

Abstract
Entry into host cells is required for many bacterial pathogens to effectively disseminate within a host, avoid immune detection and cause disease. In recent years, many ostensibly extracellular bacteria have been shown to act as opportunistic intracellular pathogens. Among these are strains of uropathogenic Escherichia coli (UPEC), the primary causative agents of urinary tract infections (UTIs). UPEC are able to transiently invade, survive and multiply within the host cells and tissues constituting the urinary tract. Invasion of host cells by UPEC is promoted independently by distinct virulence factors, including cytotoxic necrotizing factor, Afa/Dr adhesins, and type 1 pili. Here we review the diverse mechanisms and consequences of host cell invasion by UPEC, focusing also on the impact of these processes on the persistence and recurrence of UTIs.
Keywords: adhesion, bladder, cystitis, fimbriae, invasion, lipid rafts, pili, uroplakin
The world of bacterial pathogens is often categorically divided into pathogens that can and those that cannot invade and survive within host eukaryotic cells (e.g. (1)). In recent years this division has become increasingly blurred as more and more so-called extracellular pathogens are found to have alternative intracellular lifestyles. Bacteria previously supposed to be strictly extracellular pathogens include, but are not limited to, group A streptococcus (2,3), Burkholderia cepacia (4), Helicobacter pylori (5), Haemophilus influenzae (6), and uropathogenic Escherichia coli (UPEC) (7). These bacteria are causative agents of, respectively, tonsillitis, pulmonary infections, gastric and duodenal ulcers, ear infections, and urinary tract infections (UTIs). Ongoing research indicates that these diverse bacteria can not only invade host cells, but may in many instances also establish long-lived intracellular reservoirs that resist clearance by both host immune defenses and antibiotic treatments (4,5,8–12). Such persistent intracellular reservoirs may contribute to the chronic and recurrent nature of a number of bacterial infections. In the case of UPEC, host cell invasion and intracellular bacterial replication are proposed to contribute to the ability of these pathogens to effectively colonize, disseminate and persist long-term within the urinary tract. Here we review recent findings regarding the mechanisms by which UPEC can bind and invade host cells, focusing also on the consequences of these events for the pathogenesis of UTIs.
A Blight upon the Bladder, a Curse upon the Kidney
The human urinary tract is normally a sterile environment, protected from pathogens by the shear flow of urine, secreted and tissue-associated antibacterial factors, and the bactericidal activities of effector immune cells. However, even in populations with these natural defenses seemingly intact, and despite the increasing use of antibiotics, bacterial infection of the urinary tract is an exceedingly common problem. About 60% of women in the United States will have at least one UTI during their lifetime, and 11% experience at least one per year (13,14). Close to 8 million physician visits per year are attributed to these often-chronic infections, making UTIs a problem of real economic and medical significance. Symptoms of an uncomplicated bladder infection (cystitis) can be relatively mild, including frequent or urgent voiding and suprapubic pain, but infection of the kidney (pyelonephritis) can also result, leading to fever, nausea, vomiting, and, in about 30% of cases, bacteremia and the potential for sepsis. Indeed, the most common source of bacteremia and sepsis are microbes that enter the bloodstream from sites within the urinary tract (15,16). The strong propensity of UTIs to recur, often within a few weeks or months after an initial acute infection, may contribute to additional problems, including renal scarring and an increased risk for developing bladder cancer (14,17–19).
The vast majority of UTIs are caused by UPEC, a surprisingly heterogeneous group of pathogens. It is believed that most UPEC are found initially in the gut, and are later introduced through the urethra to the bladder and kidneys. Recently, the first UPEC genome sequence, that of the human pyelonephritis isolate CFT073, was completed. Comparison of the CFT073 genome with that of the enterohemorrhagic E. coli (EHEC) isolate EDL933 and the K12 reference laboratory strain MG1655 has highlighted the profound diversity among E. coli strains (20). Notably, the genomes of the two pathogens, which share only 39.2% of their protein coding sequences, were found to be as different from each other as they are from the K12 genome. Further comparisons among CFT073 and the partially completed genomes of two additional UPEC isolates, J96 and 536, were also made, and indicate that present day UPEC and other extraintestinal pathogenic E. coli (ExPEC) strains probably arose from multiple clonal lineages. Many of the noted genomic differences among UPEC and other E. coli isolates likely reflect evolutionary adaptations; changes that have enabled UPEC isolates to better colonize the unique and varied environmental niches found within the urinary tract. These niches include the stratified epithelia lining the lumenal walls of the urethra, bladder, and ureters, as well as the renal pelvis and the adjoining tubules and collecting ducts of the kidneys.
UPEC isolates encode a variety of virulence factors that facilitate colonization of the urinary tract. To date, however, no single virulence factor has been shown to be specifically unique to, or definitive of, UPEC. The type III secretion systems often elaborated by enteric E. coli pathogens such as EHEC as a means of delivering bacterial products into target host cells are, for the most part, missing from UPEC strains, as are many of the toxins encoded by enteropathogenic strains (20,21). UPEC does, however, express a number of virulence factors that enteric pathogens often lack – including several secreted toxins, numerous adhesive molecules (adhesins), iron-acquisition systems, capsule-forming polysaccharides for immune evasion, and phase-switch recombinases. The search for additional virulence factors in UPEC is far from over: there are reports that up to half of all UPEC strains still have no or only one of the known virulence factors (22). Genomic subtraction, microarray analyses, genetic screens and other strategies are proving useful for the identification of additional contributors to UPEC pathogenicity, and we can anticipate more discoveries in coming years.
Of the known virulence factors associated with UPEC, the toxin cytotoxic necrotizing factor 1 (CNF1) and two adhesins (Afa/Dr and the type 1 pilus-associated adhesin FimH) have been shown to independently mediate bacterial entry into host epithelial cells. The ability of UPEC to invade host cells and to persist may help explain the enigmatic nature of the recurrent and chronic UTIs that plague many individuals throughout their lives. Current dogma dictates that recurrent UTIs arise from re-inoculation of the urinary tract by UPEC maintained within the intestinal flora or elsewhere in the environment. It is often argued that individuals prone to recurrent UTIs have defects in their immune defense mechanisms, making them more susceptible to UPEC colonization. Such arguments are buoyed by recent data showing that polymorphonucleocytes (PMNs) from individuals prone to recurrent UTIs have impaired bactericidal activity relative to PMNs from healthy individuals (23). Epidemiologic studies showing that up to 68% of recurrent UTIs are caused by bacterial strains identical to the strain responsible for the initial infection also purportedly support the notion that recurrent UTIs arise from re-inoculation of the urinary tract (24–28). However, these latter observations also mesh with the possibility that recurrent UTIs may represent, in some instances, the recrudescence of infections that are not entirely cleared from the urinary tract after overt clinical symptoms of the UTI subside. Studies using mouse UTI model systems demonstrate that intracellular UPEC have a marked survival advantage over their extracellular counterparts during infections lasting from days to months, with or without antibiotic treatments (7,11,12,29,30). The enhanced resistance of intracellular UPEC to clearance from the urinary tract implies that host cell invasion is a critical aspect of the pathogenesis of both acute and chronic UTIs. The mechanisms by which UPEC enter host epithelial cells, persist, multiply, and eventually re-emerge are now coming to light, revealing novel virulence strategies and hinting at a high degree of host–pathogen crosstalk during the course of a UTI.
CNF1-Mediated Bacterial Invasion
CNF1, a 113-kDa single chain toxin, was identified in E. coli close to 20 years ago on the basis of its necrotizing effects on rabbit skin and the promotion of multinucleation in cultured cells (31). Subsequent studies have shown that the secreted toxin, which is encoded by about one-third of UPEC strains, stimulates formation of actin stress fibers, lamellipodia, filopodia, and membrane ruffling (32–35). How CNF1, which is fairly hydrophilic and lacks a distinguishable signal sequence, is secreted from E. coli is incompletely understood. Once released, however, CNF1 is able to enter host cells via a low pH-mediated endocytic pathway by binding the laminin receptor precursor, a host membrane protein that also functions as a receptor for several viruses in addition to a cellular prion protein (36). Within host cells, CNF1 functions as a deamidase, converting specific glutamine residues on the Rho GTPases RhoA, Rac, or Cdc42 to glutamic acid. This amino acid change inhibits both the target proteins’ intrinsic GTP hydrolytic activity and GTP hydrolysis induced by GTPase-activating proteins (GAPs), thus rendering Rho GTPases constitutively active. These GTPases are key signaling molecules in a variety of cellular processes, including reorganization of the actin cytoskeleton.
Many of the observed effects of CNF-1 (such as formation of stress fibers and membrane protrusions) are certainly consistent with the toxins’ mechanism of action on Rho GTPases, but whether and how these effects play into UTI pathogenesis is not entirely clear. Rippere-Lampe and colleagues have argued that mutation of cnf1 has an attenuating effect on UPEC virulence (37). These authors performed single-strain and competition assays using isogenic cnf1+ and cnf1− pairs in a mouse model of ascending UTI. Their data vary in significance depending on the mouse and bacterial strains used and time points examined, but there is evidence that, at least in some cases, cnf1− UPEC are less able to colonize the bladder and kidneys than isogenic cnf1+ bacteria. These data are in contrast to the findings of a similar study by the Donnenberg group (38) but, as noted by Rippere-Lampe and colleagues, differences in strains and infection times may account for the discrepancies.
In non-UTI studies, results regarding the role of CNF1 as a virulence factor have also been mixed. In a study of porcine colibacillosis using colostrum-deprived germfree piglets that were orally infected with a pig enteritis E. coli isolate, Fournout et al. (39) concluded that CNF1 has no effect on cytokine responses or bacterial titers in the intestine, lungs, liver, spleen or kidneys. In contrast, Khan and colleagues (40) found that CNF1 has a notable influence on the pathogenicity of E. coli K1, an important agent of meningitis in humans. These researchers found that a cnf1 deletion mutant, relative to the wild-type K1 strain, is significantly impaired in its ability to invade cultured brain endothelial cells and is less efficient at crossing the blood–brain barrier in an experimental hematogenous meningitis animal model. E. coli that cause meningitis appear to be phylogenetically linked and share several virulence factors with those that cause acute cystitis (41), and so it is possible that this meningitis study is relevant to UTI pathogenesis as well.
In spite of contrasting evidence for a role of CNF1 in pathogenesis, there are attractive models for how CNF1 may be able to promote bacterial colonization and virulence within the urinary tract. Mills and colleagues used TUNEL staining to show that CNF1 induces apoptosis in a bladder epithelial cell line, and suggested that in vivo this may lead to bladder cell exfoliation and enhanced bacterial access to underlying tissue (35). CNF1 may also promote UPEC pathogenesis via more subversive mechanisms. Two recent papers presented data showing that the effects of CNF1 on activation of Rac are in fact only transient; a few hours after being constitutively activated by CNF1, the modified Rho GTPases are ubiquitinated and subsequently degraded by the host proteasome (42,43). This delayed degradation is arguably a safety mechanism for the host cells, in which continual Rac activation could be deleterious. However, Doye and colleagues (42) took their experiments further, demonstrating that proteasome depletion of CNF1-activated Rac in a bladder epithelial cell line promoted plasma membrane ruffling and filopodia formation, stimulated host cell motility and substantially increased bacterial internalization (Figure 1). Internalization, as noted above, can promote UPEC survival within the urinary tract and enhanced host cell motility may facilitate the dissemination of UPEC within the infected tissue. It thus seems that CNF1 provides UPEC with a means of hijacking the host proteasome degradation machinery, positively affecting bacterial invasion along with the redistribution and accessibility of target host cells.
Figure 1. CNF1 effects on bladder cell motility and bacterial entry.
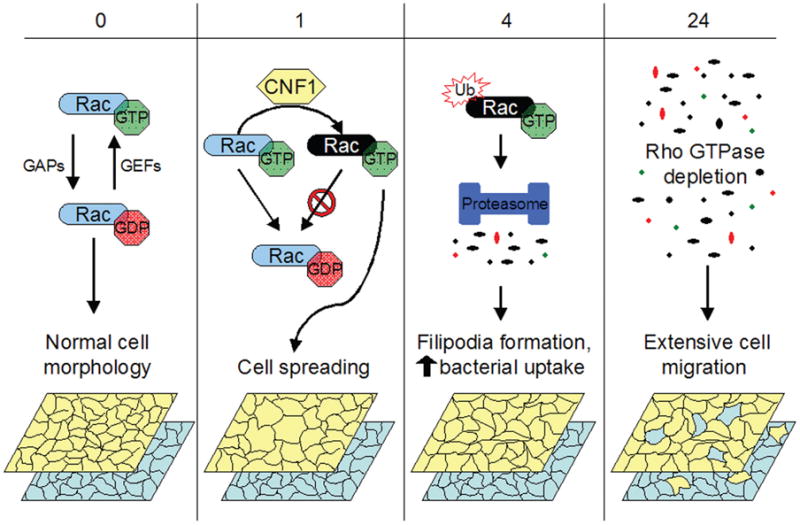
In normal uninfected bladder epithelial cells, Rho GTPases like Rac cycle between active GTP-bound and inactive GDP-bound states, regulated by GAPs and GEFs (guanine nucleotide exchange factors). CNF1 renders Rho GTPases constitutively active by catalyzing deamidation of glutamine 63 in RhoA (or glutamine 61 in Rac and Cdc42). CNF1-modified constitutively active Rac, in this example, is shown as a black oval. Activation of Rac and other Rho GTPases leads to cytoskeletal rearrangements resulting in increased cell spreading and membrane ruffling (1–4 h post-CNF1 intoxication). By 4 h postintoxication, constitutively activated Rho GTPases are ubiquitinated and targeted for proteasomal degradation. The resulting depletion of Rho GTPases promotes host cytoskeletal rearrangements, resulting in enhanced filopodia formation and stimulating bacterial uptake. Bacterial internalization continues to increase up to 24 h post-CNF1 intoxication. Rho depletion also stimulates bladder cell migration. Theoretically, this could result in redistribution of host cells within a stratified three-dimensional tissue such as the bladder epithelium, allowing UPEC to more efficiently disseminate and colonize underlying cells. The model shows the migration of host cells in the upper (yellow) and lower (blue) layers of an idealized tissue bilayer as a consequence of CNF1-stimulated Rac depletion.
Adhesin-Mediated Invasion
More direct routes of entry into host cells are afforded by some of the adhesins expressed by UPEC and other bacterial strains. Typically, adhesins are presented on bacterial surfaces as components of supramolecular proteinaceous filamentous organelles called pili (also known as fimbriae) or as afimbrial monomeric or multimeric molecules anchored within the bacterial outer membrane (44). UPEC encodes a number of adhesins that target a variety of different host receptors, including extracellular and cell surface protein structures in addition to complex and simple sugars linked to host glycoproteins or glycolipids. Adhesins commonly associated with UPEC strains include the Afa/Dr family of fimbrial and afimbrial adhesins, S/F1C, Pap (P), and type 1 pili, M hemagglutinin, and nonfimbrial adhesins 1–6. To date, only the Afa/Dr adhesins and type 1 pili have been conclusively shown to promote host cell invasion as well as bacterial attachment.
Host cell entry via Afa/Dr adhesins
The Afa/Dr family consists of the fimbrial adhesive organelles F1845, Dr and Dr-II along with other so-called afimbrial adhesins, including Afa-1 and Afa-3 (reviewed in (45)). Afa-3 and other Afa/Dr family members were initially characterized as ‘afimbrial’ based on electron microscopic analyses, but recent high-resolution structural studies and modeling indicate that at least Afa-3 may comprise fine filaments that collapse under the rigors of fixation for electron microscopy (46). The Afa/Dr adhesins are each encoded by at least five genes (A through E), with gene E typically coding for the actual adhesin (45). All Afa/Dr adhesins bind to the Dra blood group antigen present on the glycophosphatidylinositol (GPI)-anchored complement regulatory molecule CD55 (also known as decay-accelerating factor, DAF). A subclass of Afa/Dr adhesins, including F1845, Dr and Afa-3, also bind three members of the carcinoembryonic antigen-related adhesion molecules (CEACAM) family: CEA, CEACAM1, and CEACAM6 (47). In addition, Dr fimbriae, which comprise DraE and DraD subunits, bind type IV collagen and both Dr and Afa-3 adhesins recognize α5β1 integrin (48–50).
Afa/Dr adhesins are expressed by both UPEC strains and intestinal pathogens defined as diffusely adherent E. coli (DAEC) (45). Most work with the Afa/Dr adhesins has focused on their role in the pathogenesis of infections caused by DAEC. Growing evidence, however, indicates that these adhesins can contribute significantly to UPEC colonization of the urinary tract. For example, a recent study has shown that the type IV collagen-binding activity of Dr fimbriae facilitates UPEC persistence within the interstitial compartments of the kidneys in a mouse model of pyelonephritis (51). Other work implicates Afa/Dr-positive UPEC strains as major culprits in the development of gestational pyelonephritis (52,53). Up-regulation of CD55, which occurs normally in humans during pregnancy, appears to enhance colonization of the urinary tract by Afa/Dr-positive UPEC. Cell culture studies confirm that the extent of host cell colonization by Afa/Dr-positive E. coli is proportional to the level of CD55 expression (54). It has been reasoned that the host might have evolved mechanisms to down-regulate CD55 expression, in some situations, as a means of hindering bacterial colonization (55). In support of this hypothesis, it was found that nitric oxide, which is often generated in copious amounts by host nitric oxide synthases (NOS) in response to infection, inhibits CD55 mRNA and protein synthesis in endometrial cells and diminishes bacterial colonization. Nitric oxide is abundantly produced within the urinary tract in response to UPEC infection (56,57) and may interfere with CD55 expression and Afa/Dr-positive UPEC colonization in a mechanistically similar fashion.
Several recent in vitro studies, although not focused on E. coli interactions with host cells that are particularly relevant to UTIs, suggest that Afa/Dr adhesins may contribute to the pathogenesis of UTIs in additional ways. Research using intestinal and cervical epithelial cell lines demonstrate that Dr-mediated bacterial adherence to CD55 can activate phosphatidylinositol-3 (PI-3) and mitogen-activated protein kinases, eliciting downstream pro-inflammatory responses, including expression of the chemokine IL-8 and stimulation of PMN migration (58,59). It is feasible that Afa/Dr-positive UPEC strains trigger similar host responses within the urinary tract. Other recent work shows that Afa/Dr adhesin interactions with CEA can activate the Rho GTPase Cdc42 (47). This, in turn, appears to stimulate phosphorylation of the actin binding proteins ezrin/radixin/eosin (ERM), resulting in the formation of microvilli-like extensions that bind and enwrap adherent bacteria and contribute to characteristic host cytopathic effects elicited by Afa/Dr-positive DAEC and UPEC. Bacterial engagement of CEACAM receptor molecules may also simultaneously interfere with host T-cell responses. For example, Neisseria gonorrhoeae colony opacity-associated (Opa) proteins bind CEACAM1 similarly to some Afa/Dr adhesins and inhibit the activation and proliferation of CD4+ T lymphocytes that are a normal constituent of submucosal (subepithelial) compartments throughout the body (60). Similar effects elicited by CEA-CAM1-binding Afa/Dr-positive UPEC would presumably dampen host immune responses within the urinary tract while facilitating bacterial colonization and persistence.
The Afa/Dr-associated proteins AfaD, DraE, and DraD have been implicated as invasins, molecules capable of stimulating the internalization of bacteria by target host cells (48,61,62). Recognition of CD55 and/or α5β1 integrin by these Afa/Dr adhesins promotes the progressive envelopment of adherent microbes or invasin-coated beads by the host cell membrane via what has become known as a zipper-like mechanism (49,54,63). Whether CEACAM family members (some of which can mediate internalization of Opa-expressing N. gonorrhoeae (64)) promote host cell invasion by Afa/Dr-positive E. coli is unclear. The signaling and trafficking events involved in host cell invasion by AfaD- or Dr-positive E. coli are currently known in only cursory detail. Inhibitor studies coupled with microscopy indicate that internalization of Afa/Dr-positive E. coli is not especially reliant upon host F-actin rearrangements (49,62). Rather, internalization of these bacteria occurs via a process requiring dynamically unstable microtubules and, possibly, a microtubule-associated motor such as dynein. Only a few other bacterial pathogens, including Chlamydia trachomatis (65) and Campylobacter jejuni (66), have been shown to enter host cells via such a microtubule-dependent, microfilament-independent pathway. This mode of entry for Afa/Dr-positive E. coli is even more surprising when contrasted with Yersinia, a pathogen that also employs α5β1 integrin for host cell invasion via a zipper-like mechanism, but does so by a process that is fully dependent on actin filaments (67).
Microscopic analyses indicate that CD55 and α5β1 integrin cluster beneath adherent Afa/Dr-positive microbes prior to internalization (48,49,54). Lipid raft components, including GM1 ganglioside and the integral membrane protein caveolin-1, have also been shown to aggregate in association with bound Afa/Dr-positive E. coli. The makeup and potential functions of lipid rafts have been extensively studied and reviewed. In brief, lipid rafts are defined empirically as microdomains of the plasma membrane that are (i) resistant to solubilization by nonionic detergents at 4 °C and (ii) enriched in cholesterol, sphingolipids, and GPI-anchored proteins. Lipid rafts that contain caveolin-1 constitute a subclass of rafts known as caveolae that, in some cell types, form flask-shaped invaginations at the cell surface. One proposed function of lipid rafts is to serve as scaffolds for the assembly of signaling complexes. In accordance with this proposed function, it has been noted that the efficiency of Afa/Dr-mediated bacterial entry into epithelial cells depends upon the GPI anchor of the CD55 receptor, suggesting that the anchor serves to relay signals into the host cell via an apparent lipid raft-dependent process (54). Drugs such as methyl-β-cyclodextrin and filipin, which deplete or sequester host cell cholesterol and thereby disrupt raft domains, effectively inhibit Afa/Dr-mediated bacterial invasion, further supporting a role for lipid rafts in the entry process (49,63). Interestingly, cholesterol depletion does not prevent the mobilization of α5β1 integrin, CD55, GM1, or caveolin-1 around adherent Afa/Dr-positive bacteria. This observation suggests that cholesterol depletion interferes with signaling processes downstream of Afa/Dr-mediated receptor binding and clustering.
Type 1 pilus-mediated invasion
Type 1 pili are fairly ubiquitous, being encoded by the vast majority of UPEC isolates and many other pathogenic and commensal species (68,69). These peritrichous adhesive organelles are made up of a 7-nm wide rod comprising repeating FimA subunits linked to a short 3-nm-wide distal tip fibrillum structure composed of two adapter subunits, FimF and FimG, and the mannose-binding adhesin FimH (70,71). FimH consists of a C-terminal pilin domain, involved in the incorporation of FimH into the tip fibrillum, connected via a short linker peptide to an N-terminal adhesin domain with a carbohydrate-binding pocket (72). UPEC isolates commonly express FimH variants that effectively bind both monomannose and trimannose-containing glycoprotein receptors (73). In contrast, commensal E. coli isolates typically show high affinity binding to only trimannose residues. The high affinity monomannose-binding phenotype is likely selected for within the urinary tract by facilitating UPEC interactions with uroepithelial cells and allowing them to better resist clearance by the bulk flow of urine and possibly other host defenses (74). Several studies have demonstrated that type 1 pili are preeminent virulence factors within the urinary tract (7,75–78). Neutralization of FimH with antibodies or disruption of the fimH adhesin gene greatly impairs the ability of UPEC to colonize the urinary tract, particularly the bladder.
The predominant receptor for monomannose-binding FimH within the urinary tract is believed to be uroplakin 1a (UP1a) (79). In complex with UP2, UP1a forms hexameric complexes that associate with other hexamers comprising at least three additional uroplakin proteins (UP1b, UPIIIa, or UPIIIb) (80). These integral membrane protein complexes further assemble into plaques of approximately 0.5 μm in diameter that cover almost the entire lumenal surface of the bladder epithelium. Assembly of the uroplakin plaques occurs at the trans-Golgi network within large (often >150 μm in diameter), terminally differentiated superficial epithelial cells that line the bladder surface. These superficial cells overlay much smaller immature intermediate and basal epithelial cells. Together, the immature and superficial cells of the bladder epithelium create a formidable permeability barrier, keeping the unwanted constituents of urine from leaching into the host tissue and circulation. In vitro binding assays along with high-resolution microscopy have verified that FimH can bind uroplakin complexes and, more specifically, UP1a (7,79). FimH, however, can also mediate bacterial adherence to a number of other glycosylated and nonglycosylated host receptors, including the matrix-associated proteins type I and type IV collagens, laminin, and fibronectin, components of the glycocalyx that partly covers the bladder surface, the GPI-anchored protein CD48, CEACAM family members, and the highly glycosylated Tamm-Horsfall protein (THP) (81–85).
THP is constitutively released by kidney cells into human urine at high levels (~50 mg/day) where it can act as a soluble FimH receptor, obstructing bacterial–host cell interactions and limiting the ability of UPEC to colonize the urinary tract (83,86,87). Recent research indicates that the inhibitory effects of soluble THP may be countered by the dynamic physics of FimH receptor binding (88). Using computer-intensive steered molecular dynamic simulations coupled with mutational analyses and binding assays, Sokurenko and coworkers found that shear stress, such as might be generated as urine flows past adherent UPEC within the bladder, can actually enhance FimH-mediated bacterial adhesion (88). It was revealed that rapid shear force induces the extension of the FimH interdomain linker, which in turn is predicted to structurally alter the adhesin domain, resulting in increased binding (decreased off-rates) between FimH and mannosylated host receptors. As a consequence of this binding characteristic, FimH should dissociate more readily from soluble receptors that move with the bulk flow of urine and therefore contribute less to the generation of shear stress than would tissue-bound receptors. Within the urinary tract, this phenomenon would presumably favor FimH–mediated UPEC interactions with integral membrane receptors such as UP1a over soluble receptors like THP.
Like Afa/Dr-associated proteins, FimH can also act as an invasin, promoting bacterial entry into bladder epithelial cells as well as mast cells and macrophages (89–91). FimH-mediated bacterial internalization by mast cells and macrophages occurs via interactions with CD48, a GPI-anchored protein expressed primarily by cells of hematopoietic lineage (89,90,92). In contrast, the receptor involved in FimH-mediated bacterial invasion of bladder epithelial cells is currently unknown, although circumstantial evidence suggests that UP1a is a good candidate. High-resolution electron microscopy shows that the apical membrane of mouse bladder superficial cells can zipper around invading UPEC in vivo, apparently via interactions between hexameric uroplakin complexes and the FimH-containing tips of type 1 pili (7). UP1a, being the only uroplakin protein capable of binding FimH in vitro, is arguably responsible for these interactions and for mediating the ‘zipper-like’ invasion process. No study, however, has rigorously proven that UP1a facilitates FimH-mediated bacterial invasion. Indeed, FimH is capable of effectively promoting bacterial entry into a number of epithelial cell lines that do not express UP1a (unpublished observations and (91)). Given the mannose-binding specificity of FimH, it is likely that the adhesin can bind multiple glycoprotein receptors, in addition to UP1a, on host cells throughout the bladder epithelium with varying results. One intriguing possibility is that alternate FimH host receptors can direct invading UPEC into different endocytic pathways, some of which may be more or less beneficial to the intracellular survival and persistence of the microbe.
In contrast to Afa/Dr-mediated bacterial invasion of host cells, FimH-mediated invasion of bladder epithelial cells absolutely requires localized rearrangement of the host actin cytoskeleton (91). Cytochalasin D, a specific inhibitor of F-actin polymerization, blocks FimH-mediated bacterial invasion, but does not affect bacterial adherence. The signal transduction cascade that triggers localized actin cytoskeletal rearrangements during FimH-mediated invasion has been partially determined using pharmacological inhibitors and various dominant-negative mutants of known host cell signaling components (Figure 2). These studies show that FimH-mediated bacterial invasion of cultured bladder cells is dependent upon the activation of protein tyrosine kinases and phosphoinositide-3-kinases (PI-3-kinases) (91,93). PI-3-kinases catalyze the synthesis of the 3-phosphoinositides PI(3)P, PI(3,4)P2, and PI(3,4,5)P3. These lipids are important second messengers that affect host cytoskeletal changes by uncapping barbed actin filaments and by altering either directly or indirectly the cellular localization, conformation and activity of a number of different eukaryotic proteins, including the Rho GTPase Rac1. PI-3-kinase can be stimulated by interactions with both activated Rac1 and focal adhesin kinase (FAK), an important modulator of host cytoskeletal components. The phosphorylation of FAK at tyrosine 397 creates a binding site for other proteins, including Src and PI-3-kinase. In UPEC-infected bladder epithelial cells, phosphorylation of FAK correlates with the formation of transient complexes between FAK and PI-3-kinase and may help boost PI-3-kinase activity. These various processes likely work upstream of complex formation between α-actinin and vinculin, two cytoskeletal components that may help stabilize actin filaments surrounding invading UPEC. Additional Rho-GTPases, including Cdc42 and RhoA, may also facilitate UPEC invasion of bladder cells.
Figure 2. Pathway and consequences of FimH-mediated invasion.
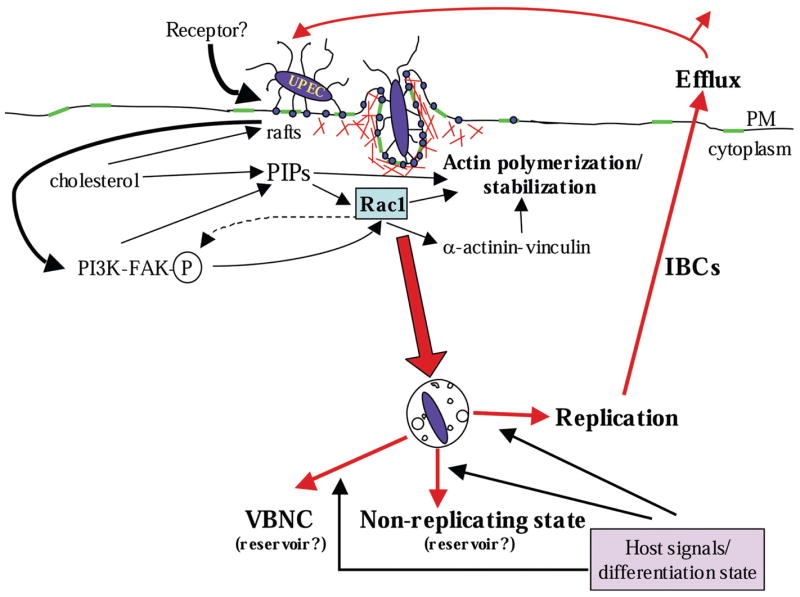
FimH, localized at the distal tips of type 1 pili, bind and cluster unspecified host receptors, possibly concentrated within lipid raft domains (green regions). This triggers a signaling cascade leading to localized rearrangements of F-actin (red), which in turn results in the envelopment and uptake of adherent UPEC. Once internalized, UPEC is trafficked into membrane-bound compartments. From here, undefined signals derived from the host environment (and possibly associated with the host cell differentiation state) trigger UPEC replication, resulting in the formation of intracellular bacterial communities (IBCs) and the eventual efflux of UPEC. Alternatively, UPEC enters a nonreplicating or viable but nonculturable (VBNC) state that can persist for days or weeks, possibly serving as a reservoir for subsequent recurrent acute infections. PIPs phosphoinositides.
As with Afa/Dr-mediated bacterial invasion of host cells, lipid rafts appear to have an important role in FimH-mediated entry. Abraham and coworkers have presented data showing that lipid raft components, including cholesterol and caveolin-1, cluster around type 1-piliated E. coli as they enter bladder epithelial cells, mast cells or macrophages (89,94,95). In addition, the FimH receptors CD48 and UP1a both appear to localize within lipid raft domains. Depletion or sequestration of cholesterol in these cell types, again using either methyl-β-cyclodextrin or filipin, also interferes with FimH-mediated bacterial entry (89,94,95). While the inhibitory effects of disturbing host cell cholesterol distribution with drugs on bacterial entry are often cited as evidence of lipid raft involvement, recent work suggests that these types of data should be interpreted with caution. For example, cholesterol depletion, in addition to disrupting lipid rafts, can alter the localization of phosphoinositides and thereby interfere with actin dynamics at the host cell surface (96,97). This effect could conceivably inhibit bacterial invasion regardless of lipid raft involvement.
Beyond the contribution of cholesterol and lipid rafts, it is not yet clear if the putative signaling pathway depicted in Figure 2 is also relevant to FimH-mediated CD48-dependent bacterial internalization by mast cells or macrophages. Even within the bladder epithelium, the signaling processes that promote FimH-mediated UPEC invasion may differ substantially between bladder epithelial cell types just as the relevant host receptors for FimH may vary. For example, FimH binding to uroplakin plaques that are apically expressed by terminally differentiated superficial bladder cells may trigger an internalization process that is distinct from that associated with less differentiated bladder cells, which often synthesize few, if any, uroplakin complexes. The uroplakin plaques that are elaborated by bladder superficial cells are dynamic structures and may provide a unique vehicle for internalizing bound UPEC. When the bladder fills with urine, it is proposed that uroplakin plaques are deposited in greater numbers on the apical surface of the superficial cells as a means of increasing the available lumenal surface area (80). As the bladder empties, plaques are endocytosed via an ATP-dependent process and subsequently trafficked into late endosomes called multivesicular bodies (MVBs). From the MVBs, most plaques appear to be targeted to lysosomes for degradation, although a few may be recycled back to the cell surface. UPEC, by virtue of FimH–UP1a interactions, may be able to hitchhike along this endocytic pathway. Alternate receptors and entry pathways, and probably alternate bacterial adhesins, may be utilized as the infection progresses and new host cell types are encountered.
Consequences of UPEC Invasion
Host cell invasion may provide UPEC with enhanced protection from both innate and adaptive host defenses, while also facilitating UPEC dissemination and access to nutrients within host tissues. However, evidence supporting the beneficial aspects (or pathogenic aspects from the human perspective) of host cell invasion by UPEC is limited in most cases. Both the Afa/Dr and FimH invasins direct E. coli into membrane-bound vacuoles, none of which has been characterized in much detail (48,54,61,89,91). It is hypothesized, however, that Afa/Dr- and type 1 pili-expressing UPEC strains, by entering host cells through caveolae-like lipid raft domains, may avoid immediate fusion with lysosomes (95). This alone should theoretically prolong intracellular survival of UPEC. Indeed, cell culture-based studies indicate that pathogenic E. coli strains that enter host epithelial cells via an Afa/Dr-dependent pathway can remain viable, sometimes as part of small bacterial clusters, for several days (48,54). Likewise, FimH-mediated entry of UPEC into macrophages can prolong UPEC intracellular survival for at least a few hours, relative to bacteria that are internalized and rapidly killed via an antibody-mediated opsonization route (89). In contrast to these examples, the consequences of FimH-mediated invasion of bladder epithelial cells by UPEC have thus far proven to be more dramatic and less hypothetical.
Intracellular replication and efflux of UPEC
Upon entry into host bladder epithelial cells, culture-based and in vivo studies indicate that UPEC isolates can both multiply and eventually exit the host cells, or they can enter a more quiescent, latent state (see Figure 2) (11,30). Examination of infected, undifferentiated cultured bladder cells using transmission electron microscopy has revealed that UPEC is trafficked into membrane-bound vacuoles where the bacteria can initiate replication and persist for days (11,91). These UPEC-containing vacuoles (which are apparently distinct from autophagosomes) are often morphologically similar to enlarged MVBs, having numerous internal membrane blebs (Figure 3). As seen with many intracellular bacterial pathogens, the UPEC-containing vacuoles are also frequently in close (sometimes intimate) association with mitochondria proximal to the host nucleus. Whether this is the consequence of a default localization pattern for large compartments and organelles within bladder epithelial cells or whether the association with mitochondria is purposeful remains to be determined.
Figure 3. Localization of UPEC within bladder epithelial cells.
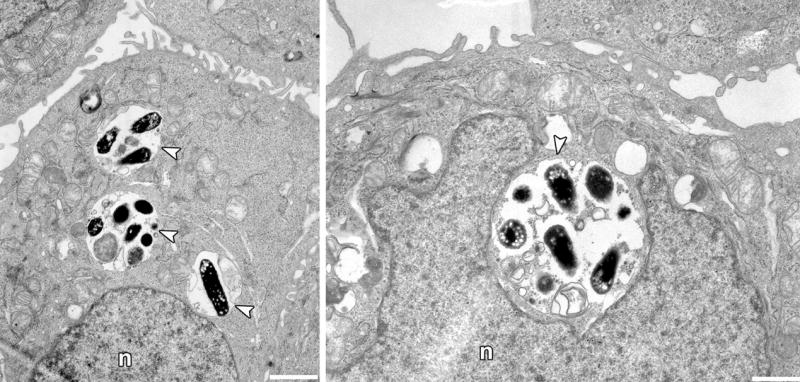
Transmission electron microscopy reveals that the human UPEC cystitis isolate UTI89 is trafficked into compartments with morphologic characteristics of MVBs (arrowheads). Images were taken 24 h postinfection of immortalized human bladder cells designated 5637. After an initial 2-h incubation, the host cell impermeable antibiotic gentamicin was added to the cell culture media to prevent extracellular bacterial growth. Scale bars, 5 μm. n, nucleus.
Bacterial replication assays using bladder cell lines indicate that intracellular growth of UPEC is closely coupled with bacterial efflux – the re-emergence of UPEC from infected host cells (see Figure 2) (11). A similar situation is observed in vivo using mouse UTI model systems (11,30). Within the terminally differentiated epithelial cells of the mouse bladder, UPEC is able to rapidly multiply, forming large inclusions having some biofilm-like characteristics (11,98). Scanning electron microscopy coupled with histologic analyses of infected mouse bladders suggested that UPEC can emerge from within infected superficial cells, as observed in vitro using bladder cell lines (11). The emerging bacteria are often partially septated and impressively long, reaching lengths of greater than 50 μm (Figure 4).
Figure 4. Efflux of filamentous UPEC from a bladder superficial epithelial cell.
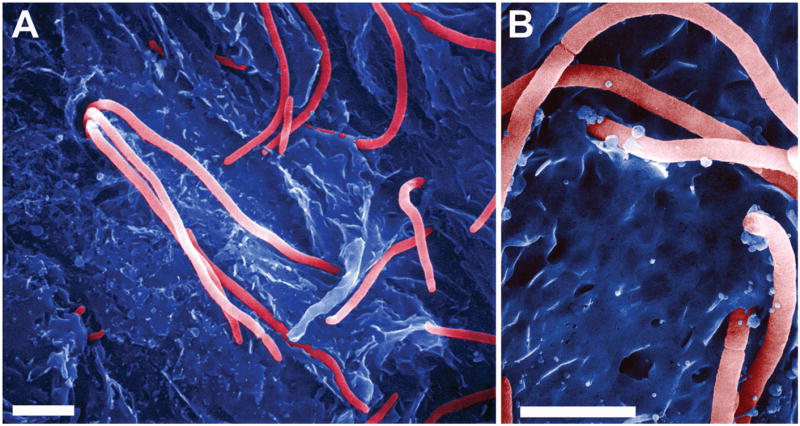
(A and B) Scanning electron microscopy shows filamentous UPEC (a cystitis isolate designated UTI89) emerging from and binding to mouse bladder superficial cells. Scale bars, (A) 5 μm and (B) 3 μm.
Recently, Justice et al. (30) verified and extended these observations using time-lapse fluorescence videomicroscopy of infected mouse bladder explants. The authors observed that UPEC undergoes morphologically and dynamically distinct developmental stages after entering host superficial bladder cells. Upon invading a superficial cell, it was noted that UPEC begins to rapidly multiply, forming large, loosely associated bacterial inclusions that the authors refer to as intracellular bacterial communities (IBCs). At this stage, UPEC within the IBCs are nonmotile, rod-shaped, and reportedly free within the host cytoplasm. Several hours later, much-enlarged IBCs containing slower growing, predominantly coccoid-shaped UPEC were observed, often consuming most of the host cell volume. After an additional 4–6 h, these coccoid-shaped bacteria reverted en masse to their prototypical rod shape and became increasingly motile as they began to stream from the infected host cells. Around this time a subpopulation of filamentous, partially septated UPEC also began to emerge. These filamentous bacteria are resistant to attack by PMNs and may facilitate UPEC dissemination within the bladder by allowing the bacteria to bridge host cells without being released into the flow of urine (11,30). Ironically, these filamentous, PMN-resistant UPEC may arise as a consequence of free radicals and/or the expression of antibacterial peptides that are stimulated as part of the host innate defenses (30,99). While the developmental stages described by Justice et al. (30) were noted at distinct times post infection, the specific timing and extent of these stages likely varies, and probably overlap, depending on inoculum size, the host and bacterial strains used, and perhaps other less defined parameters.
Balancing acts
UPEC replication and dissemination within the bladder epithelium is held in check by a number of innate and adaptive host defense mechanisms. Prominent among these defenses is the activation of Toll-like receptors (TLRs) that are expressed by epithelial and hematopoietic cells within the urinary tract (100). TLRs constitute a family of integral plasma membrane proteins that recognize conserved pathogen-associated molecular patterns, including lipopolysaccharide, peptidoglycan, and flagellin. TLR stimulation triggers downstream signaling events that result in, among other things, acute pro-inflammatory responses via activation of NF-κB and MAP kinases. Among the 11 known TLRs, TLR4 and the recently identified TLR11 have been implicated as important players in the defense of the host against UTIs. In mouse studies, TLR11 has been shown to provide specific protection against UPEC within the kidneys (101). However, the UPEC-associated molecule recognized by TLR11 is currently unknown and the functional relevance of TLR11 in humans, where the tlr11 gene appears to be nonfunctional, is questionable.
The effects of TLR4 activation, in contrast to TLR11, are apparent throughout the urinary tract. TLR4 responds to bacterial lipopolysaccharide and TLR4-deficient mice are exceptionally prone to bladder and kidney infections, being unable to effectively mount a full inflammatory response during the acute phase of infection by UPEC (100,102–104). In the absence of functional TLR4 within the bladder, cytokines are poorly expressed and PMNs, which can normally target UPEC-infected superficial cells with exquisite accuracy, fail to infiltrate into the bladder epithelium (29,100,104). The FimH adhesin, by mediating intimate association between UPEC and its associated LPS with host bladder cells, potently accentuates host inflammatory responses to UPEC (77,105). Part of the heightened inflammatory response to FimH-positive UPEC can be attributed to the ability of these bacteria to invade bladder epithelial cells (104). If FimH-mediated bacterial invasion of bladder cells is prevented by the use of cytochalasin D, host cytokine responses are significantly attenuated. Such observations suggest that FimH-mediated bacterial invasion augments TLR4 signaling and subsequent inflammatory responses, possibly as a result of TLR4 internalization and/or clustering in association with invading type 1 piliated E. coli. In addition, internalized UPEC may amplify pro-inflammatory responses by triggering activation of intracellular host pathogen pattern recognition receptors such as the Nod proteins (106). This situation raises an interesting conundrum: how do UPEC balance the benefits of invading host bladder epithelial cells with the risk of jump-starting host inflammatory responses?
The host, of course, has its own balancing acts to contend with during a UTI, one of which is to maintain the permeability barrier function of the bladder epithelium while fighting off UPEC. Normally, the bladder epithelium has an exceptionally slow turnover rate of many weeks in both humans and mice (107). However, high numbers of exfoliated bladder cells are frequently observed in urine from human UTI patients and from rodents with experimentally induced cystitis (108–111). Mouse studies have demonstrated that superficial bladder cells, when infected with sufficiently large numbers of UPEC, will exfoliate via a rapid apoptosis-like mechanism (7). This exfoliation process functions, at least in part, as a defense mechanism by working to clear infected bladder cells from the host with the flow of urine. The trigger that initiates the exfoliation process is not clear, although lipopolysaccharide has been implicated as an instigator of splotchy bladder cell exfoliation under some experimental conditions (112,113). The down-regulation of NF-κB and MAP kinase pathways in response to UPEC infection may also promote the exfoliation process (114). Independent from, but often concurrent with, bladder cell exfoliation, large numbers of PMNs infiltrate the bladder epithelium within the first several hours after initiation of a UTI (29,104). While both bladder cell exfoliation and the recruitment of PMNs are important innate host defenses, they do have their downsides. The influx of PMNs can compromise the integrity of the urothelium, possibly allowing UPEC to penetrate deeper tissues. Likewise, bladder cell exfoliation can leave underlying tissue exposed and more susceptible to infection by any bacteria that remain within the bladder or that escape from infected host cells prior to completion of the exfoliation process. Indeed, microscopic examination of infected mouse bladders indicates that UPEC can effectively invade the underlying immature cells of the bladder epithelium, although they fail to multiply appreciably within these host cells (unpublished data and (11,30,98)).
To counter the tissue-disrupting effects of bladder cell exfoliation and PMN infiltration, the bladder epithelium rapidly regenerates (7,77). Global gene expression analysis of mouse bladders indicates that shortly after infection with type 1 piliated UPEC, several wound-healing, proliferation and differentiation-associated programs kick in within the bladder epithelium, even before overt tissue damage is apparent (77). This almost preemptive up-regulation of proliferation and differentiation pathways likely ensures that the barrier function of the bladder epithelium is down for only a limited time. A potential consequence of this rapid regeneration process is that UPEC, when it does manage to penetrate deeper cell layers of the bladder epithelium, may find itself concealed within a permeability barrier. This situation may provide UPEC with a degree of protection from both antibiotic treatments and host defenses. In addition, the inability of UPEC to multiply within the immature cells of the bladder may provide further protection, since commonly used antibiotics are often only effective against replicating microbes. It is feasible that UPEC within immature bladder cells are also able to differentiate from a merely nonreplicating state to an even more quiescent status referred to as viable but non-culturable (VBNC). Such VBNC bacteria appear to be living, but fail to grow under standard conditions where they normally would. The existence of VBNC bacteria is controversial, but evidence of their presence within the urinary tract is mounting (115).
It is probable that the intracellular environment and differentiation status of the host bladder cells are primary determinants of whether UPEC can multiply following FimH-mediated internalization (see Figure 2). However, the specific factors that permit UPEC to grow within terminally differentiated bladder epithelial cells but not within their immature counterparts are not yet defined. Emerging data from our lab suggest that one potentially important regulator of UPEC intracellular replication is the host actin cytoskeleton, which differs greatly between the superficial and immature cells of the bladder (unpublished observations and (116)). Regardless of what the inhibitory signals are, the attenuation of UPEC replication within immature bladder cells is likely not permanent. Rather, these nonreplicating microbes may constitute a persistent intracellular reservoir previously speculated to be a hidden source for recurrent and chronic UTIs (11). Differentiation of infected immature bladder cells into superficial cells, as a result of either normal tissue turnover or injury or super-infecting microbes, may stimulate the resurgence of UPEC – triggering their transition out of a nonreplicating or VBNC state.
Conclusions
The data reviewed here highlight three means by which UPEC can enter host cells – via CNF1-, Afa/Dr adhesin- or type 1 pili-mediated processes. However, growing evidence suggests that UPEC strains may utilize additional strategies for host cell invasion. For example, Springall and colleagues (117) recently showed that UPEC could high-jack the host complement system as a means of gaining entry into renal cells, a process that appears to enhance UPEC virulence within the kidneys. The fact that UPEC has evolved multiple mechanisms for invading host cells supports the idea that UPEC are truly opportunistic intracellular pathogens and that an intracellular lifestyle is beneficial to the persistence and eventual dissemination of these microbes. How UPEC takes advantage of the host from within an intracellular niche, however, is poorly understood and major questions continue to arise. Answering these questions will not only shed light on UPEC virulence mechanisms, but will also provide useful insight for combating and preventing chronic and recurrent UTIs.
Acknowledgments
Research in the authors’ laboratory is funded by NIH grants DK068585-01 and DK069526-01.
References
- 1.Janeway CA, Travers P, Walport M, Capra JD. The Immune System in Health and Disease. 4. New York: Garland; 1999. Immunobiology. [Google Scholar]
- 2.LaPenta D, Rubens C, Chi E, Cleary PP. Group A streptococci efficiently invade human respiratory epithelial cells. Proc Natl Acad Sci U S A. 1994;91:12115–12119. doi: 10.1073/pnas.91.25.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osterlund A, Engstrand L. Intracellular penetration and survival of Streptococcus pyogenes in respiratory epithelial cells in vitro. Acta Otolaryngol. 1995;115:685–688. doi: 10.3109/00016489509139387. [DOI] [PubMed] [Google Scholar]
- 4.Burns JL, Jonas M, Chi EY, Clark DK, Berger A, Griffith A. Invasion of respiratory epithelial cells by Burkholderia (Pseudomonas) cepacia. Infect Immun. 1996;64:4054–4059. doi: 10.1128/iai.64.10.4054-4059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen AM, Krogfelt KA. Helicobacter pylori: an invading micro-organism? A review FEMS. Immunol Med Microbiol. 2003;36:117–126. doi: 10.1016/S0928-8244(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 6.St Geme JW., 3rd Molecular and cellular determinants of non-typeable Haemophilus influenzae adherence and invasion. Cell Microbiol. 2002;4:191–200. doi: 10.1046/j.1462-5822.2002.00180.x. [DOI] [PubMed] [Google Scholar]
- 7.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 8.Osterlund A, Popa R, Nikkila T, Scheynius A, Engstrand L. Intracellular reservoir of Streptococcus pyogenes in vivo: a possible explanation for recurrent pharyngotonsillitis. Laryngoscope. 1997;107:640–647. doi: 10.1097/00005537-199705000-00016. [DOI] [PubMed] [Google Scholar]
- 9.Podbielski A, Beckert S, Schattke R, Leithauser F, Lestin F, Gossler B, Kreikemeyer B. Epidemiology and virulence gene expression of intracellular group A streptococci in tonsils of recurrently infected adults. Int J Med Microbiol. 2003;293:179–190. doi: 10.1078/1438-4221-00253. [DOI] [PubMed] [Google Scholar]
- 10.Murphy TF. Bacterial otitis media: pathogenetic considerations. Pediatr Infect Dis J. 2000;19:S9–S15. doi: 10.1097/00006454-200005001-00003. discussion S15–16. [DOI] [PubMed] [Google Scholar]
- 11.Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69:4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schilling JD, Lorenz RG, Hultgren SJ. Effect of trimethoprim-sulfamethoxazole on recurrent bacteriuria and bacterial persistence in mice infected with uropathogenic Escherichia coli. Infect Immun. 2002;70:7042–7049. doi: 10.1128/IAI.70.12.7042-7049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foxman B, Barlow R, D’Arcy H, Gillespie B, Sobel JD. Urinary tract infection: self-reported incidence and associated costs. Ann Epidemiol. 2000;10:509–515. doi: 10.1016/s1047-2797(00)00072-7. [DOI] [PubMed] [Google Scholar]
- 14.Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113(Suppl 1A):5S–13S. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- 15.McBean M, Rajamani S. Increasing rates of hospitalization due to septicemia in the US elderly population, 1986–97. J Infect Dis. 2001;183:596–603. doi: 10.1086/318526. [DOI] [PubMed] [Google Scholar]
- 16.Siegman-Igra Y, Fourer B, Orni-Wasserlauf R, Golan Y, Noy A, Schwartz D, Giladi M. Reappraisal of community-acquired bacteremia: a proposal of a new classification for the spectrum of acquisition of bacteremia. Clin Infect Dis. 2002;34:1431–1439. doi: 10.1086/339809. [DOI] [PubMed] [Google Scholar]
- 17.Kantor AF, Hartge P, Hoover RN, Narayana AS, Sullivan JW, Fraumeni JF., Jr Urinary tract infection and risk of bladder cancer. Am J Epidemiol. 1984;119:510–515. doi: 10.1093/oxfordjournals.aje.a113768. [DOI] [PubMed] [Google Scholar]
- 18.La Vecchia C, Negri E, D’Avanzo B, Savoldelli R, Franceschi S. Genital and urinary tract diseases and bladder cancer. Cancer Res. 1991;51:629–631. [PubMed] [Google Scholar]
- 19.Yamamoto M, Wu HH, Momose H, Rademaker A, Oyasu R. Marked enhancement of rat urinary bladder carcinogenesis by heat-killed Escherichia coli. Cancer Res. 1992;52:5329–5333. [PubMed] [Google Scholar]
- 20.Welch RA, Burland V, Plunkett G, 3rd, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyazaki J, Ba-Thein W, Kumao T, Akaza H, Hayashi H. Identification of a type III secretion system in uropathogenic Escherichia coli. FEMS Microbiol Lett. 2002;212:221–228. doi: 10.1111/j.1574-6968.2002.tb11270.x. [DOI] [PubMed] [Google Scholar]
- 22.Srinivasan U, Foxman B, Marrs CF. Identification of a gene encoding heat-resistant agglutinin in Escherichia coli as a putative virulence factor in urinary tract infection. J Clin Microbiol. 2003;41:285–289. doi: 10.1128/JCM.41.1.285-289.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condron C, Toomey D, Casey RG, Shaffii M, Creagh T, Bouchier-Hayes D. Neutrophil bactericidal function is defective in patients with recurrent urinary tract infections. Urol Res. 2003;31:329–334. doi: 10.1007/s00240-003-0344-z. [DOI] [PubMed] [Google Scholar]
- 24.Brauner A, Jacobson SH, Kuhn I. Urinary Escherichia coli causing recurrent infections – a prospective follow-up of biochemical phenotypes. Clin Nephrol. 1992;38:318–323. [PubMed] [Google Scholar]
- 25.Ikaheimo R, Siitonen A, Heiskanen T, Karkkainen U, Kuosmanen P, Lipponen P, Makela PH. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin Infect Dis. 1996;22:91–99. doi: 10.1093/clinids/22.1.91. [DOI] [PubMed] [Google Scholar]
- 26.Karkkainen UM, Ikaheimo R, Katila ML, Siitonen A. Recurrence of urinary tract infections in adult patients with community-acquired pyelonephritis caused by E.coli: a 1-year follow-up. Scand J Infect Dis. 2000;32:495–499. doi: 10.1080/003655400458767. [DOI] [PubMed] [Google Scholar]
- 27.Russo TA, Stapleton A, Wenderoth S, Hooton TM, Stamm WE. Chromosomal restriction fragment length polymorphism analysis of Escherichia coli strains causing recurrent urinary tract infections in young women. J Infect Dis. 1995;172:440–445. doi: 10.1093/infdis/172.2.440. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson SH, Kuhn I, Brauner A. Biochemical fingerprinting of urinary Escherichia coli causing recurrent infections in women with pyelonephritic renal scarring. Scand J Urol Nephrol. 1992;26:373–377. doi: 10.3109/00365599209181229. [DOI] [PubMed] [Google Scholar]
- 29.Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A. 2000;97:8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, Hultgren SJ. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc Natl Acad Sci U S A. 2004;101:1333–1338. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caprioli A, Falbo V, Roda LG, Ruggeri FM, Zona C. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect Immun. 1983;39:1300–1306. doi: 10.1128/iai.39.3.1300-1306.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387:725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- 33.Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Boquet P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature. 1997;387:729–733. doi: 10.1038/42743. [DOI] [PubMed] [Google Scholar]
- 34.Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun. 1999;67:496–503. doi: 10.1128/iai.67.2.496-503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills M, Meysick KC, O’Brien AD. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect Immun. 2000;68:5869–5880. doi: 10.1128/iai.68.10.5869-5880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung JW, Hong SJ, Kim KJ, Goti D, Stins MF, Shin S, Dawson VL, Dawson TM, Kim KS. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J Biol Chem. 2003;278:16857–16862. doi: 10.1074/jbc.M301028200. [DOI] [PubMed] [Google Scholar]
- 37.Rippere-Lampe KE, O’Brien AD, Conran R, Lockman HA. Mutation of the gene encoding cytotoxic necrotizing factor type 1 cnf (1) attenuates the virulence of uropathogenic Escherichia coli. Infect Immun. 2001;69:3954–3964. doi: 10.1128/IAI.69.6.3954-3964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson DE, Drachenberg C, Lockatell CV, Island MD, Warren JW, Donnenberg MS. The role of cytotoxic necrotizing factor-1 in colonization and tissue injury in a murine model of urinary tract infection. FEMS Immunol Med Microbiol. 2000;28:37–41. doi: 10.1111/j.1574-695X.2000.tb01454.x. [DOI] [PubMed] [Google Scholar]
- 39.Fournout S, Dozois CM, Odin M, Desautels C, Peres S, Herault F, Daigle F, Segafredo C, Laffitte J, Oswald E, Fairbrother JM, Oswald IP. Lack of a role of cytotoxic necrotizing factor 1 toxin from Escherichia coli in bacterial pathogenicity and host cytokine response in infected germfree piglets. Infect Immun. 2000;68:839–847. doi: 10.1128/iai.68.2.839-847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan NA, Wang Y, Kim KJ, Chung JW, Wass CA, Kim KS. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J Biol Chem. 2002;277:15607–15612. doi: 10.1074/jbc.M112224200. [DOI] [PubMed] [Google Scholar]
- 41.Johnson JR, Delavari P, O’Bryan TT. Escherichia coli O18: K1. H7 isolates from patients with acute cystitis and neonatal meningitis exhibit common phylogenetic origins and virulence factor profiles. J Infect Dis. 2001;183:425–434. doi: 10.1086/318086. [DOI] [PubMed] [Google Scholar]
- 42.Doye A, Mettouchi A, Bossis G, Clement R, Buisson-Touati C, Flatau G, Gagnoux L, Piechaczyk M, Boquet P, Lemichez E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell. 2002;111:553–564. doi: 10.1016/s0092-8674(02)01132-7. [DOI] [PubMed] [Google Scholar]
- 43.Lerm M, Pop M, Fritz G, Aktories K, Schmidt G. Proteasomal degradation of cytotoxic necrotizing factor 1-activated rac. Infect Immun. 2002;70:4053–4058. doi: 10.1128/IAI.70.8.4053-4058.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulvey MA, Hultgren SJ. Encyclopedia of Microbiology. London: Academic Press; 2000. Adhesion, Bacterial; pp. 42–52. [Google Scholar]
- 45.Nowicki B, Selvarangan R, Nowicki S. Family of Escherichia coli Dr adhesins: decay-accelerating factor receptor recognition and invasiveness. J Infect Dis. 2001;183(Suppl 1):S24–S27. doi: 10.1086/318846. [DOI] [PubMed] [Google Scholar]
- 46.Anderson KL, Billington J, Pettigrew D, Cota E, Simpson P, Roversi P, Chen HA, Urvil P, du Merle L, Barlow PN, Medof ME, Smith RA, Nowicki B, Le Bouguenec C, Lea SM, et al. An atomic resolution model for assembly, architecture, and function of the Dr adhesins. Mol Cell. 2004;15:647–657. doi: 10.1016/j.molcel.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 47.Berger CN, Billker O, Meyer TF, Servin AL, Kansau I. Differential recognition of members of the carcinoembryonic antigen family by Afa/Dr adhesins of diffusely adhering Escherichia coli (Afa/Dr DAEC) Mol Microbiol. 2004;52:963–983. doi: 10.1111/j.1365-2958.2004.04033.x. [DOI] [PubMed] [Google Scholar]
- 48.Plancon L, Du Merle L, Le Friec S, Gounon P, Jouve M, Guignot J, Servin A, Le Bouguenec C. Recognition of the cellular beta1-chain integrin by the bacterial AfaD invasin is implicated in the internalization of afa-expressing pathogenic Escherichia coli strains. Cell Microbiol. 2003;5:681–693. doi: 10.1046/j.1462-5822.2003.00308.x. [DOI] [PubMed] [Google Scholar]
- 49.Guignot J, Bernet-Camard MF, Pous C, Plancon L, Le Bouguenec C, Servin AL. Polarized entry of uropathogenic Afa/Dr diffusely adhering Escherichia coli strain IH11128 into human epithelial cells: evidence for α5β1 integrin recognition and subsequent internalization through a pathway involving caveolae and dynamic unstable microtubules. Infect Immun. 2001;69:1856–1868. doi: 10.1128/IAI.69.3.1856-1868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westerlund B, Kuusela P, Risteli J, Risteli L, Vartio T, Rauvala H, Virkola R, Korhonen TK. The O75X adhesin of uropathogenic Escherichia coli is a type IV collagen-binding protein. Mol Microbiol. 1989;3:329–337. doi: 10.1111/j.1365-2958.1989.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 51.Selvarangan R, Goluszko P, Singhal J, Carnoy C, Moseley S, Hudson B, Nowicki S, Nowicki B. Interaction of Dr adhesin with collagen type IV is a critical step in Escherichia coli renal persistence. Infect Immun. 2004;72:4827–4835. doi: 10.1128/IAI.72.8.4827-4835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goluszko P, Niesel D, Nowicki B, Selvarangan R, Nowicki S, Hart A, Pawelczyk E, Das M, Urvil P, Hasan R. Dr operon-associated invasiveness of Escherichia coli from pregnant patients with pyelonephritis. Infect Immun. 2001;69:4678–4680. doi: 10.1128/IAI.69.7.4678-4680.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hart A, Nowicki BJ, Reisner B, Pawelczyk E, Goluszko P, Urvil P, Anderson G, Nowicki S. Ampicillin-resistant Escherichia coli in gestational pyelonephritis: increased occurrence and association with the colonization factor Dr adhesin. J Infect Dis. 2001;183:1526–1529. doi: 10.1086/320196. [DOI] [PubMed] [Google Scholar]
- 54.Selvarangan R, Goluszko P, Popov V, Singhal J, Pham T, Lublin DM, Nowicki S, Nowicki B. Role of decay-accelerating factor domains and anchorage in internalization of Dr-fimbriated Escherichia coli. Infect Immun. 2000;68:1391–1399. doi: 10.1128/iai.68.3.1391-1399.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang L, Nowicki BJ, Urvil P, Goluszko P, Nowicki S, Young SL, Yallampalli C. Epithelial invasion by Escherichia coli bearing Dr fimbriae is controlled by nitric oxide-regulated expression of CD55. Infect Immun. 2004;72:2907–2914. doi: 10.1128/IAI.72.5.2907-2914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundberg JO, Ehren I, Jansson O, Adolfsson J, Lundberg JM, Weitzberg E, Alving K, Wiklund NP. Elevated nitric oxide in the urinary bladder in infectious and noninfectious cystitis. Urology. 1996;48:700–702. doi: 10.1016/S0090-4295(96)00423-2. [DOI] [PubMed] [Google Scholar]
- 57.Poljakovic M, Svensson ML, Svanborg C, Johansson K, Larsson B, Persson K. Escherichia coli-induced inducible nitric oxide synthase and cyclooxygenase expression in the mouse bladder and kidney. Kidney Int. 2001;59:893–904. doi: 10.1046/j.1523-1755.2001.059003893.x. [DOI] [PubMed] [Google Scholar]
- 58.Tieng V, Le Bouguenec C, du Merle L, Bertheau P, Desreumaux P, Janin A, Charron D, Toubert A. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A. 2002;99:2977–2982. doi: 10.1073/pnas.032668099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Betis F, Brest P, Hofman V, Guignot J, Bernet-Camard MF, Rossi B, Servin A, Hofman P. The Afa/Dr adhesins of diffusely adhering Escherichia coli stimulate interleukin-8 secretion, activate mitogen-activated protein kinases, and promote polymorphonuclear transepithelial migration in T84 polarized epithelial cells. Infect Immun. 2003;71:1068–1074. doi: 10.1128/IAI.71.3.1068-1074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boulton IC, Gray-Owen SD. Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat Immunol. 2002;3:229–236. doi: 10.1038/ni769. [DOI] [PubMed] [Google Scholar]
- 61.Jouve M, Garcia MI, Courcoux P, Labigne A, Gounon P, Le Bouguenec C. Adhesion to and invasion of HeLa cells by pathogenic Escherichia coli carrying the afa-3 gene cluster are mediated by the AfaE and AfaD proteins, respectively. Infect Immun. 1997;65:4082–4089. doi: 10.1128/iai.65.10.4082-4089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goluszko P, Popov V, Selvarangan R, Nowicki S, Pham T, Nowicki BJ. Dr fimbriae operon of uropathogenic Escherichia coli mediate microtubule-dependent invasion to the HeLa epithelial cell line. J Infect Dis. 1997;176:158–167. doi: 10.1086/514018. [DOI] [PubMed] [Google Scholar]
- 63.Kansau I, Berger C, Hospital M, Amsellem R, Nicolas V, Servin AL, Bernet-Camard MF. Zipper-like internalization of Dr-positive Escherichia coli by epithelial cells is preceded by an adhesin-induced mobilization of raft-associated molecules in the initial step of adhesion. Infect Immun. 2004;72:3733–3742. doi: 10.1128/IAI.72.7.3733-3742.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCaw SE, Liao EH, Gray-Owen SD. Engulfment of Neisseria gonorrhoeae: revealing distinct processes of bacterial entry by individual carcinoembryonic antigen-related cellular adhesion molecule family receptors. Infect Immun. 2004;72:2742–2752. doi: 10.1128/IAI.72.5.2742-2752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clausen JD, Christiansen G, Holst HU, Birkelund S. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol Microbiol. 1997;25:441–449. doi: 10.1046/j.1365-2958.1997.4591832.x. [DOI] [PubMed] [Google Scholar]
- 66.Oelschlaeger TA, Guerry P, Kopecko DJ. Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc Natl Acad Sci U S A. 1993;90:6884–6888. doi: 10.1073/pnas.90.14.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Young VB, Falkow S, Schoolnik GK. The invasin protein of Yersinia enterocolitica: internalization of invasin-bearing bacteria by eukaryotic cells is associated with reorganization of the cytoskeleton. J Cell Biol. 1992;116:197–207. doi: 10.1083/jcb.116.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brinton CC. Non-flagellar appendages of bacteria. Nature. 1959;183:782–786. doi: 10.1038/183782a0. [DOI] [PubMed] [Google Scholar]
- 69.Buchanan K, Falkow S, Hull RA, Hull SI. Frequency among Enterobacteriaceae of the DNA sequences encoding type 1 pili. J Bacteriol. 1985;162:799–803. doi: 10.1128/jb.162.2.799-803.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Russell PW, Orndorff PE. Lesions in two Escherichia coli type 1 pilus genes alter pilus number and length without affecting receptor binding. J Bacteriol. 1992;174:5923–5935. doi: 10.1128/jb.174.18.5923-5935.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones CH, Pinkner JS, Roth R, Heuser J, Nicholes AV, Abraham SN, Hultgren SJ. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc Natl Acad Sci U S A. 1995;92:2081–2085. doi: 10.1073/pnas.92.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choudhury D, Thompson A, Stojanoff V, Langermann S, Pinkner J, Hultgren SJ, Knight SD. X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science. 1999;285:1061–1066. doi: 10.1126/science.285.5430.1061. [DOI] [PubMed] [Google Scholar]
- 73.Sokurenko EV, Courtney HS, Maslow J, Siitonen A, Hasty DL. Quantitative differences in adhesiveness of type 1 fimbriated Escherichia coli due to structural differences in fimH genes. J Bacteriol. 1995;177:3680–3686. doi: 10.1128/jb.177.13.3680-3686.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sokurenko EV, Chesnokova V, Dykhuizen DE, Ofek I, Wu XR, Krogfelt KA, Struve C, Schembri MA, Hasty DL. Pathogenic adaptation of Escherichia coli by natural variation of the FimH adhesin. Proc Natl Acad Sci U S A. 1998;95:8922–8926. doi: 10.1073/pnas.95.15.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Connell H, Agace W, Klemm P, Schembri M, Marild S, Svanborg C. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci USA. 1996;93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, Hultgren SJ. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science. 1997;276:607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- 77.Mysorekar IU, Mulvey MA, Hultgren SJ, Gordon JI. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J Biol Chem. 2002;277:7412–7419. doi: 10.1074/jbc.M110560200. [DOI] [PubMed] [Google Scholar]
- 78.Bahrani-Mougeot FK, Buckles EL, Lockatell CV, Hebel JR, Johnson DE, Tang CM, Donnenberg MS. Type 1 fimbriae and extracellular polysaccharides are preeminent uropathogenic Escherichia coli virulence determinants in the murine urinary tract. Mol Microbiol. 2002;45:1079–1093. doi: 10.1046/j.1365-2958.2002.03078.x. [DOI] [PubMed] [Google Scholar]
- 79.Zhou G, Mo WJ, Sebbel P, Min G, Neubert TA, Glockshuber R, Wu XR, Sun TT, Kong XP. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J Cell Sci. 2001;114:4095–4103. doi: 10.1242/jcs.114.22.4095. [DOI] [PubMed] [Google Scholar]
- 80.Apodaca G. The uroepithelium: not just a passive barrier. Traffic. 2004;5:117–128. doi: 10.1046/j.1600-0854.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- 81.Pouttu R, Puustinen T, Virkola R, Hacker J, Klemm P, Korhonen TK. Amino acid residue Ala-62 in the FimH fimbrial adhesin is critical for the adhesiveness of meningitis-associated Escherichia coli to collagens. Mol Microbiol. 1999;31:1747–1757. doi: 10.1046/j.1365-2958.1999.01311.x. [DOI] [PubMed] [Google Scholar]
- 82.Kukkonen M, Raunio T, Virkola R, Lahteenmaki K, Makela PH, Klemm P, Clegg S, Korhonen TK. Basement membrane carbohydrate as a target for bacterial adhesion: binding of type I fimbriae of Salmonella enterica and Escherichia coli to laminin. Mol Microbiol. 1993;7:229–237. doi: 10.1111/j.1365-2958.1993.tb01114.x. [DOI] [PubMed] [Google Scholar]
- 83.Pak J, Pu Y, Zhang ZT, Hasty DL, Wu XR. Tamm-Horsfall protein binds to type 1 fimbriated Escherichia coli and prevents E. coli from binding to uroplakin Ia and Ib receptors. J Biol Chem. 2001;276:9924–9930. doi: 10.1074/jbc.M008610200. [DOI] [PubMed] [Google Scholar]
- 84.Sauter SL, Rutherfurd SM, Wagener C, Shively JE, Hefta SA. Identification of the specific oligosaccharide sites recognized by type 1 fimbriae from Escherichia coli on nonspecific cross-reacting antigen, a CD66 cluster granulocyte glycoprotein. J Biol Chem. 1993;268:15510–15516. [PubMed] [Google Scholar]
- 85.Sokurenko EV, Courtney HS, Abraham SN, Klemm P, Hasty DL. Functional heterogeneity of type 1 fimbriae of Escherichia coli. Infect Immun. 1992;60:4709–4719. doi: 10.1128/iai.60.11.4709-4719.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bates JM, Raffi HM, Prasadan K, Mascarenhas R, Laszik Z, Maeda N, Hultgren SJ, Kumar S. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: rapid communication. Kidney Int. 2004;65:791–797. doi: 10.1111/j.1523-1755.2004.00452.x. [DOI] [PubMed] [Google Scholar]
- 87.Mo L, Zhu XH, Huang HY, Shapiro E, Hasty DL, Wu XR. Ablation of the Tamm-Horsfall protein gene increases susceptibility of mice to bladder colonization by type 1-fimbriated Escherichia coli. Am J Physiol Renal Physiol. 2004;286:F795–F802. doi: 10.1152/ajprenal.00357.2003. [DOI] [PubMed] [Google Scholar]
- 88.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109:913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 89.Baorto DM, Gao Z, Malaviya R, Dustin ML, van der Merwe A, Lublin DM, Abraham SN. Survival of FimH-expressing enterobacteria in macrophages relies on glycolipid traffic. Nature. 1997;389:636–639. doi: 10.1038/39376. [DOI] [PubMed] [Google Scholar]
- 90.Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- 91.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19:2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Malaviya R, Gao Z, Thankavel K, van der Merwe PA, Abraham SN. The mast cell tumor necrosis factor alpha response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proc Natl Acad Sci U S A. 1999;96:8110–8115. doi: 10.1073/pnas.96.14.8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martinez JJ, Hultgren SJ. Requirement of Rho-family GTPases in the invasion of Type 1-piliated uropathogenic Escherichia coli. Cell Microbiol. 2002;4:19–28. doi: 10.1046/j.1462-5822.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- 94.Duncan MJ, Li G, Shin JS, Carson JL, Abraham SN. Bacterial penetration of bladder epithelium through lipid rafts. J Biol Chem. 2004;279:11088–11095. doi: 10.1074/jbc.M400769200. [DOI] [PubMed] [Google Scholar]
- 95.Shin JS, Abraham SN. Caveolae as portals of entry for microbes. Microbes Infect. 2001;3:755–761. doi: 10.1016/s1286-4579(01)01423-x. [DOI] [PubMed] [Google Scholar]
- 96.Grimmer S, Van Deurs B, Sandvig K. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J Cell Sci. 2002;115:2953–2962. doi: 10.1242/jcs.115.14.2953. [DOI] [PubMed] [Google Scholar]
- 97.Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci U S A. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 99.Rosenberger CM, Gallo RL, Finlay BB. Interplay between antibacterial effectors: a macrophage antimicrobial peptide impairs intracellular Salmonella replication. Proc Natl Acad Sci U S A. 2004;101:2422–2427. doi: 10.1073/pnas.0304455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schilling JD, Martin SM, Hung CS, Lorenz RG, Hultgren SJ. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2003;100:4203–4208. doi: 10.1073/pnas.0736473100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 102.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun. 1998;66:2798–2802. doi: 10.1128/iai.66.6.2798-2802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg Eden C. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun. 1984;46:839–844. doi: 10.1128/iai.46.3.839-844.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schilling JD, Mulvey MA, Vincent CD, Lorenz RG, Hultgren SJ. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J Immunol. 2001;166:1148–1155. doi: 10.4049/jimmunol.166.2.1148. [DOI] [PubMed] [Google Scholar]
- 105.Schilling JD, Martin SM, Hunstad DA, Patel KP, Mulvey MA, Justice SS, Lorenz RG, Hultgren SJ. CD14- and Toll-like receptor-dependent activation of bladder epithelial cells by lipopolysaccharide and type 1 piliated Escherichia coli. Infect Immun. 2003;71:1470–1480. doi: 10.1128/IAI.71.3.1470-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip. intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 2003;5:581–592. doi: 10.1046/j.1462-5822.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 107.Jost SP. Cell cycle of normal bladder urothelium in developing and adult mice. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;57:27–36. doi: 10.1007/BF02899062. [DOI] [PubMed] [Google Scholar]
- 108.Fukushi Y, Orikasa S, Kagayama M. An electron microscopic study of the interaction between vesical epitherlium and E. coli. Invest Urol. 1979;17:61–68. [PubMed] [Google Scholar]
- 109.Elliott TS, Reed L, Slack RC, Bishop MC. Bacteriology and ultrastructure of the bladder in patients with urinary tract infections. J Infect. 1985;11:191–199. doi: 10.1016/s0163-4453(85)92997-4. [DOI] [PubMed] [Google Scholar]
- 110.McTaggart LA, Rigby RC, Elliott TS. The pathogenesis of urinary tract infections associated with Escherichia coli, Staphylococcus saprophyticus and S. epidermidis. J Med Microbiol. 1990;32:135–141. doi: 10.1099/00222615-32-2-135. [DOI] [PubMed] [Google Scholar]
- 111.McTaggart LA, Elliott TS. What makes microbes stick? Lancet. 1989;1:324. doi: 10.1016/s0140-6736(89)91329-9. [DOI] [PubMed] [Google Scholar]
- 112.Aronson M, Medalia O, Amichay D, Nativ O. Endotoxin-induced shedding of viable uroepithelial cells is an antimicrobial defense mechanism. Infect Immun. 1988;56:1615–1617. doi: 10.1128/iai.56.6.1615-1617.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jezernik K, Medalia O, Aronson M. A comparative study of the desquamation of urothelial cells during gestation and in adults mice following moderate stress or endotoxin treatment. Cell Biol Int. 1995;19:887–893. doi: 10.1006/cbir.1995.1026. [DOI] [PubMed] [Google Scholar]
- 114.Klumpp DJ, Weiser AC, Sengupta S, Forrestal SG, Batler RA, Schaeffer AJ. Uropathogenic Escherichia coli potentiates type 1 pilus-induced apoptosis by suppressing NF-κB. Infect Immun. 2001;69:6689–6695. doi: 10.1128/IAI.69.11.6689-6695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anderson M, Bollinger D, Hagler A, Hartwell H, Rivers B, Ward K, Steck TR. Viable but nonculturable bacteria are present in mouse and human urine specimens. J Clin Microbiol. 2004;42:753–758. doi: 10.1128/JCM.42.2.753-758.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Romih R, Veranic P, Jezernik K. Actin filaments during terminal differentiation of urothelial cells in the rat urinary bladder. Histochem Cell Biol. 1999;112:375–380. doi: 10.1007/s004180050419. [DOI] [PubMed] [Google Scholar]
- 117.Springall T, Sheerin NS, Abe K, Holers VM, Wan H, Sacks SH. Epithelial secretion of C3 promotes colonization of the upper urinary tract by Escherichia coli. Nat Med. 2001;7:801–806. doi: 10.1038/89923. [DOI] [PubMed] [Google Scholar]