

Abstract
An enantioselective synthetic route to the enediyne antibiotic N1999A2 (1) is described, proceeding in 21 steps (0.4% yield, 77% average yield per step) from (R)-(+)-glycidol. The route involves the convergent assembly of three components: a (1-iodovinyl) stannane (2), a 1,5-hexadiyne-3,4-diol derivative (3), and a substituted naphthoic acid (4). Important transformations in the synthetic sequence include the palladium-catalyzed coupling of 2 and 3, an intramolecular oxidative cyclization of a terminal bis-acetylene, and a transannular anionic (bi)cyclization of a cyclic bromoenetriyne. The careful selection and manipulation of protective groups throughout the sequence proved to be critical to the development of the synthetic route, where all late-stage intermediates were unstable and could not be concentrated. In the final step of the sequence, three protective groups were removed in a single operation, providing synthetic N1999A2 (1) in 76% yield. Conditions were found that, for the first time, led to the precipitation of 1 as a solid.
N1999A2 (1) is a 9-membered “enediyne” natural product with extraordinary antiproliferative effects in human cancer cells.1 Although it is structurally similar to neocarzinostatin chromophore,2 N1999A2 (1) lacks an aminoglycoside residue and was isolated without an associated carrier protein.1a Also, as Hirama and co-workers established in their prior synthetic studies,1b,1c N1999A2 (1) is epimeric with neocarzinostatin chromophore at positions 4, 5, and 13. Here, we describe a second, enantioselective synthetic route to N1999A2 (1) that proceeds by the convergent assembly of the (1-iodovinyl) stannane 2, the 1,5-hexadiyne-3,4-diol derivative 3, and the substituted naphthoic acid 4. Each of these starting materials resulted from an empirical selection process, not detailed here,3 that was necessary to achieve the appropriate balance of stability and lability of each protective group. In this way, for example, it was possible to remove three protective groups in one operation in the final step of the synthetic route.
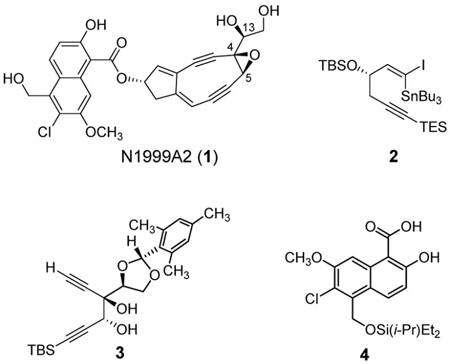
Synthesis of the (1-iodovinyl) stannane 2 began by epoxide opening of the known (R)-(+)-glycidol derivative 5 with lithium (triethylsilyl)acetylide to furnish the secondary alcohol 6 (Scheme 1).4,5 Silyl ether formation (TBSCl, imidazole, giving 7)6 followed by tetrahydropyranyl protective group removal ((CH3)2AlCl, THF, −78→23 °C)7 afforded the primary alcohol 8. Oxidation of 8 under the Parikh-Doering conditions produced the aldehyde 9.8 Treatment of 9 with carbon tetrabromide and triphenylphosphine in dichloromethane then provided the dibromoolefin 10 (95% ee, determined by Mosher ester analysis of a derivative, see Supporting Information),9 which was transformed into the terminal alkyne 11 in one operation (NaHMDS, THF, −78 °C, then n-BuLi).10 The latter product (11) was stannylated (n-BuLi; Bu3SnCl, THF, −78→23 °C) and the resulting alkynyl tributylstannane was treated with Cp2Zr(H)Cl (Schwartz’s reagent) in THF; iodination of the vinyl zirconium intermediate that was formed then produced the (1-iodovinyl) stannane 2.11
Scheme 1.
(a) (Triethylsilyl)acetylene, LHMDS, −78 °C; BF3·OEt2, then 5; (b) TBSCl, imidazole, THF, 23 °C, 95% (2 steps); (c) (CH3)2AlCl, THF, −78→23 °C, 93%; (d) pyridine sulfur trioxide complex, Et3N, CH2Cl2, DMSO, 0 °C; (e) CBr4, PPh3, CH2Cl2, 77% (2 steps); (f) NaHMDS, THF, −78 °C; n-BuLi, 99%; (g) n-BuLi, Bu3SnCl, THF, −78→23 °C; (h) Cp2Zr(H)Cl, THF, 23 °C; I2, THF, 69% (2 steps).
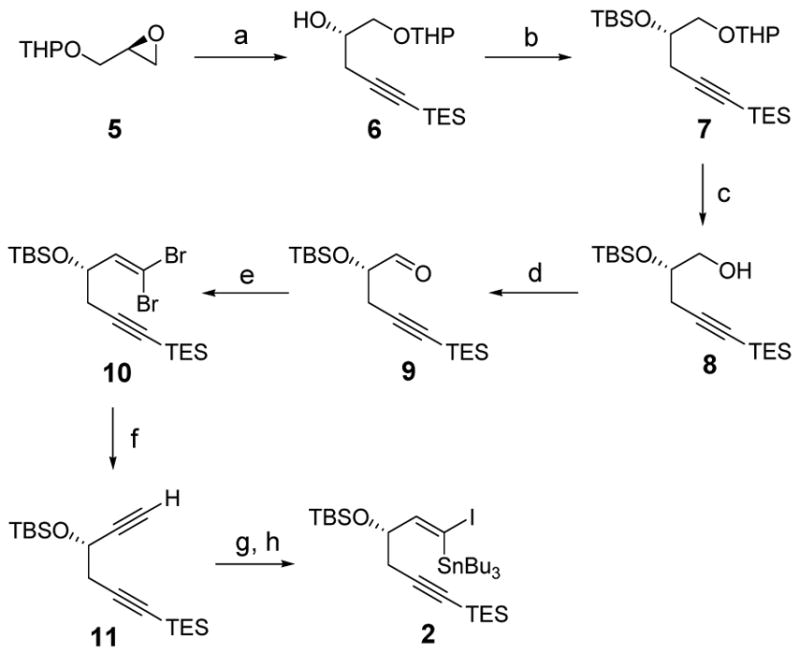
Synthesis of the 1,5-hexadiyne-3,4-diol derivative 3 began with the known propargylic ketone 13, available in two steps from (S)-glyceraldehyde acetonide (12) (Scheme 2).12,13 In the course of synthetic studies of neocarzinostatin chromophore, conditions had been developed for an E-selective Wittig reaction between 13 and the phosphonium salt 14 (KHMDS, THF, −78 °C, E:Z = 3:1).13 In this work we report that Z-selective coupling of 13 and 14 occurs in the presence of lithium halide additives (5 equiv, deprotonation of 14 with n-butyllithium). Lithium iodide was superior to other lithium halide additives (Z:E ratios: I, 5.54; Br, 3.72; Cl, 2.20; F, 1.98).14 Wittig reaction of 13 and 14 modified by the inclusion of lithium iodide as an additive typically provided the pure (Z)-olefin 15 in 74% yield after flash-column chromatography. Treatment of the latter product (15) with potassium carbonate in methanol at 0 °C selectively removed the trimethylsilyl protective group, providing the enediyne 16. We conducted a screen of several different osmylation reagents to achieve a diastereoselective dihydroxylation of the enediyne 16 and thus identified as optimal Sharpless’ catalytic system involving 1,4-bis(9-O-dihydroquinidine)diphenylpyrimidine ((DHQD)2PYR) as ligand (20 mol%), K2OsO4·2H2O as osmium source (4 mol%), and potassium ferricyanide as stoichiometric oxidant (5 equiv).15,16 The diol diastereomer 17 was produced with >95:5 selectivity; pure 17 was obtained in 53% yield after flash-column chromatography. The acetonide protective group of the dihydroxylation product 17 was cleaved quantitatively (FeCl3·6H2O, CH3CN, 23 °C)17 to provide the corresponding tetraol (18).18 Selective protection of the latter substance was achieved in the presence of mesitaldehyde dimethyl acetal (19)19 and camphorsulfonic acid. The diastereomeric mixture of acetals that was formed was separated by flash-column chromatography (3.9:1 dr, 55% isolated yield of the major diastereomer 3, stereochemistry determined by nOe analysis). Only the major diastereomer (3) was used in the subsequent coupling reaction.
Scheme 2.
(a) 14, n-BuLi, LiI, THF, −78 °C; then 13, 76% (Z-olefin); (b) K2CO3, MeOH, 0 °C, 95%; (c) (DHQD)2PYR (20 mol%), K2OsO4·2H2O (4 mol%), K3Fe(CN)6 (5.0 equiv), K2CO3 (5.0 equiv), CH3SO2NH2 (3.0 equiv), t-BuOH, H2O, 0 °C, 53%; (d) FeCl3·6H2O, CH3CN, 23 °C, 100%; (e) 19, CSA, THF, 23 °C, 3.9:1 dr, 55% (major diastereomer).
The (1-iodovinyl) stannane 2 and the 1,5-hexadiyne-3,4-diol 3 were coupled in the presence of a palladium catalyst, forming the enetriyne diol 20 (Scheme 3). One O-silyl and two C-silyl protective groups of the coupling product 20 were cleaved in the presence of tetrabutylammonium fluoride in THF. Two new O-diethylisopropylsilyl protective groups were then introduced selectively (the tertiary alcohol remained free); bromodestannylation with N-bromosuccinimide then afforded the substrate for intramolecular oxidative acetylene coupling (21). Attempts to cyclize 21 under modified Eglinton conditions (Cu(OAc)2, CuI, pyridine, THF), proven to be optimal in a closely analogous system, met with varied success.20,21 Further experiments revealed that by simply omitting CuI from the cyclization reaction the cyclic bromoenetriyne 22 could be produced in high yield and, importantly, in a reproducible manner. Solutions of this and all subsequent intermediates could not be concentrated without extensive decomposition. As a general practice, these unstable intermediates were purified by flash-column chromatography, fractions containing pure intermediates were pooled, and the combined fractions were diluted with a deuterated solvent (C6D6 or CD3CN). The resulting solution was then partially concentrated to allow for 1H NMR analysis; yields were determined using an internal standard (see Supporting Information). In the key step of the sequence, a solution of the substrate 22 in THF–toluene (1:1, stirred with 4-Å molecular sieves, 23 °C, 15 min) was treated at −78 °C with a solution of lithium hexamethyldisilazide (LHMDS) in THF (1.0 M, 1.1 equiv) to deprotonate the tertiary hydroxyl group. Subsequent addition of a solution of tert-butyllithium in pentane (1.7 M, 4.0 equiv) at −78 °C, followed immediately (<3 s) by quenching with a solution of acetic acid (30 equiv) in THF, afforded the transannular cyclization product 23 in 30–40% yield.21 Typically, the latter reaction was performed on scales of 20–25 mg; larger-scale reactions were less efficient. Selective removal of the allylic diethylisopropylsilyl ether group within the transannular product 23 was achieved by treatment of 23 with an excess of triethylamine trihydrofluoride in acetonitrile at −25 °C. The desilylated product was then coupled with the naphthoic acid 422 in the presence of N,N′-dicyclohexylcarbodiimide (DCC) in THF, providing the ester 24 (44%, 2 steps). Addition of tetrabutylammonium fluoride (6.0 equiv) to a solution of the ester 24 and the buffering agent o-nitrophenol (6.0 equiv) in THF at 0 °C cleanly removed the propargylic silyl ether within 24; subsequent protection of the phenolic hydroxyl group as the corresponding triethylsilyl ether (TESCl, Et3N, CH2Cl2, −78 °C) then afforded the highly unstable diol 25. The trans-diol function within the latter product was transformed to the corresponding epoxide by tosylation under basic conditions (TsCl, DABCO, CH2Cl2, 23 °C), providing N1999A2 in fully-protected form (26), a more stable intermediate relative to others (22 and beyond) in the sequence. During the latter transformation (25 → 26) the tosylate intermediate was observable by TLC analysis, but it did not accumulate, being transformed to 26 as it was formed. Global deprotection of the epoxide 26 was accomplished with trifluoroacetic acid in a mixture of THF and water at 0 °C.1c In this reaction both silyl groups were cleaved within 2 h, and the mesitylene protective group was removed within 5–8 h. To isolate synthetic N1999A2 particular work-up conditions were necessary. For example, addition of saturated sodium bicarbonate aqueous solution led to extensive decomposition of the product. By using pH 7 aqueous phosphate buffer as a quenching solution decomposition was minimized. The product was extracted into ether and the ethereal solution was dried (Na2SO4), then concentrated to a volume of 4–8 mL. Since N1999A2 is an appreciably polar compound, a rather polar eluent (5% CD3CN–ether) was necessary for its purification by flash-column chromatography. Although some decomposition did occur during chromatography, the product obtained was estimated to be >90% pure by 1H NMR analysis. A much simpler and more convenient purification procedure was found serendipitously. When pentane (5 mL) was added to a solution of synthetic N1999A2 (~5 mg) in ether (8 mL), a light-yellow solid precipitated from the solution; solvents were removed and the resulting solid was triturated with a 5:1 mixture of ether and pentane. N1999A2 had not previously been reported to be a solid. 1H NMR and UV spectra of synthetic (solid) N1999A2 were identical to published spectra.1b,1c Thus, for the second time, the stereostructure of N1999A2 proposed by Hirama and co-workers has been confirmed by an unambiguous synthetic route.1b,1c A high-resolution, “exact” mass spectrum of synthetic N1999A2 was also obtained using a Micromass LCT/TOF spectrometer equipped with an electrospray ionization source (calcd for C27H25ClNO8 [M+NH4]+ 526.1269, found 526.1286). Synthetic N1999A2 was surprisingly stable. In the solid state, N1999A2 could be left in the air for 10 minutes at ambient temperature without evident decomposition by subsequent 1H NMR analysis, and solutions of N1999A2 in ether or DMSO have been stored at −25 °C for 2 months without detectable decomposition by HPLC analysis.
Scheme 3.

(a) Pd(PPh)3, CuI, Et3N, benzene, 23 °C, 64%; (b) TBAF, THF, 0 °C, 91%; (c) DEIPSCl, imidazole, DMF, 0 °C, 89%; (d) NBS, CH2Cl2, 23 °C, 89%; (e) Cu(OAc)2, THF–pyridine (1:1), 60 °C, 75%; (f) LHMDS, THF–toluene (1:1), −78 °C; t-BuLi (4.0 equiv), then HOAc, 30 – 40%; (g) Et3N·3HF, CH3CN,.−25 °C; (h) 4, DCC, THF, 0 °C, 44% (2 steps); (i) TBAF (6.0 equiv), o-nitrophenol (6.0 equiv), THF, 0 °C; (j) TESCl, Et3N, CH2Cl2, −78 °C; (k) TsCl, DABCO, CH2Cl2, 23 °C, 33% (3 steps); (l) TFA, THF, H2O, 0 °C, 5 h, 76%.
Supplementary Material
Detailed experimental procedures and tabulated spectroscopic data (1H and 13C NMR, FT-IR, and HRMS) for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Generous financial support from the National Institutes of Health, and Pfizer, Inc., is gratefully acknowledged.
References
- 1.(a) Ando T, Ishii M, Kajiura T, Kameyama T, Miwa K, Sugiura Y. Tetrahedron Lett. 1998;39:6495. [Google Scholar]; (b) Kobayashi S, Reddy RS, Sugiura Y, Sasaki D, Miyagawa N, Hirama M. J Am Chem Soc. 2001;123:2887. doi: 10.1021/ja003982g. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi S, Ashizawa S, Takahashi Y, Sugiura Y, Nagaoka M, Lear MJ, Hirama M. J Am Chem Soc. 2001;123:11294. doi: 10.1021/ja011779v. [DOI] [PubMed] [Google Scholar]; (d) Miyagawa N, Sasaki D, Matsuoka M, Imanishi M, Ando T, Sugiura Y. Biochem Biophys Res Comm. 2003;306:87. doi: 10.1016/s0006-291x(03)00925-2. [DOI] [PubMed] [Google Scholar]
- 2.(a) Edo K, Mizugaki M, Koide Y, Seto H, Furihata K, Otake N, Ishida N. Tetrahedron Lett. 1985;26:331. [Google Scholar]; (b) Myers AG, Proteau PJ, Handel TM. J Am Chem Soc. 1988;110:7212. [Google Scholar]; (c) Maeda H, Edo K, Ishida N, editors. Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug. Springer; Tokyo: 1997. [Google Scholar]
- 3.Ji N. PhD Thesis. Harvard University; Cambridge, MA: 2006. [Google Scholar]
- 4.For preparation of 5, see: Meng D, Bertinato P, Balog A, Su DS, Kamenecka T, Sorensen EJ, Danishefsky SJ. J Am Chem Soc. 1997;119:10073.
- 5.For the use of alkynyl trifluoroborates in epoxide-opening reactions, see: Yamaguchi M, Hirao I. Tetrahedron Lett. 1983;24:391.
- 6.Corey EJ, Venkateswarlu A. J Am Chem Soc. 1972;94:6190. [Google Scholar]
- 7.Ogawa Y, Shibasaki M. Tetrahedron Lett. 1984;25:663. [Google Scholar]
- 8.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 9.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- 10.Grandjean D, Pale P, Chuche J. Tetrahedron Lett. 1994;35:3529. [Google Scholar]
- 11.Dabdoub MJ, Dabdoub VB, Baroni ACM. J Am Chem Soc. 2001;123:9694. doi: 10.1021/ja0114660. [DOI] [PubMed] [Google Scholar]
- 12.(S)-Glyceraldehyde acetonide (12) was synthesized in one step from (R)-(−)-2,2-dimethyl-1,3-dioxolane-4-methanol (Alfa Aesar) by the method of Janson et al. See: Janson M, Kvarnström I, Svensson SCT, Samuelsson BCB. Synthesis. 1993:129.
- 13.Myers AG, Hammond M, Wu Y, Xiang JN, Harrington PM, Kuo EY. J Am Chem Soc. 1996;118:10006. doi: 10.1021/ja012487x. [DOI] [PubMed] [Google Scholar]
- 14.In the absence of any lithium halide additive, Wittig coupling of 13 and 14 using n-butyllithium as base afforded a 1.5:1 mixture of Z- and E-olefins, respectively. The undesired E-isomer could be transformed into a 55:45 mixture of E- and Z-isomers, respectively, by UV irradiation. See Supporting Information for details.
- 15.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]
- 16.Brummond and co-workers have reported that the (DHQD)2PYR ligand was optimal for the enantioselective dihydroxylation of an achiral enyne substrate. See: Brummond KM, Lu J, Petersen J. J Am Chem Soc. 2000;122:4915.
- 17.In this application, acetonitrile was found to be a more effective solvent than dichloromethane, the solvent used by Sen et al. See: Sen SE, Roach SL, Boggs JK, Ewing GH, Magrath J. J Org Chem. 1997;62:6684.
- 18.The acetonide protective group proved to be too robust to remove without decomposition in later-stage synthetic intermediates and was therefore modified. Model studies established that the mesitylene protective group was an ideal replacement, being readily removed under acidic conditions (TFA, THF, H2O, 0 °C).1c
- 19.Mesitaldehyde dimethyl acetal (19) was synthesized in one step from mesitaldehyde as follows: camphursulfonic acid (10 mg) was added to a solution of mesitaldehyde (3.00 g, 20.2 mmol, 1 equiv) and trimethyl orthoformate (3.22 g, 30.4 mmol, 1.5 equiv) in methanol (30 mL) at 23 °C. After stirring at 23 °C for 12 h, the reaction mixture was partitioned between ether (30 mL) and saturated aqueous sodium bicarbonate solution (30 mL). The aqueous layer was separated and further extracted with ether (30 mL). The organic extracts were combined and the combined solution was dried over anhydrous sodium sulfate. The dried solution was filtered and the filtrate was concentrated to provide mesitaldehyde dimethyl acetal (19) as a colorless oil (3.90 g, 99%).
- 20.Eglinton G, Galbreath AR. J Chem Soc. 1959:889. [Google Scholar]
- 21.(a) Myers AG, Goldberg SD. Angew Chem Int Ed. 2000;39:2932. doi: 10.1002/1521-3773(20000804)39:15<2732::aid-anie2732>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (b) Myers AG, Hogan PC, Hurd AR, Goldberg SD. Angew Chem Int Ed. 2002;41:1062. doi: 10.1002/1521-3773(20020315)41:6<1062::aid-anie1062>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 22.The diethylisopropylsilyl-protected naphthoic acid 4 was synthesized in 2 steps from the corresponding triisopropylsilyl-protected naphthoic acid, see Supporting Information. For synthesis of the triisopropylsilyl-protected naphthoic acid, see: Ji N, Rosen BM, Myers AG. Org Lett. 2003;6:4551. doi: 10.1021/ol048075l.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures and tabulated spectroscopic data (1H and 13C NMR, FT-IR, and HRMS) for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.