies. Eur J Cell Biol. 1986;71:259–269. [Google Scholar]
- Geisow MJ, D’Arcy Hart P, Young MR. Temporal changes of lysosome and phagosome pH during phagolysosome formation in macrophages, studies by fluorescence spectroscopy. J Cell Biol. 1981;89:645–652. doi: 10.1083/jcb.89.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Sakaki Y. Dideoxy sequencing method using denatured plasmid templates. Anal Biochem. 1986;152:232–238. doi: 10.1016/0003-2697(86)90403-3. [DOI] [PubMed] [Google Scholar]
- Ishida M, Aihara MS, Allen RD, Fok AK. Osmoregulation in Paramecium: the locus of fluid segregation in the contractile vacuole complex. J Cell Sci. 1993;106:693–702. doi: 10.1242/jcs.106.2.693. [DOI] [PubMed] [Google Scholar]
- Ishida M, Aihara MS, Allen RD, Fok AK. Acidification of the young phagosomes of Paramecium is mediated by proton pumps derived from the acidosomes. Protoplasma. 1997;196:12–20. [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Martin TFJ. Stages of regulated exocytosis. Trends Cell Biol. 1997;7:271–276. doi: 10.1016/S0962-8924(97)01060-X. [DOI] [PubMed] [Google Scholar]
- Martindale DW. Codon usage in Tetrahymena and other ciliates. J Protozool. 1989;36:29–34. doi: 10.1111/j.1550-7408.1989.tb02679.x. [DOI] [PubMed] [Google Scholar]
- Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- Metchnikoff, E. (1893). Lectures on the Comparative Pathology of Inflammation, London: Kegan Paul Trench, Truber and Co.
- Nakai K, Kanehisa M. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Schneider C, Roland A, Newman D, Sutherland R, Asser U, Greaves MF. A one step purification of membrane proteins using a high efficiency immunomatrix. J Biol Chem. 1982;257:10766–10767. [PubMed] [Google Scholar]
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Sturgill-Koszycki S, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- Yamauchi K. The sequence flanking translational initiation site in protozoa. Nucleic Acids Res. 1991;19:2715–2720. doi: 10.1093/nar/19.10.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K, Mukai M, Ochiai T, Usuki I. Molecular cloning of the cDNA for the major hemoglobin component from Paramecium caudatum. Biochem Biophys Res Commun. 1992;182:195–200. doi: 10.1016/s0006-291x(05)80130-5. [DOI] [PubMed] [Google Scholar]
- Young RA, Davis RW. Efficient isolation of genes by using antibody probes. Proc Natl Acad Sci USA. 1983;80:1194–1198. doi: 10.1073/pnas.80.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]