

Abstract
A straightforward approach toward the total synthesis of frondosin C is described. This strategy involves a key one-pot, microwave-assisted 5-exo cyclization-Claisen rearrangement sequence that was used for the expedient assembly of the frondosic C scaffold. Subsequent manipulation of the tetracyclic core allowed the synthesis of an advanced intermediate bearing the characteristic diene moiety in the B ring.
Five novel sesquiterpene hydroquinone derivatives, frondosins A-E (Figure 1), were recently isolated from the Micronesian marine sponge Dysidea frondosa.1 Frondosins A and D, having opposite optical rotations compared to those present in Dysidea frondosa, have also been found in another sponge, Euryspongia sp.2 All members of the frondosin family are antagonists of interleukin-8 (IL-8) and inhibitors of protein kinase C (PKC) in the low micromolar range.1 IL-8 is a neutrophil-activating peptide, which is produced by several cell types in response to inflammatory stimuli.3 It is now known to also play an important role in tumor progression and metastasis in several human cancers, 4 including lung cancers.4b Thus, IL-8 antagonists hold therapeutic potential as novel anti-inflammatory agents for the treatment of several acute and chronic inflammatory disorders, such as rheumatoid arthritis, psoriasis and many lung diseases, including acute respiratory distress syndrome, chronic obstructive pulmonary disease and asthma. In addition, IL-8 represents a potential new target for antiretroviral therapy against HIV-1,2,4b,5 and inhibitors of IL-8 action may prove useful therapeutic agents against cancer as inhibitors of tumorigenesis and proangiogenesis.4
Figure 1.
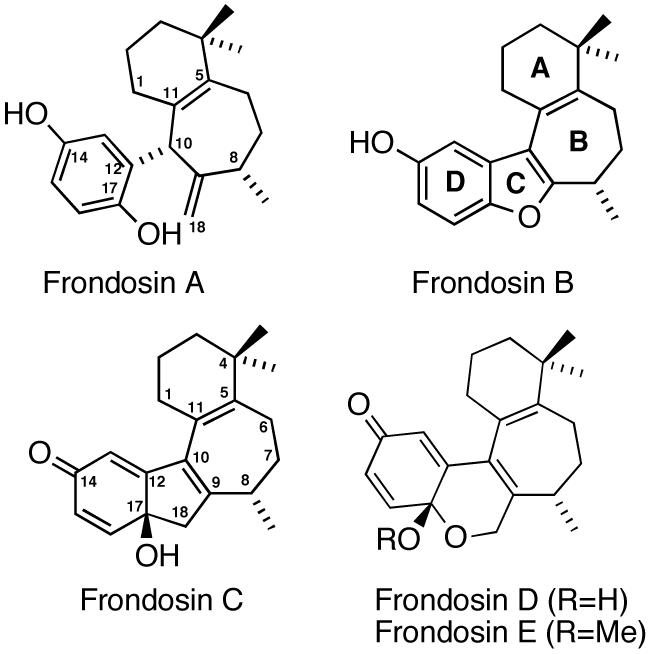
Structures of frondosins A-E.
Total synthesis of frondosin B was first achieved by Danishefsky et al.6 in 2000 and, more recently, by the Trauner7 and Flynn8 groups. Other members of the frondosin family, however, have not yet been synthesized.
We recently reported the first known approach to the tetracyclic frondosin C ring system.9 This approach is based on our ongoing investigations involving a base-catalyzed tandem cyclization/Claisen rearrangement as a convenient route to cycloheptane-containing polycyclic ring structures.10 The reaction sequence involves an initial 5-exo dig cyclization of an appropriately substituted 4-alkyn-1-ol system followed by in situ microwave-assisted Claisen rearrangement of the intermediate 2-alkylidene tetrahydrofuran derivative.11
Herein, we wish to report our progress toward the total synthesis of frondosin C. At the outset of the current investigation, it was envisaged that tetracycle 2, previously synthesized from the tertiary alcohol 1 (Scheme 1),9 could be manipulated to frondosin C in a sequence of steps involving α-methylation, generation of the requisite B ring diene functionality, demethylation of the methoxy group and oxidation of the resulting phenol system to the p-quinol moiety.
Scheme 1.
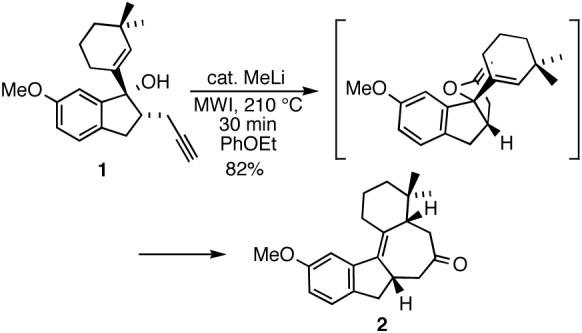
Synthesis of frondosin C tetracyclic core.
According to this plan, the requisite C8 methyl group was first introduced by treatment of 2 with NaHMDS followed by addition of MeI. It was anticipated that, due to the proximity of the A ring gem dimethyl moiety to C6, methylation at C8 would predominate in this reaction. In fact, this turned out to be the case although the observed regioselectivity was found to be rather modest; the desired ketone 3 and the C8 methylated 4 were formed as a 2.4 to 1 mixture of regioisomers (Scheme 2).12 It is noteworthy that stereoselectivity of the alkylation affording 3 was high, consistent with delivery of the electrophile from the less hindered top face of the intermediate enolate anion.
Scheme 2.
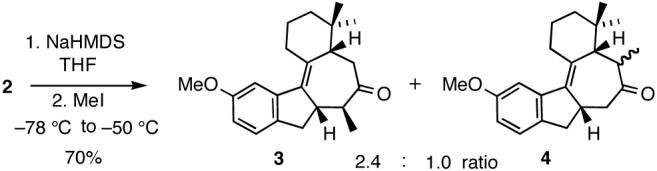
Methylation of ketone 2.
All attempts to improve regioselectivity of the methylation, including through the use of different bases, were unsuccessful and this strategy was ultimately abandoned. Instead, a different approach involving introduction of the requisite methyl group early on in the sequence was implemented. According to this strategy, readily separable isomeric ketones 6 and 7, each bearing a methyl group at the propargylic position, were prepared in 70% overall yield from commercially available 6-methoxy-1-indanone as shown in Scheme 3.
Scheme 3.
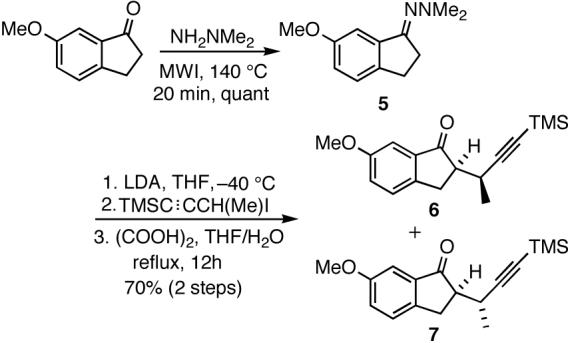
Introduction of the C8 methyl group.
The subsequent coupling reaction involving 1-iodo-3,3-dimethylcyclohexene10e and one of the diastereomeric ketones, randomly assigned as 6, was effected employing our usual protocol10 to provide tertiary alcohol 8 in 85% yield (Scheme 4).
Scheme 4.
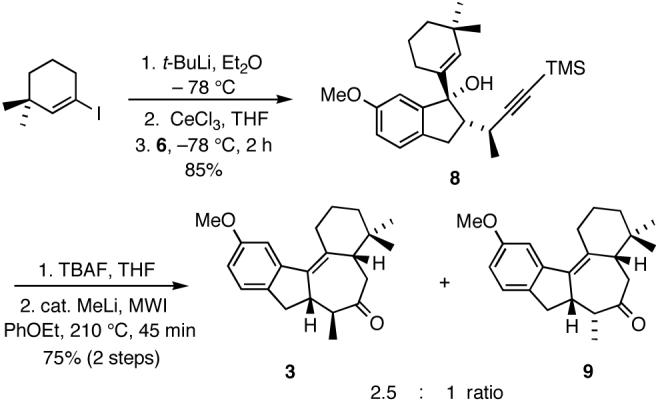
Preparation of tetracyclic ketones 3 and 9.
Following treatment with TBAF and exposure to microwave irradiation (MWI) in the presence of catalytic MeLi (∼0.1 equiv), 8 was smoothly converted to a 2.5:1 mixture of tetracyclic ketones 3 and 9 (Scheme 4). Given that the cyclization/Claisen rearrangement sequence involves a single diastereomer derived from 8, the formation of a mixture of isomeric ketones 3 and 9 in the process is intriguing and most likely arises from the interconversion of exocyclic intermediates 10 and 12 via the endocyclic intermediate 11 as depicted in Scheme 5.13
Scheme 5.
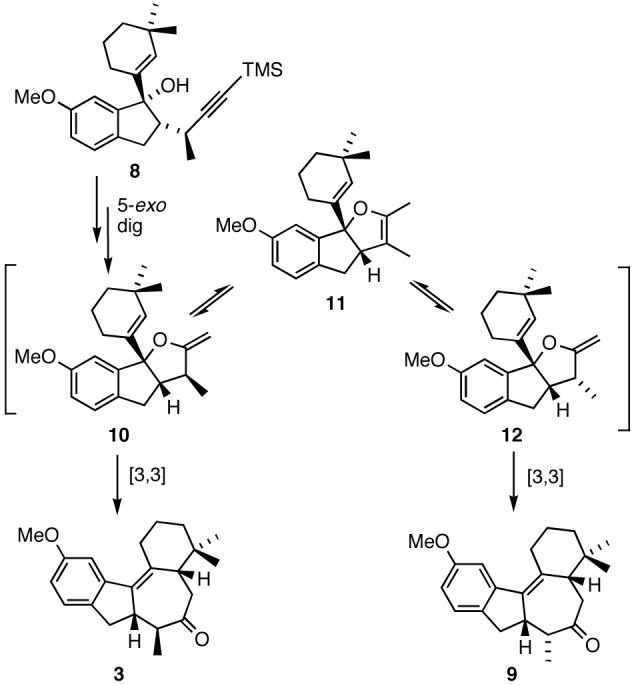
Proposed interconversion of intermediates 10 and 12.
Further evidence for the suggested mechanism is provided by the observation that an identical diastereomer ratio of tetracyclic ketones 3 and 9 is obtained when a 1:1 mixture of isomeric alcohols 13 and 14 is subjected to catalytic MeLi and MWI (Scheme 6). Indeed, the entire reaction sequence depicted in Schemes 3 and 4 may be conducted with the same end result starting with hydrazone 5 and performing the subsequent steps without prior separation of the diastereomeric intermediates.
Scheme 6.
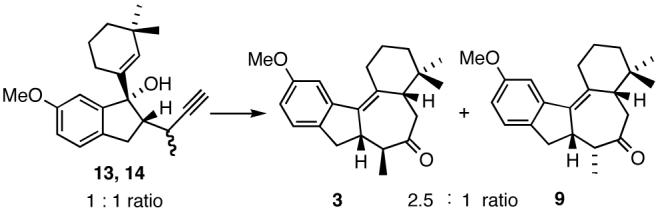
Cyclization/Claisen rearrangement sequence involving a diastereomeric mixture of acetylenic alcohols 13 and 14.
Calculation of minimum energy conformations for compounds 3 and 9 revealed an energy difference of 8.57 kJ/mol in favor of 3.14 This being the case, it was envisioned that isomerization of the mixture could provide an opportunity to increase the 3/9 ratio further.
The initial isomer ratio of 2.5 to 1 was significantly improved in favor of 3 without loss of product material when the mixture was treated with several different bases. The highest observed ratio of 14.6 to 1 (94% de) resulted from refluxing a mixture of 3 and 9 in t-BuOK/t-BuOH for 3.5h. Stereochemistry of the major isomer was confirmed by a combination of 2D NMR and 1D NOESY techniques and the experimental observations are in good agreement with the calculated trends.
Removal of the carbonyl functionality was achieved by a three-step sequence involving an initial borohydride reduction, then conversion of the resulting alcohol 15 to the corresponding mesylate and treatment of the mesylate with lithium triethylborohydride (Scheme 7).
Scheme 7.
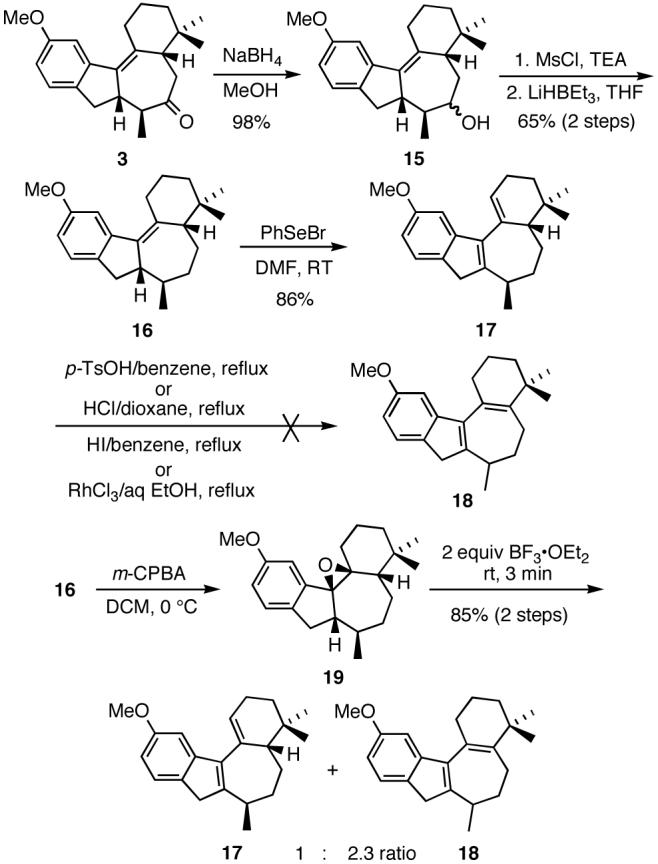
Initial strategies for the synthesis of 18.
It should be noted that several other deoxygenation methods that were attempted, including Ra-Ni reduction of a thioacetal, prepared from 3 and 1,2-ethanedithiol, as well as NaCNBH3 treatment of a tosylhydrazone derived from 3, were overall less satisfactory in providing 16.
Somewhat surprisingly, treatment of 16 with PhSeBr in DMF resulted in direct and rapid generation of diene 17 in 86% yield. Although it was anticipated that 17 could be readily converted to the desired diene 18, this turned out not to be the case (Scheme 7) and all attempts at effecting isomerization of the trisubstituted double bond in 17 failed. However, it was found that 18 could be produced as the major product along with 17 (2.3:1 ratio) in a two-step sequence involving reaction of 16 with m-CPBA, followed by treatment of the resulting unstable epoxide 19 with 2.0 equivalents of BF3·OEt2 (Scheme 7).
In an attempt to find a more reliable method to install the desired diene moiety in the B ring, tetracyclic ketone 3 was subjected to DDQ oxidation.18 Gratifyingly, this resulted in direct formation of diene 20 in 90% yield (Scheme 8). Borohydride reduction of the carbonyl group followed by reaction with mesyl chloride and triethylamine resulted in in situ elimination of the intermediate mesylate, affording triene 22 in high yield. Alternatively, 22 could be obtained in a comparable yield by subjecting alcohol 21 to phosphorous oxychloride in pyridine. Finally, diimide reduction of 22 afforded racemic diene 18 in 70% yield.
Scheme 8.
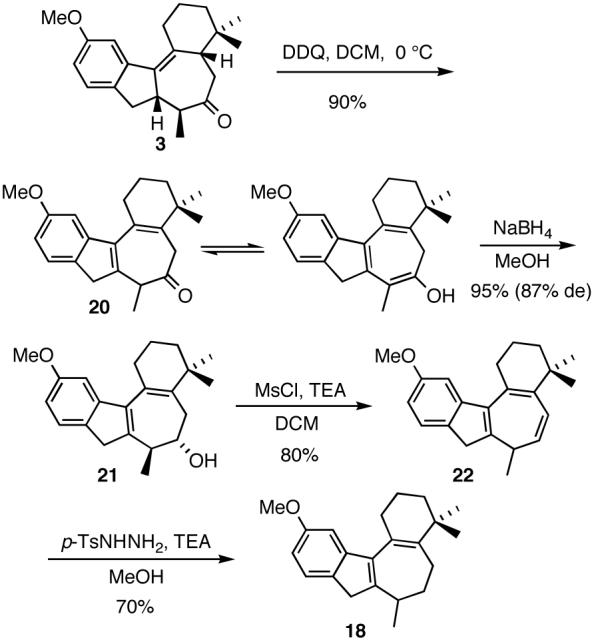
Improved synthesis of 18.
In summary, we have achieved the synthesis of an advanced intermediate 18 bearing most of the characteristic structural features of frondosin C. Efforts to complete the total synthesis of this natural product as well as other members of the frondosin family are currently underway in our laboratories.
Supplementary Material
Figure 2.
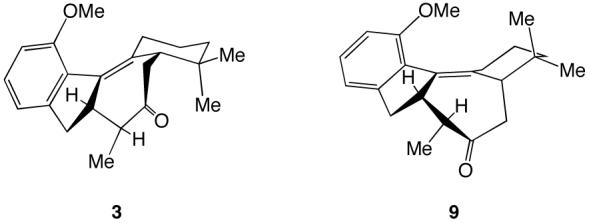
Calculated minimum energy conformations of 3 and 9.
Acknowledgment
This research was supported by grants from the National Institutes of Health (NIGMS), and the Camille and Henry Dreyfus Foundation (Scholar-Fellow Program). T.V.O also gratefully acknowledges support from the Hans and Ella McCollum-Vahlteich ‘21 endowment. R.E.K. gratefully acknowledges support from Bristol-Myers Squibb (summer undergraduate fellowship).
References
- 1.Patil AD, Freyer AJ, Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047. [Google Scholar]
- 2.Hallock YF, Cardellina JH, II, Boyd MR. Nat. Prod. Lett. 1998;11:153. [Google Scholar]
- 3(a).Seitz M, Dewald B, Gerber N, Baggiolini M. J. Clin. Invest. 1991;87:463–469. doi: 10.1172/JCI115018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller EJ, Cohen AB, Nagao D, Griffith RJ, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Am. Rev. Respir. Dis. 1992;146:247. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 4(a).Brat DJ, Bellail AC, Van Meir EG. Neuro-oncol. 2005;7:122. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu YM, Webster SJ, Flower D, Woll PJ. Br. J. Cancer. 2004;91:1970. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan A, Chen JJ, Yao PL, Yang PC. Front. Biosci. 2005:853. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 5.Lane BR, Lore K, Bock PJ, Andersson J, Coffey MJ, Strieter RM, Markovitz DM. J. Virol. 2001;75:8195. doi: 10.1128/JVI.75.17.8195-8202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6(a).Danishefsky SJ, Inoue M, Frontier AJ. Angew. Chem., Int. Ed. 2000;39:761. [PubMed] [Google Scholar]; (b) Inoue M, Carson MW, Frontier AJ, Danishefsky SJ. J. Am. Chem. Soc. 2001;123:1878. doi: 10.1021/ja0021060. [DOI] [PubMed] [Google Scholar]
- 7(a).Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:1569. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (b) Hughes CC, Trauner D. Tetrahedron. 2004;60:9675. [Google Scholar]
- 8.Kerr DJ, Willis AC, Flynn BL. Org. Lett. 2004;6:457. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]
- 9.Martinez I, Alford PE, Ovaska TV. Org. Lett. 2005;7:1133. doi: 10.1021/ol050144o. [DOI] [PubMed] [Google Scholar]
- 10(a).Ovaska TV, Roark JL, Shoemaker CM. Tetrahedron Lett. 1998;39:5705. [Google Scholar]; (b) Ovaska TV, Roses JB. Org. Lett. 2000;2:2361. doi: 10.1021/ol006139w. [DOI] [PubMed] [Google Scholar]; (c) Ovaska TV, Reisman SE, Flynn MA. Org. Lett. 2001;3:115. doi: 10.1021/ol006823a. [DOI] [PubMed] [Google Scholar]; (d) Ovaska TV, Ravi Kumar JS, Hulford CA, O’Sullivan MF, Reisman SE. Tetrahedron Lett. 2002;43:1939. [Google Scholar]; (e) McIntosh CE, Martinez I, Ovaska TV. Synlett. 2004:2579. [Google Scholar]
- 11.For an early report of this sequence, see Marvell EN, Titterington D. Tetrahedron Lett. 1980:2123.
- 12.The stereochemistry at C8 of compound 4 could not be determined with certainty.
- 13.Rhoads SJ, Brandenburg CF. J. Am. Chem. Soc. 1971;93:5805. [Google Scholar]
- 14.The minimum energy conformations shown were calculated using MacroModel v. 9.0015 with the MM3* forcefield and a GB/SA chloroform solvent model.15 Structures were generated by a 10,000 step large scale low-mode conformational search.16,17
- 15.Still WC, Tempczyk A, Hawley RC, Hendrickson T. J. Am. Chem. Soc. 1990;112:6127. [Google Scholar]
- 16.Keseru GM, Kolossvary I. J. Am. Chem. Soc. 2001;123:12708. doi: 10.1021/ja0160086. [DOI] [PubMed] [Google Scholar]
- 17.Keseru GM, Kolossvary I. J. Comput. Chem. 2001;22:21. [Google Scholar]
- 18.Baran PS, Guerrero CA, Corey EJ. J. Am. Chem. Soc. 2003;125:5628. doi: 10.1021/ja034491+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.