

Abstract
Protein microarrays are the format of choice for high-throughput, high-content protein interaction analysis. In most of the array formats, reporter molecules are used in multi-step detection of the protein interactions. Among the few existing label-free detection approaches, Surface Plasmon Resonance (SPR) and Mass Spectrometry stand out as most promising for utilization in protein microarrays, albeit both have been used only sporadically for high-content protein arrays. Shown here for the first time is the combination of SPR and MS detection on a single high-content protein microarray. Antibodies to five human plasma proteins were arrayed in a 10 × 10 spot arrangement on a chemically activated gold-coated glass chip. Binding of proteins to their corresponding antibodies was monitored via SPR imaging across the entire surface of the chip. Following protein affinity retrieval, the chip was overlaid with MALDI matrix and MS analyzed, producing protein-specific mass spectra from distinct spots on the array. The SPR-MS dual detection is well suited for high-content protein microarrays and comprehensive protein analysis – from quantitative assessment of the protein concentration to detection of structural protein variants arising from genetic variations and post expression processing.
Proteomics approaches capable of high-throughput analysis and delivering high-content data are needed to address the many facets of the human proteome. Currently there are two approaches that meet these objectives: mass spectrometry (MS) -based methods, and protein microarrays. While MS enables rapid structural characterization and identification of hundreds of proteins on a large scale 1, 2, protein microarrays are effectively becoming the format of choice for high-throughput protein quantification and protein interactions screening 3, 4. Nonetheless, neither method is an all-in-one solution for protein assaying. MS-based detection usually generates highly convoluted, content-rich, and quantification-challenging data that often times requires powerful informatics approaches for protein data interpellation. Microarrays, on the other hand, except for the high-throughput and miniaturization benefits, rarely provide more content than traditional methods of protein quantification (i.e., structural modifications are not assessed in typical reporter molecule-based detection schemes). It is thus beneficial to create high-throughput, high-content protein microarrays capable of delivering a complete protein analysis – from quantitative assessment of the protein concentration, to detection of structural protein variants. To achieve this goal, multiplex detection has to be utilized because no single technique is capable of providing such all-inclusive information. Combination of a quantitative protein detection method with qualitative mass spectrometry analysis would deliver the desired outcome. Surface plasmon resonance (SPR) is a label-free method of quantitative protein analysis that is ideally suited for combination with MS detection: it doesn't introduce additional variables (e.g. labels) that might interfere with downstream MS analysis, and it is non-destructive, leaving the proteins intact and unmodified. SPR exploits the interactions of light photons with free electrons (surface plasmons) on a metal surface to quantify the changes in protein concentration on the same metal surface 5. Given proper preparation and treatment, the same surface that is used to capture and quantify the proteins via SPR, can be used as a probe for subsequent MALDI-TOF MS analysis. MS analysis of proteins directly from SPR sensor chip surfaces has been well established 6-12. However, the current SPR-MS approach is low in throughput, typically involving analysis of 2-4 sites on a single SPR chip 13. To overcome this limitation, a true SPR-MS array platform was developed in this work. Simultaneous monitoring of protein binding to multiple spots on a high-content protein array was enabled by SPR Imaging 14, 15. In SPR Imaging the change in the light intensity reflected from the metal surface is measured at a fixed angle and wavelength. Hence, arrays of molecules can be analyzed in parallel over the entire array surface. For the proof of principle experiments, antibodies to five proteins were arrayed in a 10×10 arrangement on an activated-gold chip surface. Binding of the proteins to their corresponding antibody spots was monitored via SPR Imaging, and was followed by MALDI-TOF MS analysis of specific spots on the array. The results obtained demonstrate the feasibility and the high-throughput capability of the combined SPR-MS protein array platform.
Experimental Section
Reagents
Rabbit anti-human polyclonal antibodies to beta 2 microglobulin (B2m, A0072, 11 g/L), cystatin C (CysC, A0451, 17 g/L), C-reactive protein (CRP, A0073, 8.3 g/L), transferrin (TRFE, A0061, 14 g/L), and transthyretin (TTR, A0002, 3.9 g/L) were obtained from DAKO (Carpinteria, CA, USA). Prior to arraying, the TTR antibody was diluted 10-fold, and the b2m, CysC, CRP, and TRFE antibodies 20-fold in 10 mM sodium acetate (pH 5.0). Purified beta-2-microglobulin (Cat. No. 475823), cystatin C (Cat. No. 240896), CRP (Cat. No. 236603) and transthyretin (Cat. No. 529577) antigens were purchased from Calbiochem (San Diego, CA, USA). Purified transferrin antigen (Cat. No. T2252) was obtained from Sigma (St. Louis, MO, USA). All antigens were diluted to a final concentration of 100 nM with 10 mM HEPES buffer (pH 7.4) containing 150 mM NaCl, 3 mM EDTA, and 0.005% v/v polysorbate (HBS buffer).
Chips
Plain-gold chips (SPR-1000-050) were obtained from GWC Technologies (Madison, WI, USA). The chips were washed with ethanol and incubated overnight in a solution of 10 mg/mL 11-mecraptoundecanoic acid (Aldrich), in ethanol, to allow for the formation of self-assembled monolayers. The surface carboxyl groups were then activated with 1,1″-carbonyldiimidazole (CDI, Aldrich, overnight incubation in a 10 mg/mL acetone solution). Prior to the arraying, the chips were rinsed in acetone and dried under nitrogen stream. The chips were arrayed immediately after their activation was completed.
Antibody Arraying
The chips were arrayed in a SpotBot Protein Edition Personal Microarrayer (Telechem International Inc., Sunnyvale, CA), equipped with humidity control apparatus and megasonic wash station. During the arraying, the humidity was controlled at 40-55%, and the platen temperature was set at 15° C by a thermal control bath. The four-pin print head of the arrayer was fitted with a single SMP6 Stealth microspotting pin (Telechem International), producing spots with 200 μm diameter. The activated chips were positioned in a pre-determined area on the platen, and fixed by an adhesive tape. Ten μl aliquot of an antibody solution was placed in a single well of a 384-well microtiter plate positioned within the arrayer enclosure. The antibodies were arrayed in a single chip using one arraying run consisting of five spotting cycles (one per antibody). A single cycle consisted of: drawing 0.25 μL of the antibody solution with the SMP6 pin, arraying 20 spots in 2 columns of 10 spots on the chip (with 450 μm spacing between each spot), and washing and drying the pin four times in preparation for the next cycle. A total of 100 spots (10 × 10) were arrayed in a single run on an individual chip, in ∼10 min, covering an area of 4.25 × 4.25 mm in the middle of the chip. The arrayed chips were then rinsed with three 200 μL-water aliquots, dried under nitrogen, and inserted into the SPR Imager.
SPR imaging
The SPR Imaging was performed on an SPR Imager II instrument (GWC Technologies), at ambient temperature (23-24°C). Individual chips were placed into the sample holder/flow cell assembly, and the flow cell was filled with HBS buffer via peristaltic pump driven flow through the inlet tubing to the flow cell (0.5 mL was needed to completely fill the tubing and flow cell with buffer or analyte). The buffer flow was then stopped and the chip was incubated with the buffer for ∼1 h to allow for complete CDI inactivation. Next, the background pixel intensity of the array spots was adjusted to the optimum level for protein binding detection (∼1/3 of the maximum value) with the help of the Digital Optics V++ Version 4 software (Auckland, New Zealand). A 30-frames image was taken (with the buffer in the flow cell) to serve as a reference (pre-binding) image. The inlet tubing of the flow cell was then placed in a vial containing 100 mM solution of the specific protein(s), and the flow cell was filled with the protein(s) solution. After 10 min incubation, the inlet tube was placed in a HBS buffer vial, the flow cell was filled with buffer, the buffer flow was stopped, and a 30-frames post-binding image was taken. The reference pre-binding image was subtracted from the post-binding image to obtain a difference image that shows specific protein binding. To regenerate the chip surface, a solution of 60 mM HCL was incubated in the flow cell for 5 min.
Mass Spectrometry
Following the final protein binding step, the arrayed chip was removed from the SPR imaging instrument, washed with three 200 μL aliquots of water, and dried under stream of nitrogen. MALDI matrix was applied to the surface of the chips as a thin mist via an aerosol-spraying device, covering the entire surface of the chip, desorbing the proteins from their respective antibody spots and, on rapid drying, redepositing the matrix-protein mixture on the same spots 16. Aqueous solution of α-cyano-4-hydroxycinnamic acid (ACCA), in 33% (v/v) acetonitrile, 0.2 % (v/v) trifluoroacetic acid was used as a MALDI matrix. The matrix was processed by powder-flash recrystallization from a low-heat saturated acetone solution of the original stock (Aldrich, Cat. No. 47,687-0). The matrix-overlaid chip array was affixed to a slightly recessed MALDI target (milled to accommodate the depth of the glass chip) with a double-sided tape, and placed into an Autoflex II MALDI-TOF mass spectrometer (Bruker Daltonics, Billerica, MA). Linear spectra were acquired with a delayed extraction mode using a 1.35 kV draw out pulse, 400 ns delay, and a full accelerating potential of 20.00 kV. Thirty-five laser shots were acquired from the same position within a spot on the array for each individual spectrum. The width of the laser was ∼ 100 μm so that each laser shot fit within the confines of the spots (200 μm diameter). Representative spectra were taken from all areas of the array. The spectra were then viewed using the Proteome Analyzer software (Intrinsic Bioprobes Inc., Tempe, AZ).
Results and Discussion
Several 100 × 100 antibody arrays were made and utilized for the SPR-MS experiments. To verify the binding activity of the arrayed antibodies, individual proteins were injected over the arrays and their binding monitored via SPR Imaging. The concentration of each protein in the test solution was 100 nM, which is well below their physiological plasma concentrations 17, 18. The difference SPR images resulting from binding of each protein to a single antibody array are shown in Fig. 1. All five proteins bound specifically to their corresponding antibodies spots. There was some cross reactivity of TTR with anti-CRP, and CRP with anti-TRFE, but these signals were negligible in comparison to the SPR signals resulting from binding to their target antibodies. The signal intensities in the SPR images in Fig. 1 are different for each antibody-antigen pair: they are lowest for b2m, and highest for TRFE. This increase in signal intensity with the molecular mass of the protein is expected because SPR detection registers the change of total mass on the chip surface. Hence, for similar mole amounts of proteins affinity retrieved on the antibody spots, the SPR signal will increase proportionally with the molecular mass of the proteins. Even thought the SPR imaging signals for b2m (and to some extent cysC) in Fig.1 appear to be relatively weak, with proper experimental design and prolonged incubation period it is possible to detect these two low molecular weight proteins via SPR Imaging at concentrations as low as 1 nM 19.
Fig.1.
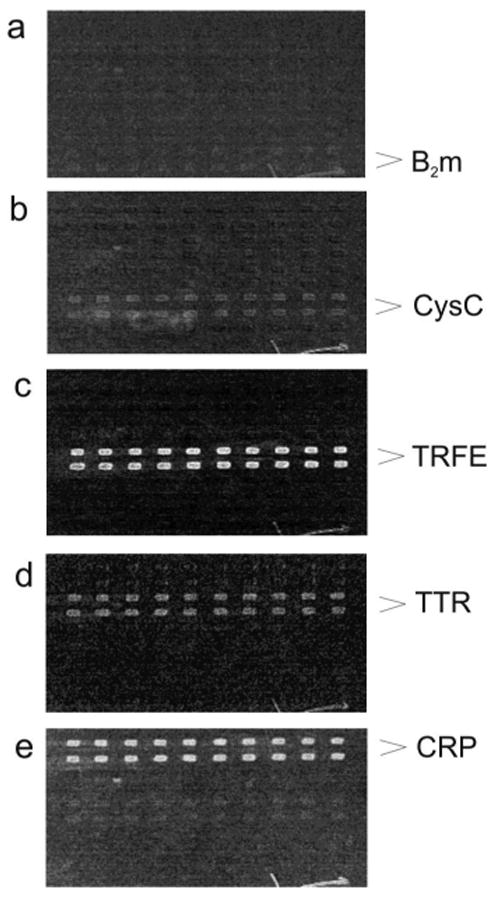
SPR images resulting from binding of (a) b2m, (b) CysC, (c) TRFE, (d) TTR, and (e) CRP (all at 100 nM concentrations) to a single 10×10 antibody array containing all five antibodies.
The durability of the arrays and reproducibility of the binding was also examined. After several days of SPR analysis on a single antibody array in the SPR Imager, and repetitive injections and regenerations with 60 mM HCl, the antibody spots retained their binding capacity, indicating robust covalent attachment of the antibodies to the CDI-activated surface, and complete regeneration of the antibody surface.
Mass spectrometry was performed on all antibody arrayed chips following affinity capture of the proteins in the SPR Imager. The MS analyses were performed on a commercially available Bruker MALDI TOF instrument, as opposed to all of the previous SPR-MS experiments from this laboratory where in-house made mass spectrometer was used. The Bruker mass spectrometer has a wide (96-well format) target which can easily accommodate the 17 × 17 mm chip. The 10×10 spots area on the chip was marked (a square was mechanically etched in the glass around the array) for easier location of the array area in the mass spectrometer (once the matrix was applied the array spots became less visible to the naked eye and to the internal camera of the Bruker instrument). A representative set of SPR-MS data obtained from a single antibody array is shown in Figure 2. An SPR image of the 10×10 antibody array taken after an injection of solutions containing all five proteins (at 100 mM each) shows binding to all spots on the array, with signal intensities similar to those observed for the injections of each individual protein. The representative mass spectra obtained from the same protein-bound antibody array show specific multiply-charged protein ions, without any trace of non-specific binding or cross reactivity. There is a noticeable trend of decreasing spectra quality with increasing MW of the protein as the MALDI matrix used (ACCA) was better suited for ionization and analysis of the smaller MW proteins (ACCA was chosen because it is better energy-absorbing matrix than sinapic acid, and is more suitable for the matrix application method used). Hence, even though the SPR imaging signals were weakest for b2m and cysC, these two small MW proteins produced the best S/N mass spectra.
Fig.2.
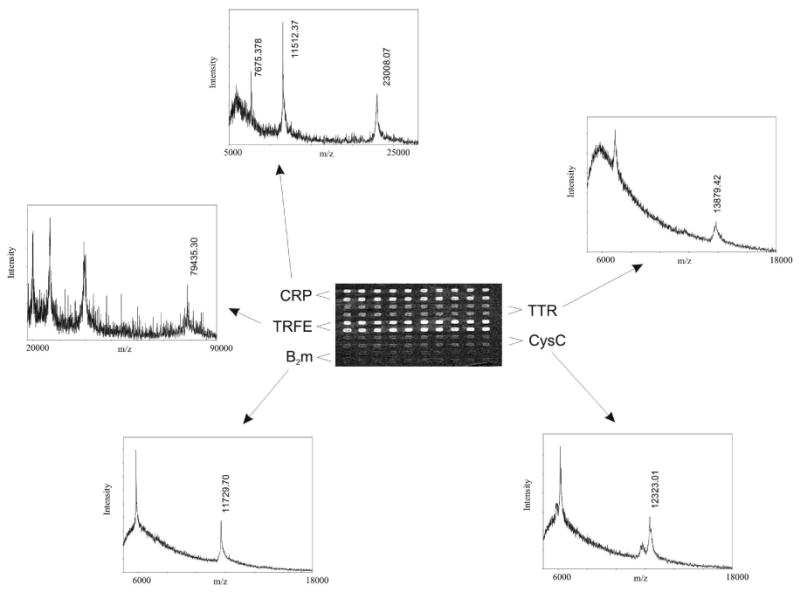
SPR image and mass spectra obtained from a single 10×10 antibody array containing all five antibodies, incubated with a solution containing the five proteins.
Mass spectra were also acquired from un-derivatized portions of the chips (results not shown), verifying that absence of non-specific binding of proteins to the carboxyl groups of the non-arrayed portions of the chip (following incubation with the running buffer, the non-arrayed, CDI-activated surface of the chip reverts back to the carboxyl groups of the mecraptoundecanoic acid monolayer). It is expected, though, that when complex biological samples are analyzed, the level of non-specific binding to the underivatized portion of the chip will significantly increase, prompting the development of specific chip chemistries (e.g., polymers, blocking reagents, etc) to suppress the non-specific binding. At this point of time it is worth mentioning that it is possible to collect SPR-MS data from the active spots on the array only. Using the software of the SPR instrument, imaging data can be obtained from pre-selected areas of the chip, effectively monitoring binding to the antibody spots only, and ignoring the possible background noise resulting from non-specific binding to the rest of the chip surface. Similarly, MS data can be obtained only from the antibody spots, by focusing the MS laser to the spots exclusively. In this way the limits of detection in SPR Imaging are being imposed solely by the specificity and affinity of the antibody.
The ability to reproducibly perform the complete SPR-MS analysis hinges on several critical steps, the most important being the MALDI matrix application to the antibody-protein array. After the SPR Imaging analysis, the proteins remain bound to the antibody spots on the array. These spots are rather small, and the MALDI matrix cannot be applied to each spot individually. Instead, the entire chip is sprayed with a fine mist of matrix solution. To maintain the resolution between the spots on the array, the fine matrix droplets must dry within few seconds of the application to prevent spreading across the chip. A MALDI matrix spraying approach perfected in this laboratory 16 was implemented for this matter and provided the desired results. The matrix application to the array remains the single most enabling step that brings the two detection approaches together.
Conclusions
Presented in this work is the first demonstration of an SPR-MS array platform. The foundation for the combined SPR-MS array approach was laid in some very recent work involving demonstration of SPR and MS arrays independently 19, 20. Once the two component technologies were optimized, their combination into one seamless approach was not only straightforward, but also logical. Quantitative SPR Imaging readouts can provide information about the concentration of a protein, and its adsorption strength (equilibrium constants). Mass spectrometry, on the other hand, is invaluable in delineating protein modifications. Hence, the resulting SPR-MS array platform is one of only few approaches to include both quantitative and qualitative aspects of protein analysis. With its high-throughput and high-content features, the SPR-MS arrays can facilitate the systematic study of protein modulations in population proteomics settings aimed at studying human protein diversity.
Acknowledgments
The project described was supported by Grant Number 4 R33 RR018475-02 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
References
- 1.Yates JR., 3rd Annu Rev Biophys Biomol Struct. 2004;33:297–316. doi: 10.1146/annurev.biophys.33.111502.082538. [DOI] [PubMed] [Google Scholar]
- 2.Ahn NG, Shabb JB, Old WM, Resing KA. ACS Chem Biol. 2007;2:39–52. doi: 10.1021/cb600357d. [DOI] [PubMed] [Google Scholar]
- 3.Kung LA, Snyder M. Nat Rev Mol Cell Biol. 2006;7:617–622. doi: 10.1038/nrm1941. [DOI] [PubMed] [Google Scholar]
- 4.Jones RB, Gordus A, Krall JA, MacBeath G. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 5.Homola J, Yee SS, Gauglitz G. Sensors actuat B. 1999;54:3–15. [Google Scholar]
- 6.Nelson RW, Nedelkov D, Tubbs KA. Anal Chem. 2000;72:404A–411A. [PubMed] [Google Scholar]
- 7.Nelson RW, Nedelkov D, Tubbs KA. Electrophoresis. 2000;21:1155–1163. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1155::AID-ELPS1155>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Nedelkov D, Nelson RW. Am J Kidney Dis. 2001;38:481–487. doi: 10.1053/ajkd.2001.26831. [DOI] [PubMed] [Google Scholar]
- 9.Natsume T, Taoka M, Manki H, Kume S, Isobe T, Mikoshiba K. Proteomics. 2002;2:1247–1253. doi: 10.1002/1615-9861(200209)2:9<1247::AID-PROT1247>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Nedelkov D, Nelson RW, Kiernan UA, Niederkofler EE, Tubbs KA. FEBS Lett. 2003;536:130–134. doi: 10.1016/s0014-5793(03)00042-5. [DOI] [PubMed] [Google Scholar]
- 11.Grote J, Dankbar N, Gedig E, Koenig S. Anal Chem. 2005;77:1157–1162. doi: 10.1021/ac049033d. [DOI] [PubMed] [Google Scholar]
- 12.Nedelkov D, Nelson RW. Trends Biotechnol. 2003;21:301–305. doi: 10.1016/S0167-7799(03)00141-0. [DOI] [PubMed] [Google Scholar]
- 13.Nedelkov D. Expert Rev Proteomics. 2006;3:631–640. doi: 10.1586/14789450.3.6.631. [DOI] [PubMed] [Google Scholar]
- 14.Wegner GJ, Wark AW, Lee HJ, Codner E, Saeki T, Fang S, Corn RM. Anal Chem. 2004;76:5677–5684. doi: 10.1021/ac0494275. [DOI] [PubMed] [Google Scholar]
- 15.Kanda V, Kariuki JK, Harrison DJ, McDermott MT. Anal Chem. 2004;76:7257–7262. doi: 10.1021/ac049318q. [DOI] [PubMed] [Google Scholar]
- 16.Nedelkov D, Nelson RW. J Mol Recognit. 2000;13:140–145. doi: 10.1002/1099-1352(200005/06)13:3<140::AID-JMR496>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 17.Ritchie RF, editor. Serum Proteins in Clinical Medicine. Foundation for Blood Research; Scarborough, ME: 1999. [Google Scholar]
- 18.Craig WY, Ledue TB, Ritchie RF. Plasma proteins: Clinical utility and interpretation. Foundation for Blood Research; Scarborough, ME: 2001. [Google Scholar]
- 19.Lee HJ, Nedelkov D, Corn RM. Anal Chem. 2006;78:6504–6510. doi: 10.1021/ac060881d. [DOI] [PubMed] [Google Scholar]
- 20.Nedelkov D, Nelson RW. Methods Mol Biol. 2006;328:131–139. doi: 10.1385/1-59745-026-X:131. [DOI] [PubMed] [Google Scholar]