

Abstract
The first detailed study of a room temperature asymmetric Diels–Alder reaction of N-sulfonyl-1-aza-1,3-butadienes enlisting a series of nineteen enol ethers bearing chiral auxilaries is reported with many providing highly diastereoselective (endo and facial diastereoselection) reactions largely the result of an exquisitely organized [4 + 2] cycloaddition transition state. Three new, readily accessible, and previously unexplored auxilaries (18a, 19a and 37a) rationally emerged from the studies and provide remarkable selectivities (for 19a and 37a: 49:1 endo:exo and 48:1 facial selectivity) that promise to be useful in systems beyond those detailed.
Introduction
In a series of studies, we introduced and have explored the 4π participation of electron-deficient N-sulfonyl-1-aza-1,3-butadienes in intermolecular Diels–Alder reactions with electron-rich dienophiles.1 The complementary N1 substitution of an α,β-unsaturated imine with an electron-withdrawing substituent (–SO2R) accentuates the inherent electron-deficient nature of the 1-aza-1,3-butadiene accelerating its participation in [4 + 2] cycloaddition reactions with electron-rich dienophiles in LUMOdiene-controlled Diels–Alder reactions. Further, a bulky electron-withdrawing N1 substituent decelerates competitive 1,2-imine addition relative to [4 + 2] cycloaddition and stabilizes the [4 + 2] cycloaddition product (deactivated enamine) to the reaction conditions while enhancing the electron-deficient character of the diene. Based on theses features, the N-sulfonyl-1-aza-1,3-butadienes that were explored were found to participate in regiospecific and diastereoselective inverse electron demand Diels–Alder reactions with characteristics of a concerted [4 + 2] cycloaddition reaction (Figure 1).2
Figure 1. . N.
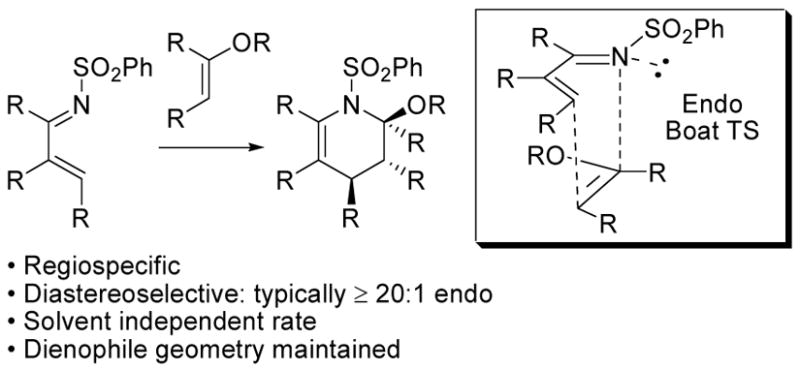
-Sulfonyl-1-azadiene Diels–Alder Reaction.
Moreover, the reactions exhibited an unusually high endo diastereoselectivity (≥20:1) that could be attributed to a [4 + 2] cycloaddition transition state which benefits not only from a conventional stabilizing secondary orbital interaction, but also one in which the nitrogen lone pair and the σ C–O bond of an enol ether dienophile lie trans periplanar to one another further benefiting from a transition state anomeric effect. A bonus of the N-sulfonyl-1-aza-1,3-butadienes was that they proved to be stable imine derivatives capable of simple isolation and purification (SiO2 or Florisil chromatography), resistant to tautomerization to the corresponding stabilized enamine, and still remarkably reactive in prototypical [4 + 2] cycloadditions that were found to proceed under unusually mild conditions (25–100 °C). The utility of these reactions has been exploited in the total synthesis of several natural products including streptonigrone,3 fredricamycin A,4 nothapodytine B and (−)-mappicine,5 (+)-camptothecin,6 and (−)-piericidin A1.7
Throughout the course of these studies, we have periodically examined approaches to conducting asymmetric variants of this [4 + 2] cycloaddition reaction. To date, we have not been successful in identifying Lewis acid catalysts (chiral or achiral) that can accelerate the [4 + 2] cycloaddition; rather they promote the nonproductive consumption of the reacting partners. By contrast and largely the result of the exquisitely organized [4 + 2] cycloaddition transition state, we have found and herein detail that their reactions with enol ether dienophiles bearing chiral auxiliaries provide highly diastereoselective (endo and facial diastereoselection) Diels–Alder cycloadditions. Three new, readily accessible, and previously unexplored auxiliaries, which rationally emerged from these studies, proved highly selective (>20:1 endo and >20:1 facial selectivity) and promise to be useful in systems beyond those that we detail.
At the onset of the studies, we explored enol ethers bearing chiral auxiliaries that have been disclosed in preceding cycloaddition reactions. Posner has shown that modest to good diastereomeric excesses are observed when chiral vinyl ethers derived from benzylic secondary alcohols undergo [4 + 2] cycloadditions with 3-sulfinyl- and 3-sulfonyl-2-pyrones both in the presence and absence of a Lewis acid catalyst.8 Dujardin utilized chiral enol ethers derived from mandelate or pantalactone to prepare enantiopure hexose derivatives and β-benzamido aldehydes via their reaction with β,γ-unsaturated-α-keto esters.9,10 Denmark has observed excellent diastereoselectivities in the Lewis acid-catalyzed reactions of a series of chiral enol ethers with nitroalkenes,11 and Pettus has extended their use in uncatalyzed cycloaddition reactions with o-quinone methides (Figure 2).12
Figure 2.
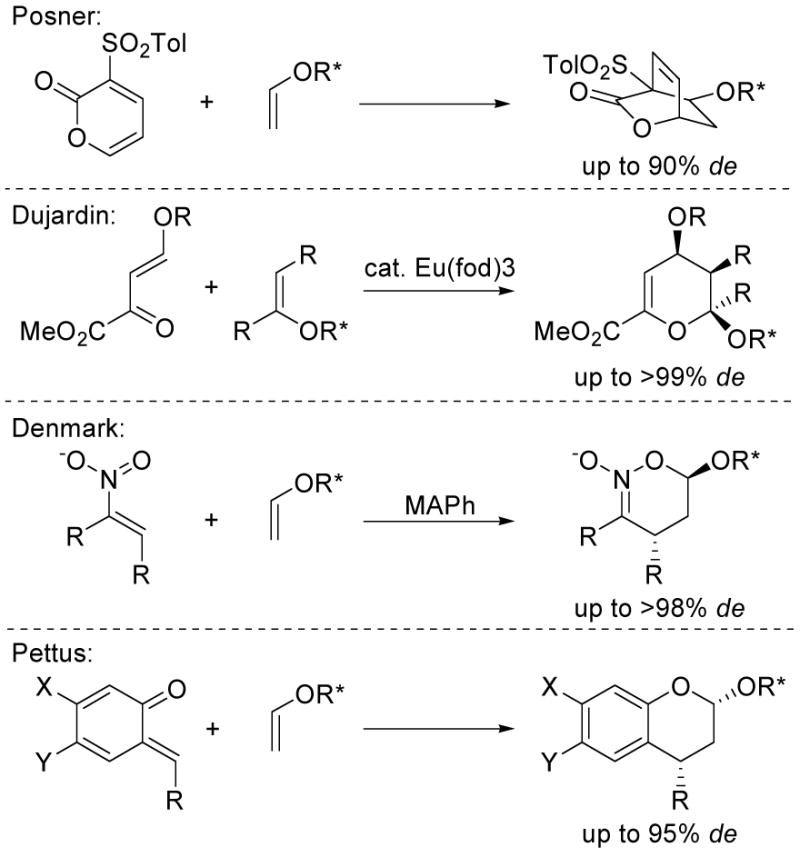
Cycloadditions of optically active enol ethers.
Dienophile Preparation
Several methods were enlisted for the preparation of the vinyl ethers examined (Scheme 1). Typically, they were prepared from ethyl vinyl ether by Hg(OAc)2-catalyzed exchange with the alcohol corresponding to the desired vinyl ether (Method A).13 A variant of this approach utilizes mercury(II) trifluoroacetate, although few examples of its use in transetherification reactions have yet to be detailed (Method B).14 Dujardin developed a two-step procedure for vinyl ether formation involving acid-catalyzed mixed acetal formation from the chiral alcohol and ethyl vinyl ether, followed by a TMSOTf promoted elimination to the desired vinyl ether (Method C).15 More recently, Ishii reported an iridium-catalyzed transetherification ([Ir(cod)Cl]2),16 and Schlaf reported analogous reactions catalyzed by palladium (Pd(DPP)(OCOCF3)2).17 In our hands, their effectiveness was found to be substrate dependent.
Scheme 1.
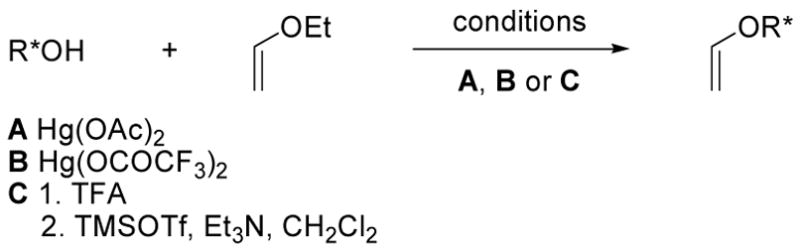
Survey of Dienophile Chiral Auxiliaries
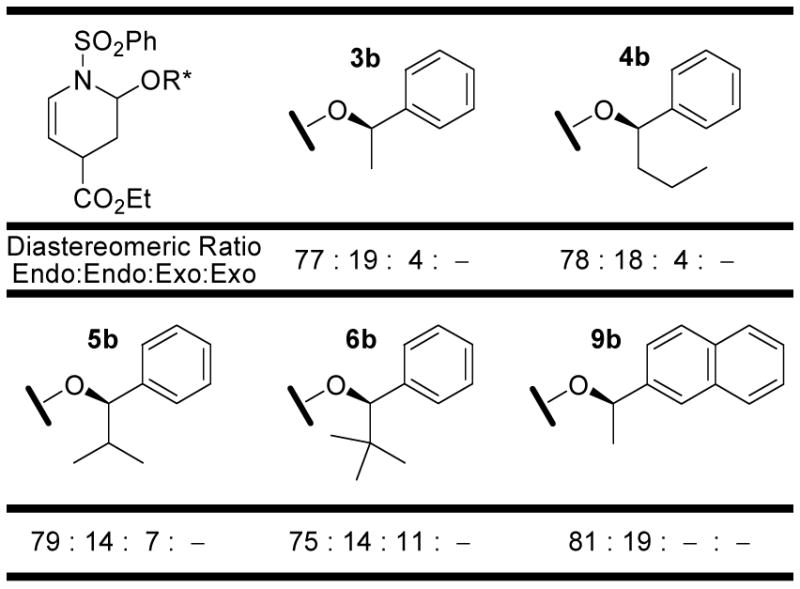
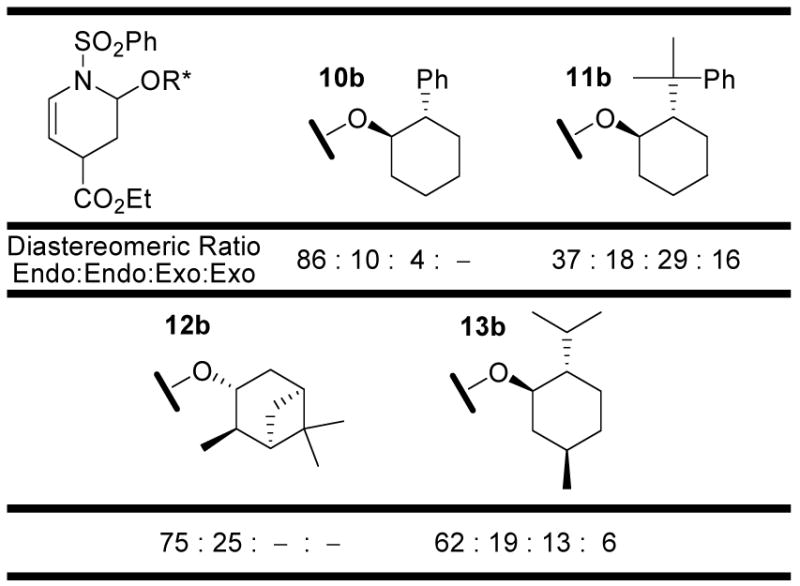
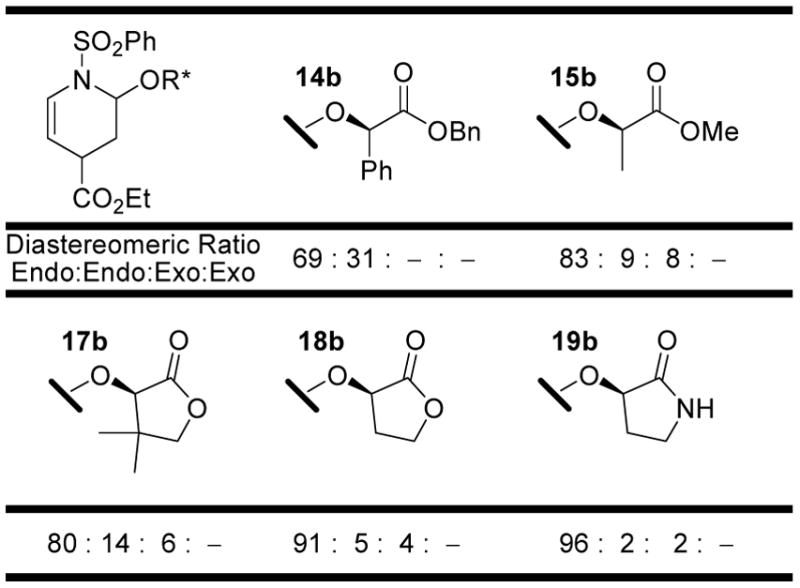
Initially, eighteen optically active enol ethers were examined with the prototypical N-sulfonyl-1-aza-1,3-butadiene 1, Figure 3 and Table 1. The distribution of cycloaddition products and their quantitation were determined by 1H NMR of the reaction mixtures which typically displayed distinguishable signals for each of the four possible products. Thus, the initial assignments and the quantitation were made by examination and integration of the C2-H and C5-H 1H NMR signals of the cycloadducts. The C5-H splitting was diagnostic of endo (ddd, J = 8.3, 5.1, 1.6 Hz) versus exo (dt, J = 8.3, 1.8 Hz) addition and both the C5-H and C2-H integrations were used for the quantitations. Representative of this, the results with dienophile 6a are illustrated in Figure 4 where C5-H of the two endo adducts are each observed as a clear ddd (5.40 and 5.26 ppm, J = 1.6, 5.1, 8.3 Hz) whereas the minor exo adduct was observed as a dt (5.32 ppm, J = 1.8, 8.3 Hz) and both the structure as well as the relative and absolute stereochemistry of the major product were established by a single-crystal x-ray structure determination.18 The diagnostic C5-H coupling in the 1H NMR spectra arises from the well-defined conformations of the endo (diaxial substituents) versus exo (axial –OR*, equatorial –CO2Et) cycloadducts and the resulting C5-H/C4-H coupling constants of ca. 5 Hz (C4-Heq) versus ca. 2 Hz (C4-Hax). In an analogous fashion, the structures of the major products derived from 3 and 6 (representative of 2–9), 12, and 17 and 19 (representative of 14–19) were established by x-ray.18
Figure 3.

Survey of optically active enol ethers.
Table 1.
Results of Survey of Optically Active Enol Ethers
| Dienophile | Time (h) | Major Adduct (% Conversion)a | Total Conv (%) | Ratio of Diastereomersb |
Endo : Exo | Endo Facial Selectivity | ||
|---|---|---|---|---|---|---|---|---|
| endo | exo | |||||||
| 2a | 60 | 2b (52)c | 97 | 54: 40 | : | 3: 3 | 16 : 1 | 1.4 : 1 |
| 3a | 60 | 3b (72) | 94 | 77: 19 | : | 4: – | 19 : 1 | 4 : 1 |
| 4a | 60 | 4b (49) | 63 | 78: 18 | : | 4: – | 24 : 1 | 4 : 1 |
| 5a | 60 | 5b (63) | 80 | 79: 14 | : | 7: – | 13 : 1 | 6 : 1 |
| 6a | 72 | 6b (48) | 64 | 75: 14 | : | 11: – | 8 : 1 | 5 : 1 |
| 7a | 60 | 7b (71) | 91 | 78: 18 | : | 4: – | 24 : 1 | 4 : 1 |
| 8a | 60 | 8b (53) | 69 | 77: 15 | : | 8: – | 12 : 1 | 5 : 1 |
| 9a | 60 | 9b (71) | 88 | 81: 19 | : | –: – | >25 : 1 | 4 : 1 |
| 10a | 60 | 10b (85) | 98 | 86: 10 | : | 4: – | 24 : 1 | 9 : 1 |
| 11a | 48 | 11b (12)c | 31 | 37: 18 | : | 29: 16 | 1 : 1 | 2 : 1 |
| 12a | 60 | 12b (41) | 55 | 75: 25 | : | –: – | >25 : 1 | 3 : 1 |
| 13a | 60 | 13b (55) | 86 | 62: 19 | : | 13: 6 | 4 : 1 | 3 : 1 |
| 14a | 60 | 14b (12) | 18 | 69: 31 | : | –: – | >25 : 1 | 2 : 1 |
| 15a | 60 | 15b (27) | 45 | 83: 9 | : | 8: – | 12 : 1 | 11 : 1 |
| 16a | 60 | 16b (32) | 38 | 85: 9 | : | 6: – | 16 : 1 | 9 : 1 |
| 17a | 60 | 17b (54)d | 68 | 80: 14 | : | 6: – | 16 : 1 | 6 : 1 |
| 18a | 60 | 18b (59)d | 65 | 91: 5 | : | 4: – | 24 : 1 | 18 : 1 |
| 19a | 48 | 19b (96)d | ≥98 | 96: 2 | : | 2: – | 49 : 1 | 48 : 1 |
| ethyl vinyl ether | 46 | (82)e | – | >25 : 1 | – | |||
Reactions performed at RT, 0.3 M with respect to diene in toluene.
Based on integration of major product C2-H versus minor diastereomer C2-H’s and diene C2-H. Some hydrolysis of diene ocassionally observed (< 5%)
Ratios based on 1H NMR integration of C2-H of products unless otherwise noted. Endo and exo compounds assigned based on 1H NMR splitting patterns. Some peaks could not be located due to overlap, small size or low conversion.
Integration based on C5-H.
Anhydrous CHCl3 as solvent, 1 M with respect to diene.
Yield from reference 1c.
Figure 4.
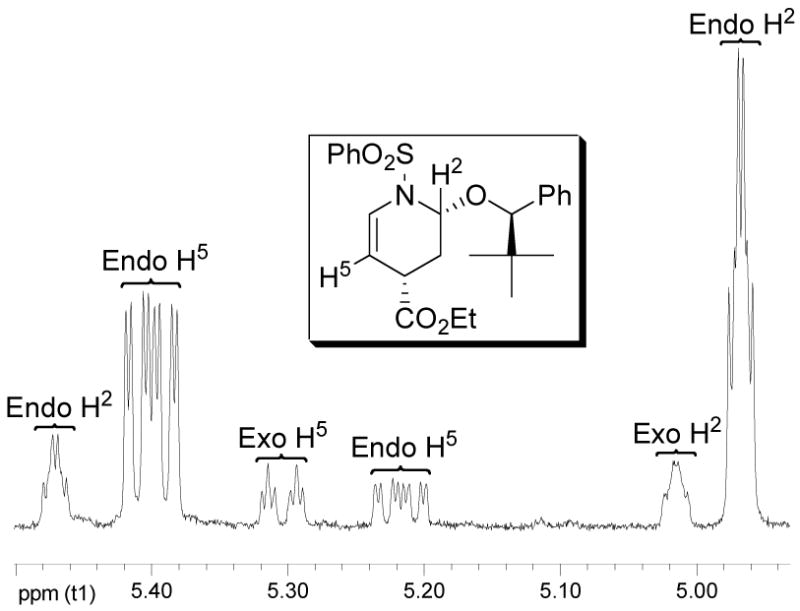
Diagnostic cycloadduct 1H NMR signals.
Clear trends emerged in the examination of the series 2–9a inspired by the Posner studies where the role of the aryl ring was probed with 2 versus 3 and the size of the dienophile alkyl substituent was systematically increased with 3–6. As the size of the alkyl group was increased (3a → 6a), the facial selectivity of the cycloaddition increased modestly (6:1 vs 4:1), but did so at the expense of the rate of the reaction and with an erosion of the superb endo diastereoselectivity (8:1 vs 19:1). Replacing the aryl ring with a saturated alkyl group (2a) or a larger naphthyl ring (9a) did not significantly impact the endo diastereoselection, and the latter did not improve the cycloaddition facial selectivity. Within this series, the amount of the major diastereomer approached the respectable level of 80% of the product for each of the dienophiles with the enhanced facial selectivity of the larger alkyl derivatives being offset by a larger proportion of exo adduct(s). As such, and because of their enhanced rates of reactions, 3a and 9a emerge as the most useful dienophiles in this series (Figure 5).
Figure 5.
Dienophile series 3–9.
Several enol ethers 10–13a bearing chiral six-membered rings were examined being inspired by the work of Denmark and Swindell (10–12a),19–21 or Posner (13a).8 Whereas the most hindered of these (11a and 12a) exhibited poor reactivities, poor endo diastereoselectivities (12a) and/or modest to poor facial selectivities, 13a and especially 10a exhibited good chemical reactivity (% conversions), outstanding (10a, 24:1) or modest (13a, 4:1) endo diastereoselectivity, and good (10a, 9:1) or modest (13a, 3:1) facial selectivity. Of these, the dienophile 10a proved most satisfactory providing conversions that slightly exceed 85% for the major diastereomer in a reaction that provides superb conversions (99%) with exceptional endo diastereoselectivity (24:1) and excellent facial selectivity (8–9:1), Figure 6.
Figure 6.
Dienophile series 10–13.
The most interesting and useful series 14–19 began with an examination of enol ethers derived from α-hydroxy esters and lactones being inspired by the studies of Dujardin.9 Thus, the mandelate-derived enol ether 14a analogous to those explored by Dujardin22 exhibited superb endo diastereoselection, but poor facial selectivity (2:1) and low conversions whereas Dujardin’s pantalactone-derived enol ether 17a9 maintained the excellent endo diastereoselection (16:1) and exhibited a substantially improved facial selectivity (6:1). Like the preceding hindered enol ethers (e.g. 6a), the reactivity of 17a was modest and likely contributing to the erosion of the endo diastereoselection. Consequently, we examined 18a with removal of the gem dimethyl substitution of 17a which has not been previously explored, and found that it was more reactive, exquisitely endo diastereoselective (24:1), and remarkably facial selective (18:1). The lactate-derived enol ethers 15a and 16a were subsequently examined to define the importance of cyclic structure of 18a and were found to be both less reactive and selective than 18a. Finally, the corresponding lactam 19a, which proved both synthetically more accessible and more stable to subsequent transformations, was examined and found to exhibit a higher reactivity and an even greater level of endo (49:1) and facial (48:1) diastereoselection than 18a, Figure 7. Thus, it is from these latter studies that two of the new and most impressive auxilaries (18a and 19a) rationally emerged exhibiting remarkable endo (>20:1) and facial (>20:1) selectivities for the [4 + 2] cycloaddition reactions with 1 which promise to be useful in systems beyond those that we detail.
Figure 7.
Dienophile series 14–19.
Preparation of 18a and 19a
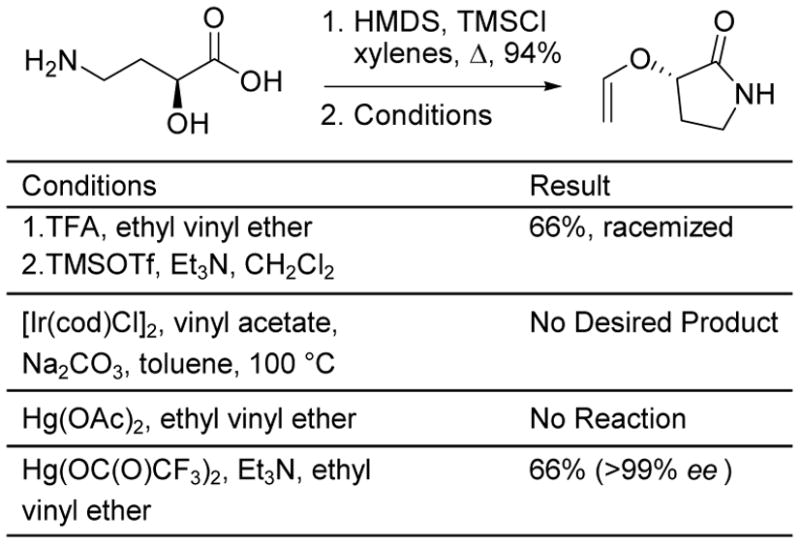
The preparation of 18a and 19a deserve a more detailed discussion as a result of their utility. Although we have not yet examined this in depth, the preparation of 18a was conducted from ethyl vinyl ether and (+)-(R)-2-hydroxy-γ-butyrolactone (both enantiomers are commercially available) utilizing Dujardin’s two step protocol15 (20%) or Ishii’s [Ir(cod)Cl]2-catalyzed16 alcohol exchange (20%), whereas the Hg(II)-catalyzed reactions led only to side product formation. This difficulty in the preparation of 18a (low conversions), coupled with its propensity to undergo competitive lactone ring opening reactions (hydrolysis, esterification) in subsequent transformations led to the examination and preferential use of the corresponding lactam 19a. The preparation of 19a, which eventually proved straightforward, also provided unexpected initial observations, Scheme 2. (−)-(S)-3-Hydroxy-2-pyrrolidinone, which is commercially available or readily prepared from (−)-(S)-4-amino-2-hydroxybutanoic acid (HMDS, TMSCl, xylenes, 94%)23 and is readily available in both enantiomeric forms,24 could be cleanly converted to 19a using ethyl vinyl ether and Dujardin’s two step protocol, but unexpectedly provided racemic product. Presumably, this arises through TMSOTf/Et3N silylation of the amide (O-silyl imidate formation) and subsequent racemization.25 Although the iridium and Hg(OAc)2-catalyzed exchange with ethyl vinyl ether did not cleanly provide 19a, the reaction catalyzed by Hg(O2CCF3)2 in the presence of Et3N effectively provides 19a (66%) without evidence of competitive racemization.25 Significantly, 19a provides beautiful, large white prisms upon recrystallization from EtOAc–hexanes providing a convenient stage and manner for ensuring its chemical and optical purity (for (S)-19a: [α]D25 –78 (c 0.77, CHCl3)).25
Scheme 2.
Cycloaddition Model
We have suggested that the exceptional endo diastereoselection observed with the N-sulfonyl-1-aza-1,3-butadienes results from not only a conventional stabilizing secondary orbital interaction, but also a transition state anomeric effect in which the nitrogen lone pair and the C–O σ bond of the dienophile enol ether lie trans periplanar to one another in the endo boat transition state.1 Regardless of the origin of stabilization for an anomeric effect (n–σ* stabilization or avoidance of electrostatic repulsion),26 the endo boat transition state embodies its trans periplanar lone pair/C–O bond arrangement whereas the exo boat transition state places them proximal and gauche to one another. We have suggested that it is the superimposition of this transition state anomeric effect on top of the conventional stabilizing secondary orbital interaction that provides the exceptional endo diastereoselection.1 Any erosion of this endo diastereoselectivity with 2–19a is derived from destabilizing steric interactions present in the endo transition state which then result in a slower rate of reaction.
Houk has shown computationally and experimentally that vinyl ethers prefer to react in inverse electron demand Diels–Alder and 1,3-dipolar cycloadditions through a s-trans conformation (Figure 8, β = 180°) even though their ground state conformation is typically s-cis (β = 0°).27 Enlisting the s-trans conformation, the cycloadditions provide products consistent with reaction through a conformation in which α is approximately 60°, providing the transition state illustrated in Figure 8 with the dienophile H oriented toward the diene and with its large group oriented away from the diene. When the dienophile approaches the diene in this s-trans conformation, the chiral auxiliary comes into proximity of the sterically demanding and electronegative sp3 sulfonyl moiety requiring that the sulfonyl phenyl group orient itself on the diene face opposite the approaching dienophile. This places the electronegative sulfonyl oxygens on the same diene face as the approaching dienophile accounting for the observation that the electronegative ester, lactone, or lactam carbonyls of 14–19, although being sterically undemanding, are similarly directed away from the sulfonyl group and behave as the most effective RL. Notably, the x-ray crystal structures of the cycloadducts,18 albeit being ground state structures of the reaction products, embody each of these features, Figure 8.
Figure 8.
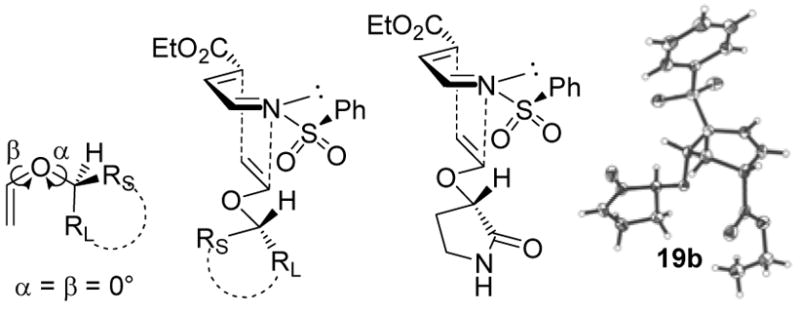
Transition state model. (S-enantiomer depicted)
Reaction Parameters
A series of reaction parameters were examined to identify those that might significantly impact the cycloaddition. As anticipated, increasing the reaction temperature lowers the reaction diastereoselectivity. However, the N-sulfonyl-1-aza-1,3-butadienes typically possess an intrinsic reactivity that permits their use at 25 °C. Nonetheless, the reaction of 1 with 19a was examined at a range of reaction temperatures and both the endo selectivity and the facial diastereoselectivity of the cycloaddition diminished as the reaction temperature increased. However, the detected changes were very small and almost within the limits of error of the detection method (NMR), Figure 9. The more sterically hindered pantalactone-derived dienophile 17a, which reacts more slowly with 1, exhibited a more discernible trend with a varied reaction temperature. Thus, as the reaction temperature was increased to drive the sluggish reaction to completion, the endo diastereoselection dropped more significantly than the endo facial selectivity, Figure 9.
Figure 9.
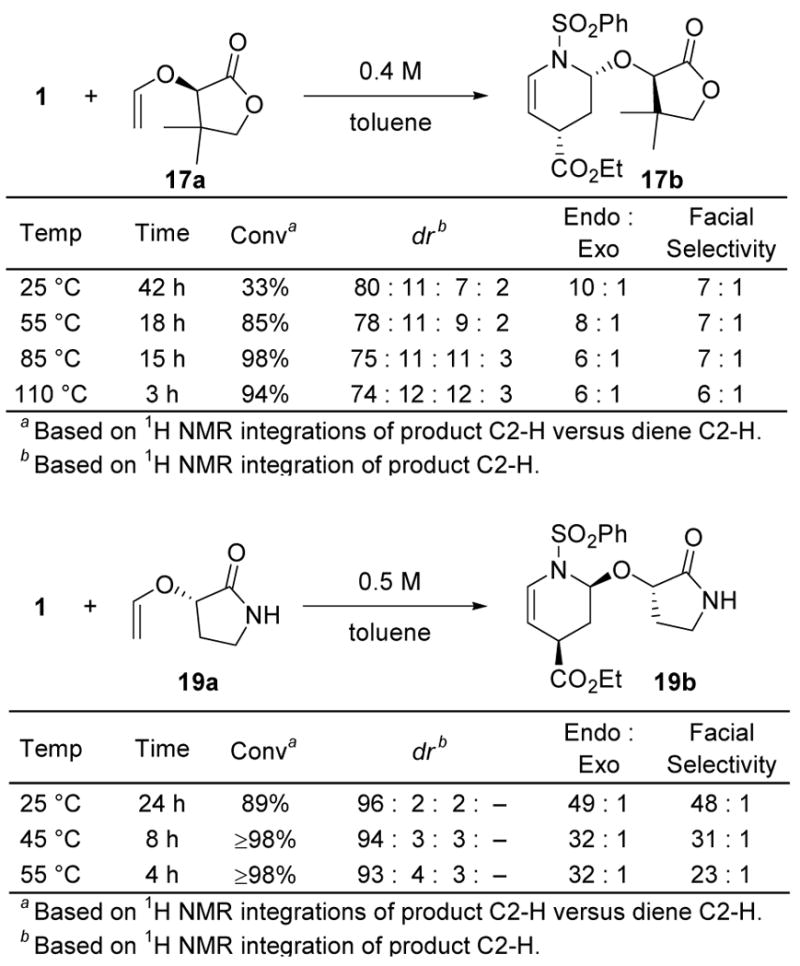
Temperature Dependence.
As anticipated, the reactions exhibit a pronounced dependence on the concentration with satisfactory rates observed at 25 °C at reactant concentrations ≥0.5 M, Figure 10.
Figure 10.
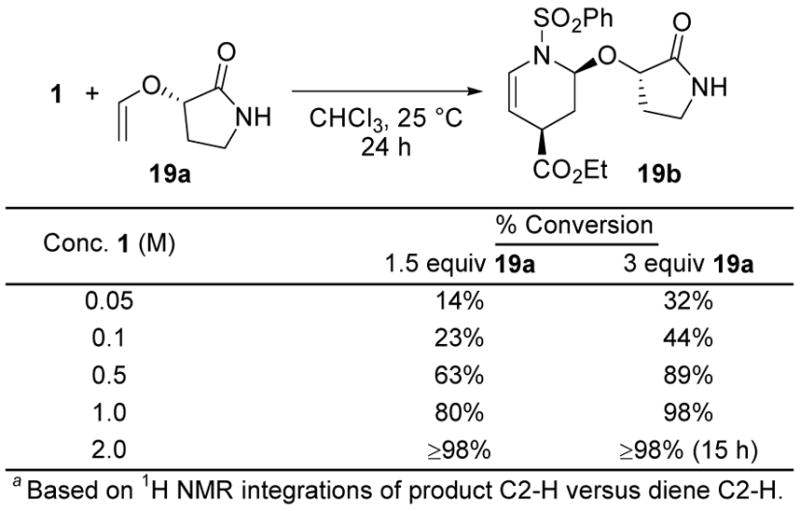
Concentration Dependence.
Consistent with expectations of a concerted [4 + 2] cycloaddition reaction, the rate of the reaction of diene 1 with dienophile 19a as well as the diastereoselectivity exhibited little solvent dependency, Figure 11. In fact, slightly faster rates of reaction were observed as the solvent polarity decreased, not increased, consistent with little or no charge buildup in the transition state of the reaction. Moreover, the slight rate enhancements as the solvent polarity decreases is consistent with the nonpolar solvent enhancement of a preferential cycloaddition transition state involving the s-trans versus s-cis enol ether conformation.27 Of all the solvents explored, CHCl3 consistently provided the cleanest reactions proceeding at good rates with outstanding diastereoselectivities accommodating a range of substrate solubilities.
Figure 11.
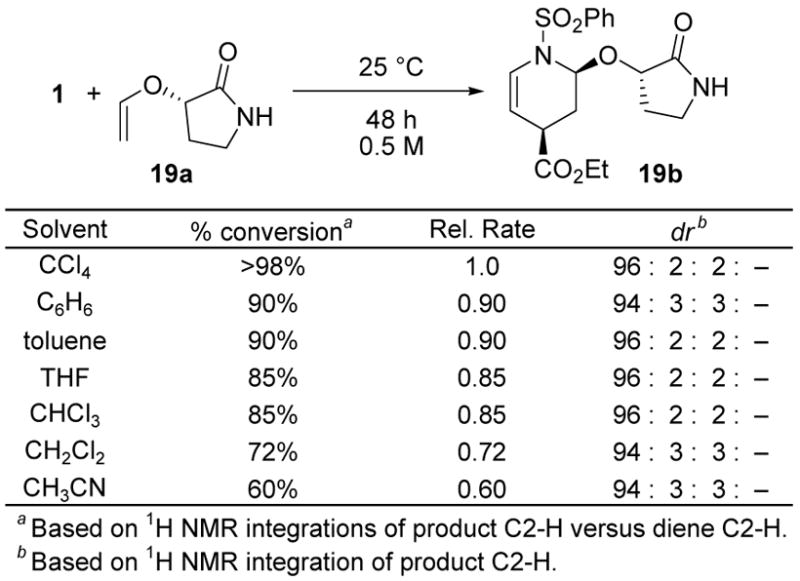
Solvent Effects.
N-Sulfonyl Substituent: Impact on Reactivity and Diastereoselectivity
Although the reactivity of the N-phenylsulfonyl azadiene 1 is excellent at 25 °C and its reaction diastereoselectivity with 19a is outstanding, alternative sulfonyl substituents were examined in efforts to establish their impact and potential importance. Moreover, in addition to a predictable impact that their electron-withdrawing properties might have on reactivity or that their steric size might have on the facial diastereoselectivity, the additional methods available for their deprotective removal (e.g., 21a/H+ or 22a and 23a/RS–)28,29 further extend the scope of the N-sulfonyl-1-aza-1,3-butadiene Diels–Alder reactions. Thus, dienes 20–25a were prepared by direct condensation of ethyl trans-4-oxo-2-butenoate with the corresponding sulfonamide (0.6 equiv of TiCl4, 2.3 equiv Et3N, CH2Cl2, 15 to 0 °C, 60–85%) and their reaction with 19a compared alongside diene 1. Represented in Figure 12 are conversions at a single time point under conditions that were suitable to distinguish between all seven dienes.
Figure 12.
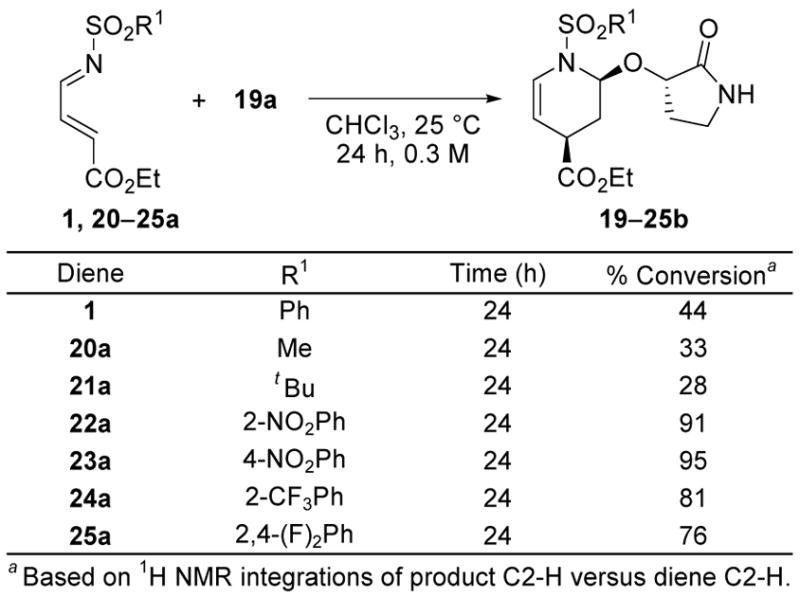
Sulfonyl Substituent.
Consistent with expectations, both 20a and 21a bearing aliphatic sulfonyl substituents were slightly less reactive than the phenylsulfonyl diene 1. Notably, the reactivity and reaction diastereoselectivity (>20:1 endo:exo; >20:1 facial selectivity) of 20a and 21a were comparable suggesting that the size of the sulfonyl substitutent has little impact on the reaction consistent with the transition state model (Figure 8). The addition of electron-withdrawing phenyl substituents (22–25a) predictably and significantly increase the diene reactivity. This smooth, predicable, and tunable reactivity of the substituted phenylsulfonyl derivatives allows the extension of these studies to 1-aza-1,3-butadienes that might possess an intrinsic lower reactivity. Each of the dienes exhibited a cycloaddition diastereoselectivity with 19a that was not distinguishable from that of 1 (>20:1 endo, >20:1 facial selectivity).
Cycloadduct Transformations
Catalytic hydrogenation of the cycloadduct double bond followed by acid-catalyzed (TFA) removal of the chiral auxiliary in the presence of water or an alcohol cleanly provides the 2-alkoxy or 2-hydroxy derivatives and this is illustrated in Scheme 3 with 17b, 19b, and 21b. Limited attempts to remove (exchange) the auxilary without prior reduction (or reaction) of the double bond led to preferential and additional alkoxy addition at C5 providing the 2,5-dialkoxy derivative. Unlike the lactam 19b, reactions of the cycloadducts 17b and 18b bearing the lactone auxilaries were often precluded by preferential ring opening reactions of the reactive five-membered lactones and this was more significant with 18b versus 17b. Finally, reduction of cycloadduct 22b (H2, Pd(OH)2/C, EtOH) followed by acid treatment of the resulting amine led to cyclization to the unique tricyclic heterocycle 38 in superb conversions.
Scheme 3.
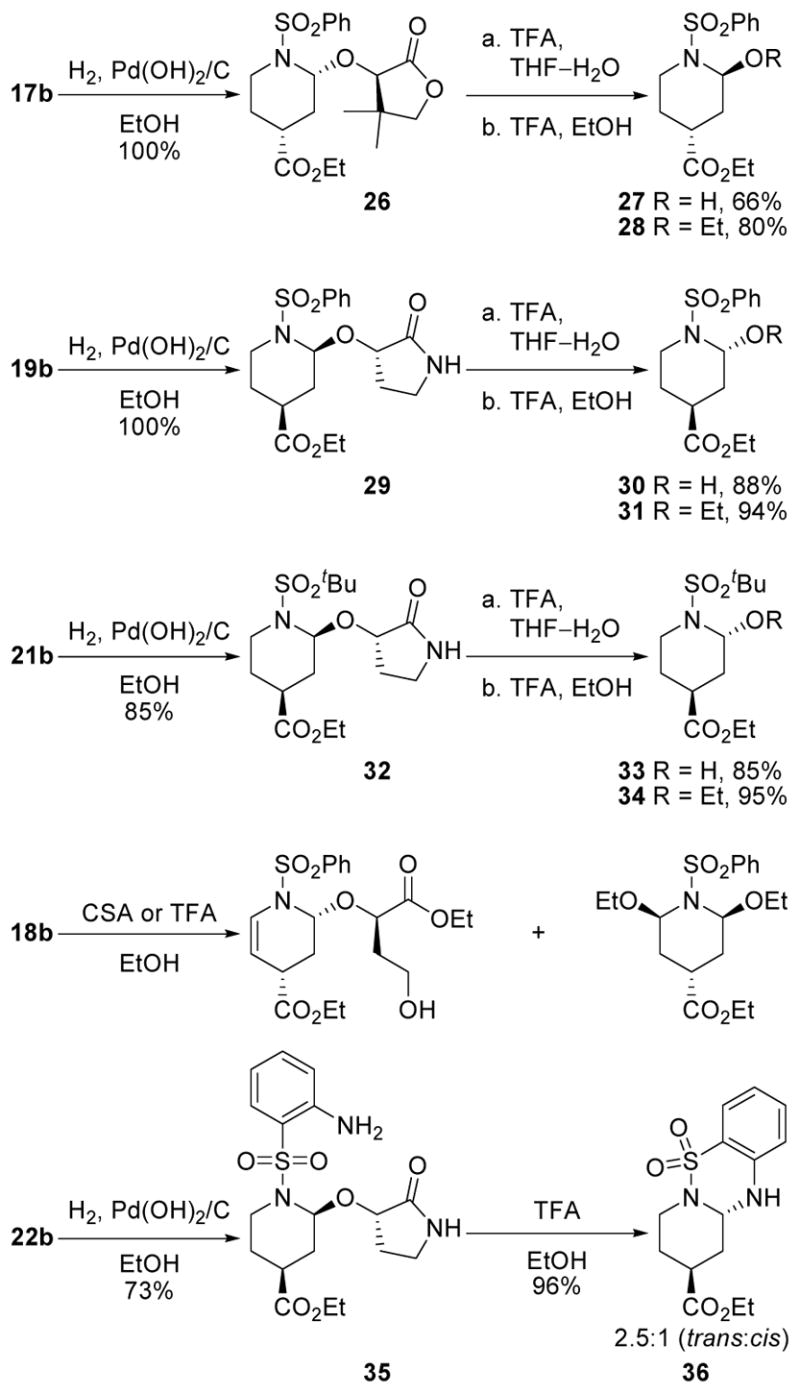
Dienophile N-Substitution
With the emergence of 19a bearing a chiral cyclic amide auxiliary as the most effective dienophile for use with the N-sulfonyl-1-azadienes, the role of the free NH of the secondary amide became important to establish. Although the comparison of 18a (the corresponding lactone) with 19a suggested little role for the amide free NH, the latter did exhibit a greater endo diastereoselectivity (49 vs 24:1) and a significantly better endo facial selectivity (48 vs 18:1). Moreover, and on occasion, competitive dimerization addition of the amide NH of 19a or its cycloadducts with the N-sulfonyl-1-aza-1,3-butadiene or dienophile enol ether was observed in the slower reactions when one of the reaction partners was enlisted in larger excesses at higher reaction concentrations or at increased reaction temperatures. As a consequence, we examined the corresponding N-methyl derivative 37a possessing a cyclic tertiary amide as the chiral auxiliary which lacks the free NH of 19a. Thus, N-methylation of 19a (1.0 equiv of NaH, 1.5 equiv of MeI, THF, 25 °C, 8 h, 95%) cleanly provided 37a ([α]D25 − 49 (c 0.58, CHCl3, >99% ee). Enol ether 37a reacted with the prototypical N-sulfonyl-1-azabutadiene 1 to provide 37b with an endo diastereoeselectivity (49:1) and endo facial selectivity (48:1) that was indistinguishable from those of 19a (Figure 13). Thus, the free NH of 19a does not contribute to the diastereoselectivity of its cycloaddition reactions and the behavior of 37a along with the transition state model in Figure 8 suggests that a wide range of amide substitutions might be accommodated. Moreover, 37a proved much more soluble in the more nonpolar solvents (toluene) than 19a where it exhibited a comparable or perhaps slightly enhanced reactivity and, unlike 19a, it is not susceptible to competitive dimerization reactions entailing reactions of the free NH.
Figure 13.
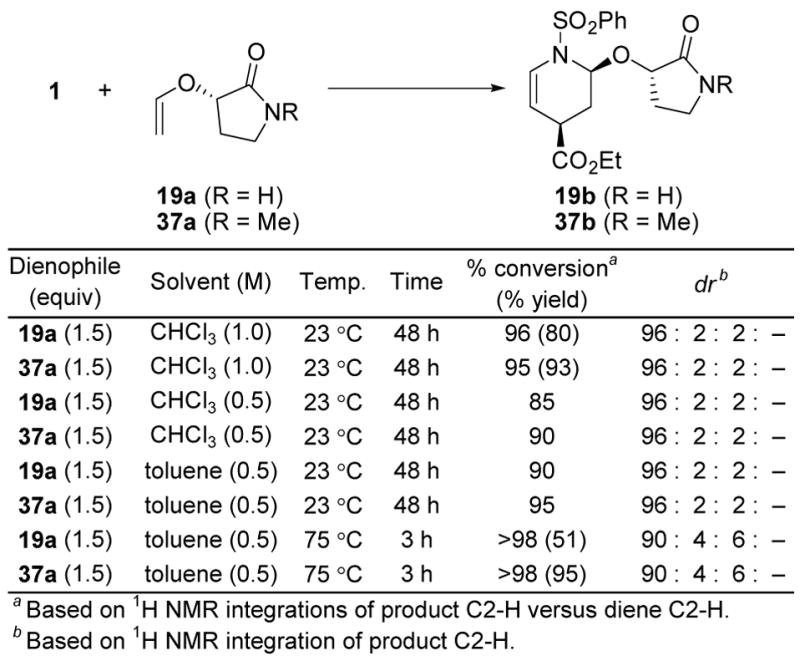
Dienophile N-Substitution.
Conclusions
A systematic examination of the [4 + 2] cycloaddition of N-sulfonyl-1-aza-1,3-butadienes with enol ethers bearing chiral auxilaries led to the delineation and optimization of factors that influence the facial selectivity of the Diels–Alder reaction while maintaining the intrinsically superb endo diastereoselectivity. The emerging transition state model of the [4 + 2] cycloaddition (Figure 8) led to the examination of three readily available, but previously unexplored auxilaries (18a, 19a and 37a) that provided remarkable selectivities (for 19a and 37a: 49:1 endo and 48:1 facial diastereoselectivity) for an uncatalyzed Diels–Alder reaction that proceeds at room temperature. Additional uses of the new vinyl ether auxilaries and applications of the azadiene Diels–Alder reaction are in progress and will be reported in due course.
Supplementary Material
Full experimental details are provided. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA 42056) and the Skaggs Institute for Chemical Biology. RCC is a Skaggs Fellow.
References
- 1.(a) Boger DL, Kasper AM. J Am Chem Soc. 1989;111:1517. [Google Scholar]; (b) Boger DL, Corbett WL, Wiggins JM. J Org Chem. 1990;55:2999. [Google Scholar]; (c) Boger DL, Curran TT. J Org Chem. 1990;55:5439. [Google Scholar]; (d) Boger DL, Corbett WL, Curran TT, Kasper AM. J Am Chem Soc. 1991;113:1713. [Google Scholar]; (e) Boger DL, Corbett WL. J Org Chem. 1993;58:2068. [Google Scholar]
- 2.(a) Boger DL, Weinreb SM. Hetero Diels–Alder Methodology in Organic Synthesis. Academic; San Diego: 1987. [Google Scholar]; (b) Boger DL. Tetrahedron. 1983;39:2869. [Google Scholar]; (c) Boger DL. Chem Rev. 1986;86:781. [Google Scholar]; (d) Boger DL. Chemtracts: Org Chem. 1996;9:149. [Google Scholar]
- 3.Boger DL, Cassidy KC, Nakahara S. J Am Chem Soc. 1993;115:10733. [Google Scholar]
- 4.Boger DL, Hüter O, Mbiya K, Zhang M. J Am Chem Soc. 1995;117:11839. [Google Scholar]
- 5.Boger DL, Hong J. J Am Chem Soc. 1998;120:1218. [Google Scholar]
- 6.Boger DL, Blagg BSJ. Tetrahedron. 2002;58:6343. [Google Scholar]
- 7.Schnermann MJ, Boger DL. J Am Chem Soc. 2005;127:15704. doi: 10.1021/ja055041f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Posner GH, Wettlaufer DG. Tetrahedron Lett. 1986;27:667. [Google Scholar]
- 9.(a) Dujardin G, Rossignol S, Brown E. Tetrahedron Lett. 1996;37:4007. [Google Scholar]; (b) Dujardin G, Rossignol S, Brown E. Synthesis. 1998:763. [Google Scholar]; (c) Gong J, Bonfand E, Brown E, Dujardin G, Michelet V, Genêt J. Tetrahedron Lett. 2003;44:2141. [Google Scholar]; (d) Gaulon C, Dhal R, Chapin T, Maisonneuve V, Dujardin G. J Org Chem. 2003;68:4338. doi: 10.1021/jo040102y. [DOI] [PubMed] [Google Scholar]
- 10.Gizecki P, Dhal R, Poulard C, Gosselin P, Dujardin G. J Org Chem. 2004;37:4007. doi: 10.1021/jo0203138. [DOI] [PubMed] [Google Scholar]
- 11.Review: Denmark SE, Thorarensen A. Chem Rev. 1996;96:137. doi: 10.1021/cr940277f.
- 12.Pettus TRR, Selenski C. J Org Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe WH, Conlon LE. J Am Chem Soc. 1957;79:2828. [Google Scholar]
- 14.Tan DS, Schreiber SL. Tetrahedron Lett. 2000;41:9509. [Google Scholar]
- 15.Dujardin G, Rossignol S, Brown E. Tetrahedron Lett. 1995;36:1653. [Google Scholar]
- 16.Okimoto Y, Sakaguchi S, Ishii Y. J Am Chem Soc. 2002;124:1590. doi: 10.1021/ja0173932. [DOI] [PubMed] [Google Scholar]
- 17.Bosch M, Schlaf M. J Org Chem. 2003;68:5225. doi: 10.1021/jo034376h. [DOI] [PubMed] [Google Scholar]
- 18.Atomic coordinates for the major cycloadducts derived from the reaction of 1 with 3a (3b, CCDC286828), 6a (6b, CCDC286829), 12a (12b, CCDC286826), 17a (17b, CCDC286830), and 19a (19b, CCDC286827, (S)-enantiomer) and of the major cycloadduct from the reaction of 21a with 19a (21b, CCDC287011, (S)-enantiomer) have been deposited with the Cambridge Crystallographic Data Center.
- 19.Denmark SE, Schnute ME. J Org Chem. 1991;56:6738. [Google Scholar]
- 20.Denmark SE, Thorarensen A. J Org Chem. 1996;61:6727. doi: 10.1021/jo960912c. [DOI] [PubMed] [Google Scholar]
- 21.Swindell CS, Tao M. J Org Chem. 1993;58:5889. [Google Scholar]
- 22.(a) Prapansiri V, Thornton ER. Tetrahedron Lett. 1991;32:3147. [Google Scholar]; (b) Dujardin G, Molato S, Brown E. Tetrahedron: Asymmetry. 1993;4:193. [Google Scholar]
- 23.Sarairi D, Maurey G. Bull Soc Chim Fr. 1987:297. [Google Scholar]
- 24.Perhaps the most expedient approach to either enantiomer of 3-hydroxy-2-pyrrolidinone is Amano PS-C lipase mediated alcoholysis of racemic 3-acetoxy-2-pyrrolidinone (>99% ee of either enantiomer at 50% conversion, er = 1057): Kamal A, Ramana KV, Ramana AV, Babu AH. Tetrahedron: Asymmetry. 2003;14:2587.
- 25.The two enantiomers of 19a are readily separable by chiral phase HPLC: 2 × 25 cm Chiralcel OD, 10% i-PrOH/hexanes, 1 mL/min, tR = 15.3 (3R) and 17.0 (3S) min, α = 1.11.
- 26.Juarist E, Cuervas G. Tetrahedron. 1992;48:5019. [Google Scholar]
- 27.Liu J, Niwayama YY, You Y, Houk KN. J Org Chem. 1998;63:1064. [Google Scholar]
- 28.Sun P, Weinreb SM, Shang M. J Org Chem. 1997;62:8604. doi: 10.1021/jo971455i. [DOI] [PubMed] [Google Scholar]
- 29.Kan T, Fukuyama T. Chem Commun. 2004:353. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details are provided. This material is available free of charge via the internet at http://pubs.acs.org.