

Abstract
Integral membrane proteins, which include many cellular effector proteins and drug targets, can be difficult to produce, purify and manipulate. Although the isolated ectodomains of many membrane proteins can be expressed as water soluble proteins, biological activity is frequently lost when these proteins are released from the membrane surface. An example is tissue factor, the integral membrane protein that triggers the blood clotting cascade and for which membrane anchoring is essential: Its isolated ectodomain (soluble tissue factor) can be expressed with high yield in bacteria but is orders of magnitude less active than the intact, membrane-anchored protein. We now report full restoration of biological activity to the isolated tissue factor ectodomain by engineering a hexahistidine tag onto its C-terminus and using it in combination with membrane bilayers containing nickel-chelating lipids. When soluble tissue factor was tethered to the membrane surface via such metal-chelating lipids, it bound factor VIIa with the same high affinity as wild-type tissue factor, and the resulting factor VIIa-tissue factor complexes supported factor X activation and factor VII autoactivation with essentially wild-type enzyme kinetic constants. Furthermore, when such bilayers were immobilized onto solid supports they efficiently captured histidine-tagged soluble tissue factor directly from crude culture supernatants - with full biological activity - obviating the need for purification or laborious membrane reconstitution procedures. This strategy is rapid, efficient, scalable, automatable, and should be applicable to other integral membrane proteins, especially those with a single transmembrane domain. Applications include high-throughput screening of mutants or drugs, flow reactors, clinical assays and point-of-care instrumentation.
Integral membrane proteins represent an estimated 20-30% of the proteins in many genomes and are a major class of current and future drug targets (1-3). They are, however, more difficult to express at high yield than are soluble proteins, and they require detergents to extract them from membranes and keep them soluble during purification, which may diminish their stability and activity. Although effective procedures have been developed to reconstitute purified integral membrane proteins into phospholipid vesicles, most are laborious, time-consuming, and expensive (4).
The isolated ectodomains of some integral membrane proteins can be expressed with very high yield in recombinant systems and are generally far easier to handle than are their membrane-anchored counterparts. However, the soluble ectodomains of some membrane proteins have little or no biological activity. An example is tissue factor (TF1) the integral membrane protein responsible for triggering the blood clotting cascade in normal hemostasis and many thrombotic diseases (5). TF tightly binds and allosterically activates the plasma serine protease, factor VIIa (fVIIa), with the resulting TF:VIIa complex activating two membrane-bound protease zymogens - factors IX and X (fX) - via limited proteolysis. Studies with truncated forms of recombinant human TF have shown that membrane anchoring is essential for procoagulant activity (6, 7). Thus, while the isolated ectodomain of TF (sTF) is highly water soluble and can be produced recombinantly with high yield (7-12), it has reduced affinity for fVIIa (8), cannot promote the conversion of zymogen factor VII (fVII) to fVIIa (13, 14), and has drastically reduced procoagulant activity (7-9, 13, 15).
We sought to develop a facile method for restoring full biological activity to the isolated ectodomains of membrane proteins while retaining all their desirable expression, solubility and handling characteristics. Recombinant human TF is an excellent subject for such investigations, since membrane-anchored TF (membTF), when incorporated into suitable phospholipid vesicles, is the active principle in recombinant thromboplastin reagents that are widely used in clinical clotting assays (16). In addition, the TF:VIIa complex has become an increasingly attractive target for developing novel, highly selective antithrombotic agents (17, 18).
Nickel-chelating lipids such as DOGS-NTA-Ni (1,2-dioleoyl-sn-glycero-3-[(N(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] nickel salt) have been used to create two-dimensional crystals of oligohistidine-tagged recombinant proteins on artificial membranes, in order to obtain structural information by electron crystallography and other approaches (19, 20). They have also been used recently to attach oligohistidine-tagged peptides to liposomes, as a delivery method for peptides to targeted cells (21); however, they have rarely been used to attach proteins to membranes for functional studies (22). We reasoned that incorporating a hexahistidine tag at the C-terminus of sTF would allow it to bind to chelated nickel ions at the membrane surface and become an effective protein cofactor for fVIIa (but at much lower surface densities than when preparing two-dimensional protein crystals!). We show here that this system restores full biological function to the isolated ectodomain of this important integral membrane protein.
EXPERIMENTAL PROCEDURES
Materials
Pooled normal human plasma was from George King Bio-Medical (Overland Park, KS). Chicken egg phosphatidylcholine (PC), porcine brain phosphatidylserine (PS), bovine liver phosphatidylethanolamine (PE) and DOGS-NTA-Ni were from Avanti Polar Lipids, Inc. (Alabaster, AL). Chromozym® t-PA substrate (N-methylsulfonyl-D-Phe-Gly-Arg-4-nitroanilide acetate) was from Roche Applied Science (Indianapolis, IN). Spectrozyme fXa substrate (methoxycarbonyl-D-cyclohexylglycyl-Gly-Arg-4-nitroanilide acetate) and recombinant human fVIIa were from American Diagnostica, Inc. (Stamford, CT). Purified plasma-derived fVII, fX and factor Xa (fXa) were from Enzyme Research Laboratories (South Bend, IN). Bio-Beads® SM-2 adsorbent was from BioRad Laboratories (Hercules, CA). Octaethylene glycol monododecyl ether (C12E8) was from Fluka and antifoam C from Sigma (Sigma-Aldrich, St. Louis, MO). Recombinant human membTF (consisting of amino acids 1-244 numbered according to Morrissey et al. (23)) and sTF (amino acids 1-219) were purified from an E. coli expression system as described (10, 24). The sTF expression construct was modified to include an oligohistidine tag (sTF-His) by inserting double-stranded oligonucleotides encoding the amino acid sequence, GGAAGHHHHHH, followed by a stop codon, immediately after residue 219 of TF. sTF-His was expressed in E. coli strain BL21(DE3) and purified by immunoaffinity chromatography as previously described for sTF (10). Crude culture supernatants containing sTF were collected by centrifugation from spent E. coli cultures that had been induced overnight at 25°C with 50 μM isopropyl-β-D-thiogalactopyranoside. Concentrations of sTF-His were determined by ELISA (10), with minor changes: TF9-5B7 IgG (a monoclonal anti-tissue factor antibody (25)) was used as the capture antibody and biotinylated HPC4 IgG (a monoclonal antibody to the HPC4 epitope tag at the N-terminus of sTF-His (10)) was used as the detecting antibody.
Liposomes
Unilamellar phospholipid vesicles were prepared using the rapid Bio-Bead method (26) with 6 mM C12E8 as the detergent. Liposome compositions were as follows: PCPS contained 20% PS, 80% PC; NiPCPS contained 15% DOGS-NTA-Ni, 65% PC, 20% PS; and NiPCPSPE contained 10% DOGS-NTA-Ni, 47.5% PC, 12.5% PS, 30% PE. Liposome compositions are given throughout as mol%, and their concentrations as the molar concentration of total lipid.
Incorporation of membTF into liposomes
Purified membTF was incorporated into unilamellar vesicles composed of PCPS, NiPCPS, or NiPCPSPE, as described (26), using 6 mM C12E8 as the detergent. The effective (available) concentration of active TF molecules incorporated into the liposomes was quantified by titration with fVIIa as described (8). Concentrations of TF in liposomes are reported here as the available TF concentrations.
fVIIa binding
Binding affinities of fVIIa for membTF incorporated into PCPS or sTF-His plus NiPCPS were quantified using the TF-dependent increase in the rate of fX activation by fVIIa as the readout for complex formation. Activation of fX was measured using a continuous chromogenic assay (13), with reaction mixtures containing a fixed concentration of fVIIa and increasing concentrations of either membTF (incorporated into PCPS liposomes) or sTF-His plus NiPCPS liposomes. The buffer was HBSAC (20 mM Hepes pH 7.4, 100 mM NaCl, 0.1% NaN3, 0.1% bovine serum albumin, 5 mM CaCl2). Reactions were initiated by adding a mixture of fX and chromogenic substrate in a 96-well plate, yielding final concentrations of 0.1 pM fVIIa, 0-500 pM membTF or sTF-His, 50 μM liposomes, 30 nM fX and 0.5 mM Spectrozyme fXa. Change in A405 was monitored at ambient temperature in a VERSAmax microplate reader (Molecular Devices Corp., Sunnyvale, CA). Initial rates of fX activation were determined by fitting a second-order polynomial to the A405 data (13, 27) and were converted to concentrations of TF:VIIa complex (8). The quadratic ligand binding equation was fitted to plots of [TF:VIIa] vs. [TF] to derive Kd values for the binding of fVIIa to TF (13).
fX activation using liposomes
Initial rates of fX activation were measured at ambient temperature in multiwell plates using a discontinuous chromogenic assay (8) modified as follows: Reaction mixtures in HBSAC contained 500 pM fVIIa, varying fX, and either 4 pM membTF in PCPS (plus 50 μM PCPS) or 4 pM sTF-His plus 50 μM NiPCPS. At varying times, 20 μl aliquots were removed into a 96-well plate containing 100 μl Stop Buffer I (40 mM Mes-NaOH pH 5.8, 12 mM EDTA, 50 mM NaCl, 0.25% Triton X-100, 0.1% NaN3, 0.012% Antifoam C) at 4°C. After warming the stopped reactions to room temperature, fXa was detected by adding 60 μl of 1.5 mM Spectrozyme fXa in HBSAC plus 0.6 M Tricine-NaOH, pH 8.4 and quantifying change in A405. Amounts of fXa generated were determined by comparison to a standard curve with purified fXa.
fX activation using immobilized lipids
All steps were conducted at ambient temperature. Lipid mixtures were dried down in a borosilicate glass test tube under a gentle stream of dry nitrogen gas to remove the solvent (chloroform), then redissolved in n-hexane at a total lipid concentration of 2 mM. Each well of a 96-well polystyrene plate (Costar 9018 high-binding plates, Corning, Inc., Corning, NY) received 60 nmol total lipid, and the hexane was allowed to evaporate in a fume cupboard. The wells were then incubated for 1 hr with 100 μl TBSA (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.02% NaN3, 1% bovine serum albumin), aspirated and washed thrice with TBS (TBSA without albumin). Wells were incubated for 1 hr with 100 μl of the indicated concentration of sTF or sTF-His in HBSA (HBSAC without calcium), then aspirated and washed thrice with TBS. Wells were incubated for 1 hr with 100 μl of 5 pM factor VIIa in HBSAC, after which reactions were initiated by adding 100 μl of 1 mM Spectrozyme fXa substrate and the indicated fX concentrations HBSAC. Change in A405 was quantified and the amount of fXa generated determined by comparison to a standard curve.
fVII autoactivation
Rates of fVII autoactivation were measured essentially as described (28). Equimolar concentrations of fVII and TF (15 nM each) were incubated in HBSAC at 37°C. At various time points, 20 μl aliquots were removed to a 96-well plate containing 60 μl Stop Buffer II (0.1 M Tricine-NaOH, pH 8.4, 6.7 mM CaCl2, 0.1% bovine serum albumin, 0.1% Triton X-100, 0.05% NaN3 and 100 nM sTF). A 20 μl aliquot of 5 mM Chromozym t-PA substrate was added and the change in A405 was monitored at ambient temperature. Two-dimensional second-order rate constants (k2D) were determined as described (28).
TF clotting assay
Thromboplastin reagents for clotting assays were prepared using membTF in PCPS, NiPCPS, or NiPCPSPE liposomes in Low Salt TBSA (TBSA containing 10 mM NaCl instead of 100 mM) to which additional appropriate liposomes were added to achieve a total lipid concentration of 100 μM. Thromboplastin reagents containing either sTF or sTF-His were likewise prepared in Low Salt TBSA plus 100 μM PCPS, NiPCPS, or NiPCPSPE liposomes. Clotting assays were performed in a STart 4 coagulometer (Diagnostica Stago, Parsippany, NJ). Briefly, 50 μl aliquots each of 25 mM CaCl2 and thromboplastin reagent were incubated together for 120 s in a coagulometer cuvette at 37°C. A 50 μl aliquot of pre-warmed pooled normal human plasma was then added, and the time to clot formation was measured. We have also discovered that nickel-chelating lipids can activate the contact, or intrinsic, pathway of blood clotting provided the lipids are preincubated with plasma for at least two minutes before adding calcium ions (Waters and Morrissey, manuscript in preparation). By modifying the clotting assay to add plasma last, activation of the contact pathway was minimized and the clotting times were dependent upon TF activity. A unit of TF activity was defined as the amount of TF in the final 150 μl clotting reaction that yields a 50 s clot time.
fXa clotting assay
The ability of fXa to initiate clotting in the presence of PCPS, NiPCPS, or NiPCPSPE liposomes was also measured. Briefly, a 50 μl aliquot of 1 nM fXa and 100 μM liposomes in Low Salt TBSA was incubated with 50μl of 25 mM CaCl2 for 120 s in a coagulometer cuvette at 37°C, after which 50 μl pooled normal plasma was added.
RESULTS AND DISCUSSION
In initial studies we tested two sTF constructs containing C-terminal oligohistidine tags (expressed in E. coli): one in which six His residues were attached directly after residue 217 and another in which a five amino acid spacer (GGAAG), followed by six His residues, was attached after residue 219. Both proteins were active but the latter construct, termed sTF-His, had higher specific activity in clotting assays when mixed with NiPCPSPE vesicles (140 U/ng for sTF-His versus 9 U/ng with the construct lacking the GGAAG spacer) and was therefore used for the rest of our study. The reason for the superior activity of sTF-His is unknown but it is possible that the peptide spacer allowed better alignment of the sTF-His protein on the membrane surface, compared to attaching the hexahistidine tag directly to the C-terminus of sTF. We next employed sTF-His in conjunction with mixed phospholipid vesicles containing DOGS-NTA-Ni, PC and PS (NiPCPS) and compared its activity to that of membTF reconstituted into PCPS liposomes. (PS was employed in both preparations because negatively charged phospholipids are required for optimal TF:VIIa activity (6).)
Wild-type TF incorporated into a suitable phospholipid bilayer will bind its primary ligand, fVIIa, with extremely high affinity (Kd < 50 pM), while sTF binds fVIIa at least one hundred times more weakly (Kd ≈ 5 nM) (8). The difference in the two binding affinities probably reflects the contribution of protein-phospholipid interactions between the fVIIa Gla domain and negatively charged phospholipids that help stabilize the TF:VIIa complex on the membrane surface; such protein-phospholipid interactions are lacking when the same complex is assembled with sTF. We therefore sought to determine if attaching the TF ectodomain to the membrane via an oligohistidine tag and metal-chelating lipids would be sufficient to restore the contribution of protein-phospholipid interactions toward stabilizing the TF:VIIa complex on the membrane surface. When fVIIa binds to TF, its rate of fX activation increases dramatically, so this can be used as a convenient readout for TF:VIIa complex formation. Using this approach, we found that fVIIa bound with extremely high affinity to the combination of sTF-His plus NiPCPS, with a Kd of 10.8 pM (Table 1). This was essentially identical to its affinity for membTF in PCPS liposomes (Kd = 10.0 pM). Therefore, when the isolated TF ectodomain was attached to the membrane surface via interaction with metal-chelating lipids, its fVIIa binding ability was indistinguishable from membTF that spanned the lipid bilayer.
Table 1.
Binding and kinetic constants for TF:VIIa complexes
| fVIIa binding |
fX activation |
fVII autoactivation |
||||
|---|---|---|---|---|---|---|
| TF | Lipid | Kd (pM) | Km (nM) | kcat (s-1) | kcat/Km (μM-1 s-1) | k2D (m2 mol-1 s-1) |
| membTFa | PCPS liposomes | 10.0 ± 4.4 | 59 ± 0.88 | 3.5 ± 0.38 | 60.2 ± 6.35 | 3.4 (± 0.15) × 107 |
| sTF-Hisa | NiPCPS liposomes | 10.8 ± 4.2 | 38 ± 3.8 | 3.3 ± 0.43 | 87.1 ± 12.3 | 2.9 (± 0.37) × 107 |
| sTF-Hisb | Immobilized NiPCPS | n.dc | 66 ± 6.4 | 4.1 ± 1.4 | 63.1 ± 22.3 | n.dc |
Purified proteins.
Captured from crude culture supernatants.
Not determined.
A stringent test of TF:VIIa function is its ability to support the activation of its natural protein substrate, fX. This requires that TF be incorporated into a suitable phospholipid membrane (i.e., one containing negatively charged phospholipids). The isolated TF ectodomain, on the other hand, supports orders of magnitude lower rates of fX activation than does membTF - even in the presence of PCPS liposomes - because sTF is not anchored in the membrane (8, 13). We therefore compared the ability of various forms of TF to support the activation of fX under equivalent concentrations of enzyme (500 pM fVIIa), cofactor (4 pM TF) and liposomes (50 μM total lipid). The combination of sTF-His and NiPCPS supported rates of fX activation by fVIIa that were comparable to those obtained using membTF in PCPS liposomes (Table 1). The kcat values for the two forms of the TF:VIIa complex were similar, while the Km of fVIIa bound to sTF-His plus NiPCPS for fX was actually lower than with membTF in PCPS, leading to a slightly higher overall catalytic efficiency (kcat/Km). The high enzymatic activity of fVIIa bound to sTF-His was dependent on both nickel-chelating lipids and the oligohistidine tag on sTF, since mixing either sTF-His with PCPS liposomes, or sTF with NiPCPS liposomes, supported negligible rates of fX activation by fVIIa (data not shown).
An additional function of TF is to promote the autoactivation of fVII in a reaction that is dependent on TF surface density (28, 29). In contrast, sTF fails to support this reaction (14). Under conditions of identical TF surface densities, both membTF in PCPS and sTF-His plus NiPCPS supported fVII autoactivation with comparable rate constants (Table 1). Together, these findings show that it is not necessary for the TF ectodomain to be covalently attached to a membrane anchor to achieve wild-type levels of TF activity; reversible attachment of sTF to the membrane surface via metal-chelating lipids is functionally equivalent to conventional membrane anchoring.
An important test of TF function is its ability to promote the clotting of plasma. The procoagulant activities of membTF, sTF, and sTF-His were therefore compared in the presence of liposomes of varying composition. As expected, both sTF and sTF-His had very poor procoagulant activities in the presence of PCPS vesicles, while membTF in PCPS had high procoagulant activity (Figure 1a). When DOGS-NTA-Ni lipids were incorporated into the liposomes (NiPCPS), sTF-His and membTF had comparable procoagulant activities, but sTF continued to exhibit very poor activity (Figure 1b). Notably, however, the specific procoagulant activities achieved with NiPCPS liposomes for both membTF and sTF-His were consistently lower than those achieved using membTF in PCPS (compare Figures 1a and 1b). Because PE is known to synergize with PS to enhance TF procoagulant activity (24), we tested the inclusion of PE in the liposomes (NiPCPSPE). This increased the specific procoagulant activity of both membTF and sTF-His to a level comparable to that of membTF in PCPS (Figure 1c).
Figure 1.

Procoagulant activities. Clotting times obtained with membTF in liposomes (diamonds), sTF plus liposomes (triangles), or sTF-His plus liposomes (open circles) are plotted versus TF concentration. Liposome compositions were: (a) PCPS, (b) NiPCPS, and (c) NiPCPSPE. Data are mean ± standard error.
The results in Table 1 demonstrated that the combination of sTF-His plus NiPCPS supported wild-type levels of fX activation and fVII autoactivation, arguing that DOGS-NTA-Ni lipids are not detrimental to TF:VIIa activity. The reduced procoagulant activity of membTF in liposomes containing DOGS-NTA-Ni therefore suggested to us that the next step in the clotting cascade (the prothrombinase complex) may have lower activity on NiPCPS surfaces compared with PCPS membranes. To test this, we measured fXa-initiated plasma clotting in the presence of liposomes of varying composition, obtaining clotting times of 10.9 ± 0.3 s with PCPS liposomes, 18.9 ± 0.6 s with NiPCPS liposomes, and 11.7 ± 0.4 s with PCPSPE liposomes. The presence of PE in the liposomes was thus able to overcome the slightly inhibitory effect of DOGS-NTA-Ni toward the procoagulant activity of fXa.
The experiments described above used purified sTF-His. We reasoned that immobilized lipid bilayers containing DOGS-NTA-Ni should be able to capture sTF-His from crude mixtures, simultaneously isolating and membrane-anchoring this protein in one quick step. Although expression of sTF-His in our E. coli expression system is targeted to the periplasmic space, significant quantities accumulate in the medium of overnight cultures: Typically, 55 μg/ml (2.0 μM) sTF-His as measured by ELISA. Crude culture supernatants were therefore diluted tenfold with buffer and incubated in the wells of a polystyrene 96-well plate that had previously been coated with a lipid mixture containing DOGS-NTA-Ni, PS and PC. After washing away unbound proteins, the wells were treated with fVIIa and the rate of fX activation was measured. Because fVIIa is a very poor activator of fX in the absence of TF and a suitable phospholipid membrane, this assay is a stringent test both of the ability of the immobilized lipids to capture sTF-His and of the functional state of the resulting TF:VIIa:membrane complexes. We found that when sTF-His was captured in this way from culture supernatants it robustly supported fX activation by fVIIa, with apparent Km and kcat values that were comparable to those of membTF in PCPS liposomes (Table 1). Under these assay conditions, the concentration of purified sTF-His required to support half-maximal rates of fX activation (EC50) was 6.2 ± 4.1 nM (Figure 2), a concentration that is more than two orders of magnitude below the sTF-His concentration in our E. coli culture supernatants. Both the oligohistidine tag on sTF and the presence of nickel-chelating lipids were required, since neither combinations of sTF-His with immobilized PCPS nor of sTF with immobilized nickel-chelating lipid mixtures yielded detectable levels of fX activation, even at sTF concentrations as high as 1 μM (Figure 2).
Figure 2.
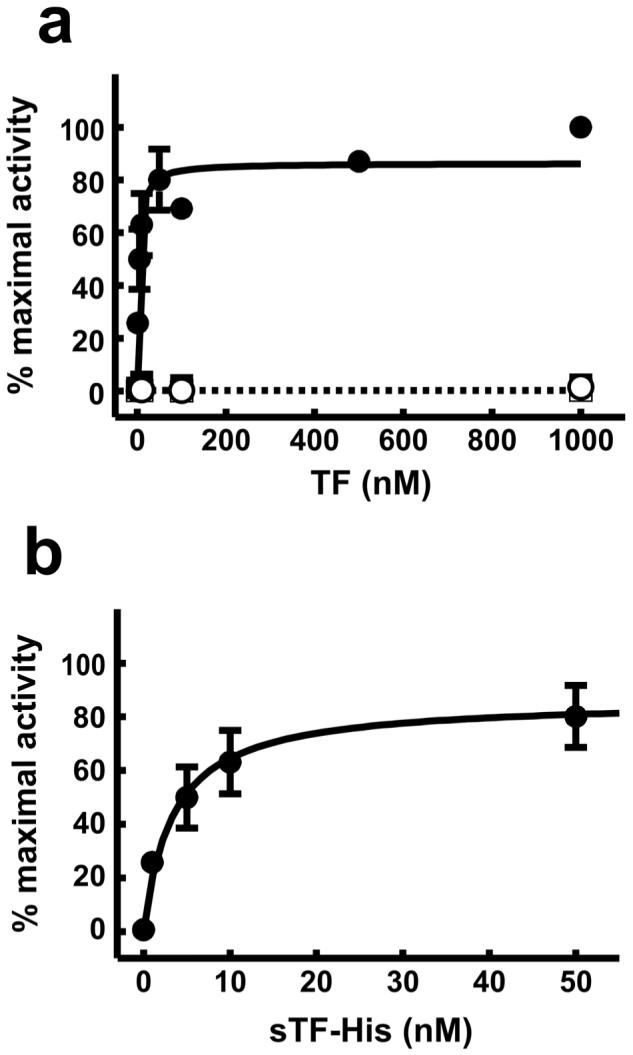
EC50 for the ability of sTF-His to enhance the rate of fX activation by fVIIa. Varying concentrations of purified sTF or sTF-His were incubated in wells of 96-well plates that had previously been coated with a mixture of either 10% DOGS-NTA-Ni, 20% PS, 70% PC (closed symbols) or 20% PS, 80% PC (open symbols). Initial rates of fXa generation were measured using 40 nM fX and are expressed as percent of the activity observed with 1 μM sTF-His in wells coated with lipid mixtures containing DOGS-NTA-Ni. (a), Concentration dependence of the ability of sTF-His (circles) or sTF (squares) to enhance the rate of fX activation by fVIIa. (b), Data for sTF-His from panel a are plotted with the x-axis expanded from 0 to 50 nM.
These studies demonstrate that full biological activity can be restored to the isolated ectodomain of an integral membrane protein by incorporating an oligohistidine tag at a suitable location on the protein, when used in conjunction with membranes containing metal-chelating lipid. They also demonstrate that membrane bilayers containing nickel-chelating lipids that have been immobilized onto a solid support can efficiently capture the histidine-tagged ectodomain from crude mixtures, simultaneously purifying the protein and anchoring it to the membrane in one simple step. Using TF as an example, the recombinant ectodomain expresses in E. coli at far higher levels than does membTF and is much easier to handle. Furthermore, reconstituting membTF into liposomes is very slow (many hours to days) and difficult to control, while mixing sTF-His with liposomes or immobilized bilayers containing DOGS-NTA-Ni is rapid and simple. This will greatly simplify not only research using TF but commercial applications of TF as well, such as the preparation of recombinant TF thromboplastin reagents (16) which otherwise require expressing recombinant membTF, purifying it, and then inserting it into liposomes in a highly controlled way. Furthermore, the ability to bind highly active preparations of sTF-His onto immobilized phospholipid bilayers containing metal-chelating lipids can facilitate the development of point-of-care clinical coagulation assays in which the activator of clotting must be attached to a chip surface. These same general approaches may also apply to the isolated ectodomains of other single-spanning integral membrane proteins. The precise number of integral membrane proteins in the human genome that pass through the membrane only once, as TF does, is unknown; however, genomic analyses reveal increasing abundance of predicted integral membrane proteins as the number of transmembrane domains decrease, suggesting that single-pass membrane proteins may be relatively common (2).
This approach can facilitate high-throughput screening methods. Using immobilized lipid bilayers containing metal-chelating lipids to capture the soluble ectodomain directly from crude mixtures such as culture supernatants, libraries of mutants of the target membrane protein can be rapidly screened for activity in a way that is easily automated. Similarly, the histidine-tagged ectodomain captured by lipid bilayers that are immobilized on multiwell plates or other surfaces can form the basis for automated screening of libraries of small molecule inhibitors. Finally, this strategy can also be used to attach the ectodomains of membrane proteins like TF to immobilized membrane surfaces in flow reactors (30, 31).
ACKNOWLEDGMENT
We thank Guoyu Li and Collin Waters for excellent technical assistance, and David Barounis for his contributions to this work.
Footnotes
- C12E8
- octaethylene glycol monododecyl ether
- DOGS-NTA-Ni
- 1,2-dioleoyl-sn-glycero-3-[(N(-amino-1-carboxypentyl) iminodiacetic acid) succinyl] nickel salt
- fVII
- coagulation factor VII
- fVIIa
- activated fVII
- fX
- factor X
- fXa
- activated fX
- membTF
- recombinant membrane-bound tissue factor (TF1-244)
- NiPCPS
- phospholipid vesicles comprising 15% DOGS-NTA-Ni, 65% PC, 20% PS
- NiPCPSPE
- phospholipid vesicles comprising 10% DOGS-NTA-Ni, 47.5% PC, 12.5% PS, 30% PE
- PC
- phosphatidylcholine
- PCPS
- phospholipid vesicles comprising 80% PC, 20% PS
- PE
- phosphatidylethanolamine
- PS
- phosphatidylserine
- sTF
- recombinant, soluble tissue factor (TF1-219)
- sTF-His
- sTF with a hexahistidine tag at the C-terminus
- TF
- tissue factor
- TF:VIIa
- enzyme complex of TF and fVIIa
Supported by grant R01 HL47014 from the National Heart Lung & Blood Institute. E.K.W. was supported in part by NIH training grant T32 GM007283.
REFERENCES
- 1.Nilsson J, Persson B, von Heijne G. Comparative analysis of amino acid distributions in integral membrane proteins from 107 genomes. Proteins. 2005;60:606–616. doi: 10.1002/prot.20583. [DOI] [PubMed] [Google Scholar]
- 2.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DT. Do transmembrane protein superfolds exist? FEBS Lett. 1998;423:281–285. doi: 10.1016/s0014-5793(98)00095-7. [DOI] [PubMed] [Google Scholar]
- 4.Seddon AM, Curnow P, Booth PJ. Membrane proteins, lipids and detergents: not just a soap opera. Biochim. Biophys. Acta. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Morrissey JH. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Colman R, Hirsh J, Marder V, Clowes A, George J, editors. Lippincott Williams and Wilkins; 2001. pp. 89–101. [Google Scholar]
- 6.Morrissey JH, Neuenschwander PF, Huang Q, McCallum CD, Su B, Johnson AE. Factor VIIa-tissue factor: functional importance of protein-membrane interactions. Thromb. Haemost. 1997;78:112–116. [PubMed] [Google Scholar]
- 7.Paborsky LR, Caras IW, Fisher KL, Gorman CM. Lipid association, but not the transmembrane domain, is required for tissue factor activity. Substitution of the transmembrane domain with a phosphatidylinositol anchor. J. Biol. Chem. 1991;266:21911–21916. [PubMed] [Google Scholar]
- 8.Neuenschwander PF, Morrissey JH. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J. Biol. Chem. 1994;269:8007–8013. [PubMed] [Google Scholar]
- 9.Waxman E, Ross JB, Laue TM, Guha A, Thiruvikraman SV, Lin TC, Konigsberg WH, Nemerson Y. Tissue factor and its extracellular soluble domain: the relationship between intermolecular association with factor VIIa and enzymatic activity of the complex. Biochemistry. 1992;31:3998–4003. doi: 10.1021/bi00131a015. [DOI] [PubMed] [Google Scholar]
- 10.Rezaie AR, Fiore MM, Neuenschwander PF, Esmon CT, Morrissey JH. Expression and purification of a soluble tissue factor fusion protein with an epitope for an unusual calcium-dependent antibody. Protein Expr. Purif. 1992;3:453–460. doi: 10.1016/1046-5928(92)90062-2. [DOI] [PubMed] [Google Scholar]
- 11.Shigematsu Y, Miyata T, Higashi S, Miki T, Sadler JE, Iwanaga S. Expression of human soluble tissue factor in yeast and enzymatic properties of its complex with factor VIIa. J. Biol. Chem. 1992;267:21329–21337. [PubMed] [Google Scholar]
- 12.Waxman E, Laws WR, Laue TM, Nemerson Y, Ross JB. Human factor VIIa and its complex with soluble tissue factor: evaluation of asymmetry and conformational dynamics by ultracentrifugation and fluorescence anisotropy decay methods. Biochemistry. 1993;32:3005–3012. doi: 10.1021/bi00063a011. [DOI] [PubMed] [Google Scholar]
- 13.Fiore MM, Neuenschwander PF, Morrissey JH. The biochemical basis for the apparent defect of soluble mutant tissue factor in enhancing the proteolytic activities of factor VIIa. J. Biol. Chem. 1994;269:143–149. [PubMed] [Google Scholar]
- 14.Neuenschwander PF, Morrissey JH. Deletion of the membrane anchoring region of tissue factor abolishes autoactivation of factor VII but not cofactor function. Analysis of a mutant with a selective deficiency in activity. J. Biol. Chem. 1992;267:14477–14482. [PubMed] [Google Scholar]
- 15.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J. Biol. Chem. 1991;266:2158–2166. [PubMed] [Google Scholar]
- 16.Tripodi A, Arbini A, Chantarangkul V, Mannucci PM. Recombinant tissue factor as substitute for conventional thromboplastin in the prothrombin time test. Thromb. Haemost. 1992;67:42–45. [PubMed] [Google Scholar]
- 17.Linkins LA, Weitz JI. New anticoagulant therapy. Annu. Rev. Med. 2005;56:63–77. doi: 10.1146/annurev.med.56.082103.104708. [DOI] [PubMed] [Google Scholar]
- 18.Lazarus RA, Olivero AG, Eigenbrot C, Kirchhofer D. Inhibitors of Tissue Factor*Factor VIIa for anticoagulant therapy. Curr. Med. Chem. 2004;11:2275–2290. doi: 10.2174/0929867043364568. [DOI] [PubMed] [Google Scholar]
- 19.Darst SA. A new twist on protein crystallization. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7848–7849. doi: 10.1073/pnas.95.14.7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubalek EW, Le Grice SF, Brown PO. Two-dimensional crystallization of histidine-tagged, HIV-1 reverse transcriptase promoted by a novel nickel-chelating lipid. J. Struct. Biol. 1994;113:117–123. doi: 10.1006/jsbi.1994.1039. [DOI] [PubMed] [Google Scholar]
- 21.Chikh GG, Li WM, Schutze-Redelmeier MP, Meunier JC, Bally MB. Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ion-chelating lipids. Biochim. Biophys. Acta. 2002;1567:204–212. doi: 10.1016/s0005-2736(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 22.Groves JT, Dustin ML. Supported planar bilayers in studies on immune cell adhesion and communication. J Immunol. Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 23.Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987;50:129–135. doi: 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 24.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: Enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34:13988–13993. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 25.Morrissey JH, Fair DS, Edgington TS. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb. Res. 1988;52:247–261. doi: 10.1016/0049-3848(88)90084-9. [DOI] [PubMed] [Google Scholar]
- 26.Smith SA, Morrissey JH. Rapid and efficient incorporation of tissue factor into liposomes. J. Thromb. Haemost. 2004;2:1155–1162. doi: 10.1111/j.1538-7836.2004.00772.x. [DOI] [PubMed] [Google Scholar]
- 27.Fiore MM, Neuenschwander PF, Morrissey JH. An unusual antibody that blocks tissue factor/factor VIIa function by inhibiting cleavage only of macromolecular substrates. Blood. 1992;80:3127–3134. [PubMed] [Google Scholar]
- 28.Neuenschwander PF, Fiore MM, Morrissey JH. Factor VII autoactivation proceeds via interaction of distinct protease-cofactor and zymogen-cofactor complexes. Implications of a two-dimensional enzyme kinetic mechanism. J. Biol. Chem. 1993;268:21489–21492. [PubMed] [Google Scholar]
- 29.Nakagaki T, Foster DC, Berkner KL, Kisiel W. Initiation of the extrinsic pathway of blood coagulation: evidence for the tissue factor dependent autoactivation of human coagulation factor VII. Biochemistry. 1991;30:10819–10824. doi: 10.1021/bi00109a001. [DOI] [PubMed] [Google Scholar]
- 30.Gemmell CH, Broze GJ, Jr., Turitto VT, Nemerson Y. Utilization of a continuous flow reactor to study the lipoprotein-associated coagulation inhibitor (LACI) that inhibits tissue factor. Blood. 1990;76:2266–2271. [PubMed] [Google Scholar]
- 31.Repke D, Gemmell CH, Guha A, Turitto VT, Broze GJ, Jr., Nemerson Y. Hemophilia as a defect of the tissue factor pathway of blood coagulation: effect of factors VIII and IX on factor X activation in a continuous-flow reactor. Proc. Natl. Acad. Sci. U. S. A. 1990;87:7623–7627. doi: 10.1073/pnas.87.19.7623. [DOI] [PMC free article] [PubMed] [Google Scholar]