

Abstract

Two distinct approaches have been developed for the synthesis of chlorins bearing formyl groups: (1) reaction of an acetal-substituted 1-acyldipyrromethane with 2,3,5,6-tetrahydro-1,3,3-trimethyldipyrrin to give upon hydrolysis a 5-formylchlorin; (2) Pd-mediated coupling of a bromochlorin with a one-carbon synthon (hydroxymethyl tributyltin or CO) to give a 13-, 15-, or 3,13-formylchlorin. The zinc chlorins exhibit long-wavelength peak absorption maxima ranging from 626 to 667 nm, indicating the wavelength tunability afforded by formyl substitution.
Formyl-substituted porphyrins have proved valuable for fundamental spectroscopic studies and as versatile synthetic intermediates.1 Formyl-substituted chlorins have provoked interest owing to the distinct spectra provided by chlorophyll b, which contains a formyl group at the 7-position, versus chlorophyll a, which contains a 7-methyl group. The formyl group also provides a valuable site for synthetic elaboration of chlorins,2-5 although the synthesis of formylchlorins has presented a number of challenges.
One popular synthetic approach has been to use chlorophyll b directly in derivatization processes, including formation of the imine,6 olefin,7 or the meso-carbon of an attached porphyrin.2 A second approach has been to formylate an intact chlorin. This approach is largely restricted to simple substituted chlorins (e.g., β-octaethylchlorin8,9 or meso-tetraarylchlorins10) so as to avoid formation of regioisomers, although one notable exception is the regioselective 20-formylation of selected naturally occurring chlorins.8,11 A third approach entails the modification of naturally occurring chlorins such as chlorophyll a. Examples include (i) oxidation of a 3-vinyl group to give a 3-formyl species,12 (ii) oxidation of an 8-vinyl group to give an 8-formyl species,13 (iii) rupture of the isocyclic ring followed by functional group transformations to give the 13-formylchlorin12,14 or 15-formylchlorin,15 and (iv) derivatization of alkyl groups at the 8- or 12-positions.5,16 The ability to place formyl groups at designated sites in chlorins would benefit from more versatile methods of synthesis, particularly methods that do not rely on naturally occurring chlorins as starting materials.
We recently prepared a family of chlorins bearing auxochromes at the 3- and 13-positions.17,18 The chlorins were prepared by de novo synthesis and contained a geminal dimethyl group in the reduced ring to ensure stability toward adventitious oxidation. The substituents were introduced at the 3,13-positions because the transition that gives rise to the long-wavelength (Qy) band is polarized along this axis. The auxochromes included phenyl, vinyl, TIPS-ethynyl, and acetyl, of which the acetyl group gave the most pronounced changes in the absorption spectra. The presence of a single acetyl group at the 13-position caused a 26 nm redshift, whereas 3,13-diacetyl substitution caused a 56 nm redshift. On the other hand, similar groups at the 15-position caused much smaller effects.19 Extension of this approach to synthetic chlorins bearing formyl groups at specific positions is of great interest because the formyl group is expected to be a more potent auxochrome than acetyl, ethynyl, or vinyl. Toward, this goal, we report herein two rational routes to chlorins that enable placement of formyl groups at designated sites, including the 5-, 13-, 15- and 3,13-positions.
Synthesis of a 5-Formylchlorin from an Acetal-dipyrromethane
The synthesis of a 5-formylchlorin was approached through use of a 1-acyldipyrromethane that contains a protected formyl group (1).20 Other acetal-chlorins have been prepared by reduction of the corresponding porphyrin21 or from the formylchlorin.22 1-Acyldipyrromethane 1 was treated with NBS23 at –78 °C for 1 h to afford the corresponding 1-bromo-9-acyldipyrromethane (2) in 79% yield (Scheme 1). Reduction of 2 with NaBH4 afforded the 1-bromo-dipyrromethane-9-carbinol (Eastern half). The latter was immediately subjected to condensation23 with tetrahydrodipyrrin 324 (Western half) in the presence of TFA to obtain the tetrahydrobilene-a derivative, which upon oxidative cyclization gave the 5-acetal-substituted zinc chlorin 4 in 16% yield. The acetal group was hydrolyzed (and the zinc chlorin was demetalated) by treatment with TFA/H2O in CH2Cl2, thereby affording the free base formylchlorin FbC-F5P10 in 84% yield. Treatment of FbC-F5P10 with Zn(OAc)2·2H2O afforded the zinc chelate ZnC-F5P10 in quantitative yield.
Scheme 1.

Synthesis of Formylchlorins from Bromochlorins
To more generally introduce formyl groups at various β-positions, we first investigated Stille coupling of a bromochlorin and hydroxymethyltributyltin25,26 to give the corresponding hydroxymethylchlorin (Scheme 2). The zinc chelate of the 13-bromochlorin (ZnC-M10Br13)17 gave limited conversion upon refluxing with THF. Demetalation of ZnC-M10Br13 with TFA afforded the crude free base chlorin. Coupling of the latter and Bu3SnCH2OH in the presence of Pd(PPh3)4 in THF for 30 h gave the 13-hydroxymethylchlorin 5 in 51% yield. The oxidation of 5 with MnO2 in toluene at room temperature afforded the 13-formylchlorin FbC-M10F13 in 91% yield. The free base formylchlorin FbC-T5M10F13 was obtained similarly (Scheme 2). A streamlined procedure including demetalation, Pd-coupling, and oxidation gave FbC-T5M10F13 in 47% yield starting from ZnC-T5M10Br13.
Scheme 2.

We next investigated the more direct method of Pd-mediated reductive carbonylation.27,28 A short survey of conditions (solvent, palladium source, and amount of HCO2Na) identified effective conditions. Thus, treatment of ZnC-M10Br13 (10 mM) with sodium formate (20 mM) in the presence of (PPh3)2PdCl2 (20 mol%) and PPh3 (20 mol%) in DMF at 108 °C under an atmosphere of CO afforded the 13-formylchlorin ZnC-M10F13 in 68% yield. This Pd-mediated carbonylation method was successfully applied to introduce the formyl group into the chlorin macrocycle at the 13-, 3,13-, and 15-positions (Table 1).
Table 1.
Scope of Pd-Mediated Carbonylation of Chlorins
Absorption Spectra of Formylchlorins
The absorption spectra of the formylchlorins (in toluene at room temperature) are summarized in Table 2. Three classes are noted: zinc chlorins bearing a 10-mesityl group (ZnC-M10 series); zinc chlorins bearing a 5-p-tolyl group and a 10-mesityl group (ZnC-T5M10 series); and various free base chlorins (FbC series). Selected absorption spectra of the ZnC-M10 series are shown in Figure 1 (see Supporting Information for additional spectra).
Table 2.
Spectral Properties of Chlorins.a
| Compoundb | λB (fwhm) in nm | λQy (fwhm) in nm | IB/IQc | Δυ (cm-1)d |
|---|---|---|---|---|
| ZnC-M10 | 405 (13) | 606 (12) | 4.3 | --- |
| ZnC-P10 | 405 (13) | 605 (11) | 4.1 | --- |
| ZnC-M10A13 | 418 (18) | 632 (14) | 2.1 | 680 |
| ZnC-M10F13 | 418 (20) | 634 (12) | 2.2 | 730 |
| ZnC-F5P10 | 425 (29) | 650 (36) | 6.2 | 1100 |
| ZnC-F3M10F13 | 439 (20) | 667 (17) | 1.4 | 1500 |
| ZnC-A3M10A13 | 436 (21) | 662 (18) | 1.5 | 1400 |
|
| ||||
| ZnC-T5M10 | 412 (13) | 608 (11) | 5.0 | --- |
| ZnC-T5M10F15 | 417 (16) | 626 (20) | 5.6 | 470 |
| ZnC-T5M10F13 | 424 (15) | 637 (13) | 2.6 | 750 |
|
| ||||
| FbC-M10 | 400 (34) | 637 (9) | 2.7 | --- |
| FbC-P10 | 403 (34) | 637 (9) | 2.8 | --- |
| FbC-M10F13 | 414 (35) | 659 (11) | 1.9 | 520 |
| FbC-T5M10F13 | 422 (36) | 663 (11) | 2.2 | 620 |
| FbC-F5P10 | 416 (41) | 672 (27) | 5.0 | 820 |
In toluene at room temperature.
All compounds that are not synthesized herein are described in ref. 19.
Ratio of the intensities of the B and Qy bands.
The redshift of the Qy band relative to that of the parent chlorin (ZnC-M10, ZnC-T5M10 or FbC-M10).
Figure 1.
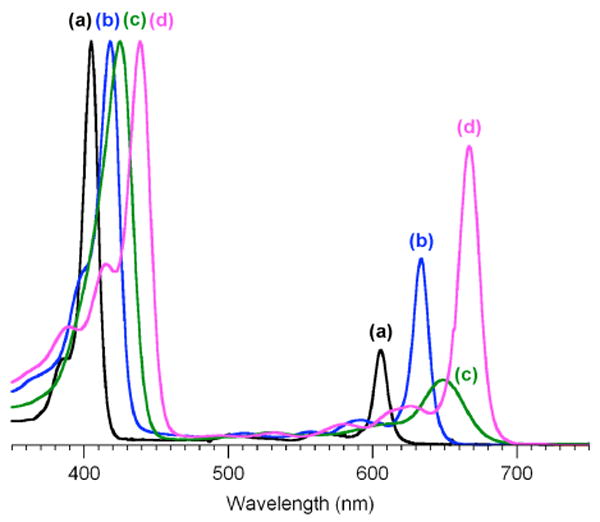
Absorption spectra (normalized) in toluene at room temperature of ZnC-M10 (trace a, λQy 606 nm),17ZnC-M10F13 (trace b, λQy 634 nm), ZnC-F5P10 (trace c, λQy 650 nm), and ZnC-F3M10F13 (trace d, λQy 667 nm).
The presence of a formyl group at the 3-, 5-, 13-, or 15-positions causes a redshift of the long-wavelength absorption (Qy) band. However, the magnitude of the redshift, the sharpness of the Qy band, and the intensity of the Qy band relative to the band in the blue region (B band) depend on the position of substitution. The major observations are as follows:
A 13-formyl group causes a redshift of 730 cm-1 (28 nm; ZnC-M10F13 versus ZnC-M10), to be compared with only 680 cm-1 (26 nm) for the corresponding 13-acetylchlorin17 (ZnC-M10A13). The relative intensity of the Qy band increases by approximately two-fold for ZnC-M10F13 versus ZnC-M10 while the sharpness (fwhm) remains relatively constant (12 nm). Note that the 10-mesityl and 10-phenyl groups give essentially identical spectra.19
3,13-Diformyl groups cause a redshift of 1500 cm-1 (61 nm; ZnC-F3M10F13 versus ZnC-M10), to be compared with 1400 cm-1 (56 nm) for the corresponding 3,13-diacetylchlorin17 (ZnC-A3M10A13). A further increase in relative intensity of the Qy band occurs (IB/IQ = 1.4 for ZnC-F3M10F13 versus 4.3 for ZnC-M10) while the sharpness (fwhm) decreases only modestly (17 nm).
A 5-formyl group causes a strong redshift of 1100 cm-1 (44 nm; ZnC-F5P10 versus ZnC-P10), which is more pronounced than at any other single site examined herein. However, the Qy band is relatively broad (fwhm = 36 nm).
A 15-formyl group causes a modest redshift of 470 cm-1 (18 nm; ZnC-T5M10F15 versus ZnC-T5M10), band broadening (fwhm = 20 nm), and no increase in the relative intensity of the Qy band (IB/IQ = 5.6 for ZnC-T5M10F15 versus 5.0 for ZnC-T5M10).
Thus, the formyl group is a slightly more potent auxochrome than the acetyl group at those positions where comparisons can be made (13- or 3,13-positions), causing bathochromic and hyperchromic effects without substantial band broadening. The bathochromic shift increases in order of positions 5 < 13 < 15, whereas the band-broadening effect increases in order of positions 13 < 15 < 5. Similar trends were noted in the free base chlorins where comparisons can be made.
In summary, we have developed rational approaches for the synthesis of formylchlorins. Pd-mediated formylation is attractive where suitable bromochlorins are available (positions 3, 13, 15, etc.), otherwise a complementary route entails construction of a chlorin with use of an acetal-substituted precursor (e,g., 5-position). Both approaches were successfully implemented to give milligram quantities of formylchlorins. The absorption spectra of zinc chelates of 5-, 13-, 3,13- and 15-formylchlorins revealed a significant effect of the formyl group depending on the position of substitution. The long-wavelength absorption band can now be tuned over the range of 606 – 667 nm. For applications in flow cytometry, the chlorins with distinct and sharp bands could provide a panel of fluorophores. For applications in solar energy conversion, the collection of wavelength-tunable chlorins, including the chlorins with broad bands, could provide enhanced solar coverage. Taken together, the routes described herein afford versatile access to formylchlorins for use in fundamental spectroscopic studies and further synthetic elaboration.
Experimental Section
Carbonylation Procedure: Zn(II)-3,13-Diformyl-17,18-dihydro-18,18-dimethyl-10-mesitylporphyrin (ZnC-F3M10F13)
Following a procedure for CO-mediated formylation,28 a mixture of ZnC-Br3M10Br13 (13.8 mg, 0.0203 mmol), (PPh3)2PdCl2, (2.85 mg, 0.00406 mmol, 20 mol%), PPh3 (1.06 mg, 0.00406 mmol, 20 mol%) and sodium formate (3.50 mg, 0.0507 mmol) was dried in a Schlenk flask for 1 h. DMF (1.2 mL) was added, and CO gas was bubbled through the reaction mixture for 2 h at 108 °C. Then, the reaction mixture was heated at 108 °C under a balloon containing CO for 24 h. After cooling to room temperature, the reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, washed (water, brine), dried (Na2SO4) and concentrated. The resulting residue was chromatographed [silica, hexanes/CH2Cl2 (1:2) → CH2Cl2 → CH2Cl2/ethyl acetate (7:1)] to afford a trace amount (∼5% of the total) of a mono-formylated chlorin followed by the title compound as a green solid (6.1 mg, 52%): 1H NMR (300 MHz) δ 1.82 (s, 6H), 2.03 (s, 6H), 2.60 (s, 3H) 4.53 (s, 2H), 7.23 (s, 2H), 8.38 (d, J = 4.5 Hz, 1H), 8.62 (s, 1H), 8.87 (d, J = 4.5 Hz, 1H), 9.00 (s, 1H), 9.26 (s, 1H), 9.61 (s, 1H), 10.3 (s, 1H), 10.88 (s, 1H), 11.15 (s, 1H); 13C NMR δ 188.8, 188.7, 172.8, 167.4, 162.1, 161.5, 160.8, 144.0, 141.9, 140.2, 138.9, 137.9, 137.5, 135.8, 135.7, 131.9, 130.6, 128.2, 128.1, 110.3, 107.7, 98.3, 96.6, 94.5, 50.5, 44.3, 31.0, 21.4, 21.2; LDMS obsd 576.8; FAB-MS obsd 576.1499, calcd 576.1504 (C33H28N4O2Zn); λabs (toluene) 439, 667 nm.
Streamlined Procedure via Hydroxymethylation: 13-Formyl-17,18-dihydro-18,18-dimethyl-10-mesityl-5-p-tolylporphyrin (FbC-T5M10F13)
A sample of chlorin ZnC-T5M10Br13 (7.50 mg, 0.0108 mmol) was treated with TFA (25.0 μl, 0.324 mmol) in CH2Cl2 (1.0 mL). The reaction mixture was stirred at room temperature for 3 h. The reaction mixture was quenched by the addition of a mixture of saturated aqueous NaHCO3 and CH2Cl2. The organic layer was separated, washed (water, brine), dried (Na2SO4) and concentrated to afford the free base chlorin. Following a procedure for Stille coupling with chlorins,17 a mixture of free base chlorin (∼0.0108 mmol) and Pd(PPh3)4 (2.50 mg, 0.00216 mmol) was dried in a Schlenk flask for 30 min. Hydroxymethyltributyltin (13.8 mg, 0.0432 mmol) in THF (1.0 mL) was added, and the reaction mixture was refluxed for 30 h. The reaction mixture was concentrated. The resulting residue was dissolved in toluene (0.20 mL), and MnO2 (47.0 mg, 0.540 mmol) was added. The reaction mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated and chromatographed [silica, hexanes → hexanes/CH2Cl2 (1:1) → (2:8)] to afford a purple solid (3.0 mg, 47%) with characterization data (1H NMR, 13C NMR, LD-MS, FAB-MS, UV-Vis) consistent with those for the product obtained via individual stepwise procedures (see Supporting Information).
Supplementary Material
Spectral data (absorption, 1H NMR, LD-MS) for all new chlorins, and additional experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the NIH (GM36238).
References
- 1.Ponomarev GV. Chem Heterocyclic Compounds. 1994;30:1444–1465. [Google Scholar]
- 2.Wasielewski MR. Chem Rev. 1992;92:435–461. [Google Scholar]
- 3.Jaquinod L, Nurco DJ, Medforth CJ, Pandey RK, Forsyth TP, Olmstead MM, Smith KM. Angew Chem Int Ed Engl. 1996;35:1013–1016. [Google Scholar]
- 4.Wang JJ, Shim YK, Jiang GJ, Imafuku K. J Heterocyclic Chem. 2003;40:1075–1079. [Google Scholar]
- 5.Li G, Dobhal MP, Shibata M, Pandey RK. Org Lett. 2004;6:2393–2396. doi: 10.1021/ol0492290. [DOI] [PubMed] [Google Scholar]
- 6.Losev A, Mauzerall D. Photochem Photobiol. 1983;38:355–361. [Google Scholar]
- 7.Tamiaki H, Kouraba M. Tetrahedron. 1997;53:10677–10688. [Google Scholar]
- 8.Smith KM, Bisset GMF, Bushell MJ. Bioorg Chem. 1980;9:1–26. [Google Scholar]
- 9.Kalisch WW, Senge MO, Ruhlandt-Senge K. Photochem Photobiol. 1998;67:312–323. [Google Scholar]
- 10.Mironov AF, Moskalchuk TV, Shashkov AS. Russ J Bioorg Chem. 2004;30:261–267. [Google Scholar]
- 11.Ando T, Irie K, Koshimizu K, Takemura T, Nishino H, Iwashima A, Nakajima S, Sakata I. Tetrahedron. 1990;46:5921–5930. [Google Scholar]
- 12.Kunieda M, Tamiaki H. J Org Chem. 2007;72:2443–2451. doi: 10.1021/jo0623282. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki SI, Tamiaki H. Bull Chem Soc Jpn. 2004;77:797–800. [Google Scholar]
- 14.Ma L, Dolphin D. Tetrahedron Lett. 1995;36:7791–7794. [Google Scholar]
- 15.Wray V, Jürgens U, Brockmann H., Jr Tetrahedron. 1979;35:2275–2283. [Google Scholar]
- 16.Zheng G, Potter WR, Camacho SH, Missert JR, Wang G, Bellnier DA, Henderson BW, Rodgers MAJ, Dougherty TJ, Pandey RK. J Med Chem. 2001;44:1540–1559. doi: 10.1021/jm0005510. [DOI] [PubMed] [Google Scholar]
- 17.Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M, Lindsey JS. J Org Chem. 2006;71:4092–4102. doi: 10.1021/jo060208o. [DOI] [PubMed] [Google Scholar]
- 18.Kee HL, Kirmaier C, Tang Q, Diers JR, Muthiah C, Taniguchi M, Laha JK, Ptaszek M, Lindsey JS, Bocian DF, Holten D. Photochem Photobiol. 2007;83 doi: 10.1111/j.1751-1097.2007.00151.x. in press. [DOI] [PubMed] [Google Scholar]
- 19.Kee HL, Kirmaier C, Tang Q, Diers JR, Muthiah C, Taniguchi M, Laha JK, Ptaszek M, Lindsey JS, Bocian DF, Holten D. Photochem Photobiol. 2007;83 doi: 10.1111/j.1751-1097.2007.00151.x. in press. [DOI] [PubMed] [Google Scholar]
- 20.Balakumar A, Muthukumaran K, Lindsey JS. J Org Chem. 2004;69:5112–5115. doi: 10.1021/jo049819b. [DOI] [PubMed] [Google Scholar]
- 21.Balaban TS, Linke-Schaetzel M, Bhise AD, Vanthuyne N, Roussel C. Eur J Org Chem. 2004:3919–3930. [Google Scholar]
- 22.Tamiaki H, Kubo M, Oba T. Tetrahedron. 2000;56:6245–6257. [Google Scholar]
- 23.Taniguchi M, Ra D, Mo G, Balasubramanian T, Lindsey JS. J Org Chem. 2001;66:7342–7354. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]
- 24.Ptaszek M, Bhaumik J, Kim HJ, Taniguchi M, Lindsey JS. Org Process Res Dev. 2005;9:651–659. doi: 10.1021/op050087e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a Kosugi M, Sumiya T, Ogata T, Sano H, Migita T. Chem Lett. 1984:1225–1226. [Google Scholar]; b Kosugi M, Sumiya T, Ohhashi K, Sano H, Migita T. Chem Lett. 1985:997–998. [Google Scholar]
- 26.a Åhman J, Somfai P. Synth Commun. 1994;24:1117–1120. [Google Scholar]; b Danheiser RL, Romines KR, Koyama H, Gee SK, Johnson CR, Medich JR. Org Synth. 1993;71:133–139. [Google Scholar]
- 27.Pri-Bar I, Buchman O. J Org Chem. 1984;49:4009–4011. [Google Scholar]
- 28.Okano T, Harada N, Kiji J. Bull Chem Soc Jpn. 1994;67:2329–2332. [Google Scholar]
- 29.Laha JK, Muthiah C, Taniguchi M, Lindsey JS. J Org Chem. 2006;71:7049–7052. doi: 10.1021/jo0608265. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi M, Kim MN, Ra D, Lindsey JS. J Org Chem. 2005;70:275–285. doi: 10.1021/jo048440m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectral data (absorption, 1H NMR, LD-MS) for all new chlorins, and additional experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.