

Abstract
The second WW domain in mammalian Salvador protein (SAV1 WW2) is quite atypical, as it forms a β-clam-like homodimer. The second WW domain in human MAGI1 (membrane associated guanylate kinase, WW and PDZ domain containing 1) (MAGI1 WW2) shares high sequence similarity with SAV1 WW2, suggesting comparable dimerization. However, an analytical ultracentrifugation study revealed that MAGI1 WW2 (Leu355–Pro390) chiefly exists as a monomer at low protein concentrations, with an association constant of 1.3 × 102 M−1. We determined its solution structure, and a structural comparison with the dimeric SAV1 WW2 suggested that an Asp residue is crucial for the inhibition of the dimerization. The substitution of this acidic residue with Ser resulted in the dimerization of MAGI1 WW2. The spin-relaxation data suggested that the MAGI1 WW2 undergoes a dynamic process of transient dimerization that is limited by the charge repulsion. Additionally, we characterized a longer construct of this WW domain with a C-terminal extension (Leu355–Glu401), as the formation of an extra α-helix was predicted. An NMR structural determination confirmed the formation of an α-helix in the extended C-terminal region, which appears to be independent from the dimerization regulation. A thermal denaturation study revealed that the dimerized MAGI1 WW2 with the Asp-to-Ser mutation gained apparent stability in a protein concentration-dependent manner. A structural comparison between the two constructs with different lengths suggested that the formation of the C-terminal α-helix stabilized the global fold by facilitating contacts between the N-terminal linker region and the main body of the WW domain.
Keywords: WW domain, dimerization, stability, dynamics, ultracentrifuge, NMR
The WW domain, which consists of about 40 amino acid residues, is a monomeric protein-binding module with a three-stranded antiparallel β-sheet motif. The name WW is derived from the presence of two highly conserved tryptophan residues with 20–23 amino acid spacing in the polypeptide sequence. They are located on opposite faces of the twisted β-sheet, and the N-terminal tryptophan residue contacts the highly conserved proline residue within a hydrophobic cluster network at the C-terminal region (Jäger et al. 2001). The hydrophobic core typically involves the highly conserved “Leu–Pro” motif, 3–4 residues preceding the tryptophan residue, which induces a “hook” structure in the N-terminal region. The C-terminal tryptophan residue is exposed to the solvent and contacts the conserved aromatic residue (typically tyrosine or phenylalanine) on the second β-strand to form a hydrophobic surface that is used for interactions with proline-rich peptides. The WW domain family members are typically classified into four groups, based on the type of proline-rich peptide ligand (Kay et al. 2000; Kato et al. 2002, 2004).
The eukaryotic Salvador proteins have two tandem WW domains, and we recently reported that the second WW domain of mouse Salvador homolog protein 1 (SAV1 WW2) forms a β-clam-like homodimer, with the peptide-binding site buried in the dimeric interface (Ohnishi et al. 2007). This was the first report of homodimerization by a WW domain, and amino acid sequence analyses may identify more dimeric WW domains. Figure 1A shows the sequence classification of the human WW domains; SAV1 WW2 belongs to a subgroup containing the second WW domain from MAGI1 (membrane associated guanylate kinase, WW and PDZ domain containing 1), which has two tandem WW domains. As shown in Figure 1B, the sequence alignment of the members in this subgroup highlights two atypical sites: the common “aromatic–glycine–deletion” motif prior to the second β-strand, and the substitution of the C-terminal conserved tryptophan immediately after the third β-strand. In the SAV1 WW2 structure, these sites are critical for aromatic packing in the homodimeric interface (Ohnishi et al. 2007). Thus, we anticipated that MAGI1 WW2 would dimerize in a similar manner as the SAV1 WW2 homodimer.
Figure 1.

(A) The neighbor-joining (NJ) tree dendrogram of the WW domains from human origin. The WW domain sequences from human origin were obtained from the Pfam server (http://www.sanger.ac.uk/Software/Pfam/), and the redundancy was removed using the 100% sequence identity filter. The sequences thus selected were analyzed using the program CLUSTAL W (Thompson et al. 1994). (Magenta) The members of the subclass that contains SAV1 WW2 and MAGI1 WW2. (B) Comparative representation of the LOGO plot of the representative WW domain sequences and the sequences of the members of the subclass that contains SAV1 WW2 and MAG1 WW2. (Yellow boxes) Polar residues of E242 and S246 in SAV1 WW2, as well as those of their counterparts in MAGI1 WW2, E366 and D370. (Red) Acidic residues, (blue) basic residues.
In our NMR high-throughput pipeline, we typically design the construct of a protein domain target based on sequence comparisons with the reference entries with known structures. Secondary structure prediction provides additional clues for the construct design. In the present case of the second WW domain of human MAGI1, we designed two constructs: Leu355–Pro390 (MAGI1 WW2 355–390), based on the structural region of the SAV1 WW2, and Leu355–Glu401 (MAGI1 WW2 355–401), as multiple secondary structure prediction algorithms suggested a high probability of α-helix formation for the region between Val391 and Glu401. Both constructs were expressed well in the cell-free protein synthesis system, and their foldabilities were confirmed with the 2D 1H-15N HSQC screening tests (Kigawa et al. 1999).
Here, we report the structural determination and the physicochemical characterization of these MAGI1 WW2 constructs. To our surprise, this atypical WW domain exists as a monomer, which was not affected by changing the construct length. A structural comparison with SAV1 WW2, as well as a subsequent mutational study, revealed a key amino acid residue that regulates homodimer formation. We also performed spin relaxation and thermal denaturation studies to describe the molecular basis of the dynamics and the stability of this atypical WW domain.
Results
Oligomerization state of MAGI1 WW2
We designed the human MAGI1 WW2 construct using the segment from Leu355 to Pro390 (MAGI1 WW2 355–390), which has a residue length comparable to that of the mouse SAV1 WW2 construct (Pro231 to Pro266), for which the structure was determined in our previous study (Ohnishi et al. 2007). Interestingly, the present construct eluted in size exclusion chromatography (HPLC) with a retention volume corresponding to that of the WW domain monomer (data not shown). Indeed, an analytical ultracentrifugation study yielded a molecular weight as 5611 when the monodisperse protein system was assumed, while the theoretical molecular weight for the monomer is 5580 (Fig. 2). When the monomer–dimer equilibrium was assumed, we obtained an association constant, Ka, of 1.3 × 102 M−1. Thus, the molecular association is weak, and the monomer fraction predominates at total protein concentrations <1 mM. This is contrary to what we anticipated from the sequence analysis (Fig. 1).
Figure 2.

Equilibrium analytical ultracentrifugation of MAGI1 WW2 355–390. (Bottom panel) Radial distribution of the absorbance in the centrifuge cell at equilibrium at 16,000 rpm, with a protein concentration of 0.5 mg/mL. The solid line through the data represents the fit to a single species with a molecular weight corresponding to 5611, while the theoretical molecular weight for the single polypeptide chain is 5580. (Upper panels) Residuals for the fit.
Structural determination of MAGI1 WW2
We prepared a 0.6 mM 13C/15N-labeled MAGI1 WW2 355–390 sample for an NMR study. At this protein concentration, the monomer–dimer ratio can be estimated as ∼15 to 1, with a Ka of 1.3 × 102 M−1. Figure 3A shows a 1H-15N HSQC spectrum of the MAGI1 WW2 355–390 sample. Several signals exhibited severe line broadening, suggesting that this construct exists in chemical exchange equilibrium between multiple conformers. Using the high-throughput NMR pipeline at RIKEN Structural Genomics/Proteomics Initiative (RSGI) (Yokoyama et al. 2000), we determined the solution structure of the present construct, assuming that all of the observed NMR signals are attributable to the monomeric conformer (Fig. 3B). The statistics for the NMR structure calculations are summarized in Table 1. The Leu361–Pro390 segment forms a three-stranded antiparallel β-sheet, which is a common motif in the WW domain. The backbone structure was well-converged, and the r.m.s.d. for the backbone heavy atoms was 0.24 Å.
Figure 3.

(A) The 1H-15N HSQC spectrum of MAGI1 WW2 355–390, with labels. (*) Signals from the N- and C-terminal artificial tags of the present construct. (B) Solution structures of MAGI1 WW2 355–390, in line representations. (Cyan) Residues in the N- and C-terminal artificial tags of the present construct.
Table 1.
Statistics of the NMR structural determination

Controlling homodimerization
In order to rationalize the observation that MAGI1 WW2 chiefly exists as a monomer, we performed a detailed comparison between SAV1 WW2 and MAGI1 WW2. Their amino acid sequences are highly similar (identity 53.3%, similarity 90.0%, Fig. 1B), and their backbone structures as well as the orientations of the aromatic side chains are comparable (Fig. 4A,B). The most notable residues in this comparison are Ser246 and Glu242, which are buried in the homodimer interface of SAV1 WW2, as well as their counterparts, Asp370 and Glu366, in MAGI1 WW2. In the context of the SAV1 WW2 homodimer, a neutral polar amino acid at position 246 is crucial, since this residue contacts Glu366 in the partner molecule of the homodimer. The formation of the MAGI1 WW2 homodimer would probably be inhibited by the repulsion between the negative charges. These residues are highlighted in the sequence alignment in Figure 1B.
Figure 4.

Structural comparison between mSAV1 WW2 231–266 and hMAGI1 WW2 355–390. (A) Structure of SAV1 WW2 231–266. (Lines) Side chains of key aromatic residues (F249, Y252, H256, Y263) buried in the interface of the homodimer; (neon bars) side chains of the polar residues (E242, S246). These side chains are colored to clarify the representation of the molecular interaction. (B) Structure of MAGI1 WW2 355–390. (Lines) Side chains of the aromatic residues (Y373, Y376, H380, Y387); (neon bars) side chains of the polar residues (E366, D370).
To test this idea, we designed a single mutant, MAGI1 WW2 355–390 D370S, and examined whether it forms a dimer. The expression and purification of this mutant were straightforward, facilitating the subsequent structural characterization. As expected, it eluted in the size exclusion HPLC at a retention volume equivalent to that of the WW domain dimer (data not shown). Moreover, the analytical ultracentrifugation study yielded its molecular weight as 11,949, while the theoretical value for the monomer is 5551 (Fig. 5A). The analysis of the experimental curve with a theoretical model, assuming dimer–monomer equilibrium, yielded an association constant, Ka > 1020, and thus the mutant indeed forms a dimer. The signals in the 1H-15N and 1H-13C HSQC spectra were well-dispersed with sharp line widths, suggesting that the conformer is monodisperse and stable (data not shown). Significant changes in the chemical shift resonances were prevalent throughout β-strand 1 to β-strand 3 (Fig. 5B). Figure 5, C and D, shows the 1H-13C aromatic HSQC spectra of the wild type and the D370S mutant, respectively. The aromatic resonances of His380 (δ2 and ε1) undergo a remarkable high-field shift in the mutant (Fig. 5D), while they appear in the normal range in the wild type (Fig. 5C). Similar up-field shifts of the histidine aromatic resonances were observed in SAV1 WW2, in which the histidine residue is buried in the aromatic pocket at the homodimer interface (Ohnishi et al. 2007). Hence, it is quite likely that the single mutation, D370S, facilitates the homodimer formation by MAGI1 WW2, and that the negative charge at this position is critical for preventing the dimerization. Interestingly, there are significant numbers of homologs among the WW domain family members that conserve Glu and Asp residues at the positions equivalent to Glu366 and Asp370 in MAGI1 WW2, respectively, suggesting that there is a subset of WW domain members that employ the same mechanism to avoid homodimerization.
Figure 5.

(A) Equilibrium analytical ultracentrifugation of hMAGI1 WW2 355–390 D370S. (Bottom panel) Radial distribution at equilibrium at 16,000 rpm. The solid line through the data represents the fit to a single species with a molecular weight corresponding to 11,949, while the theoretical molecular weight for the single polypeptide chain is 5551. (Upper panel) Residuals for the fit. (B) Difference in chemical shift values of the 1HN and 15N resonances, calculated with the model by Ayed et al. (2001). (Red) Asp-to-Ser mutation at position 370; (cyan bars) residues in the β-sheet. (C,D) HSQC spectrum in the aromatic region of the wild-type hMAGI1 WW2 355–390 (C) and that of its single-point mutant, D370S (D). Red signals represent folded peaks, due to the narrow spectrum width. Resonances corresponding to the δ2 and ε1 protons of H380 are marked.
Extra α-helix formation by the C-terminal extension
In the present study, we also designed a longer construct with 11 extra C-terminal residues for the MAGI1 WW2 (Leu355–Glu401; MAGI1 WW2 355–401), as an α-helix formation for the region between Val391 and Glu401, was expected from the multiple secondary structure prediction algorithms. The longer construct displayed well-dispersed NMR signals with slightly sharper line widths than those of the MAGI1 WW2 355–390 spectrum (Fig. 6A). The elution volume of MAGI1 WW2 355–401 in the size exclusion HPLC was equivalent to that of the monomer (data not shown), as was the case for the original MAGI1 WW2 (355–390). Subsequently, we applied the structural determination protocol to the longer construct and determined its solution structure, as shown in Figure 6B. The statistics of the structural calculations are summarized in Table 1. The region between Asp356 and Gln399 was well converged, with the typical WW domain fold and an extra C-terminal α-helix (Pro390–Gln399), which sticks out on the opposite side of the peptide binding face of the β-sheet. Therefore, the C-terminal helix formation probably has little influence, if any, on the control of the homodimerization. The structure of the canonical WW domain fold moiety (Leu361–Pro390) in the longer construct was quite similar to that in the MAGI1 WW2 355–390 construct (r.m.s.d. of the main chain in this region was 0.44 Å).
Figure 6.

(A) The 1H-15N HSQC spectrum of MAGI1 WW2 355–401, with labels. (*) Signals from the N- and C-terminal artificial tags of the present construct. (B) Solution structures of MAGI1 WW2 355–401, in line representations. (Cyan) Residues in the N- and C-terminal artificial tags of the present construct.
Dynamics of the MAGI1 WW2 variants
We performed a set of 15N spin relaxation experiments for MAGI1 WW2 355–390 and the C-terminal extended construct (MAGI1 WW2 355–401), as well as that of the D370S mutant (MAGI1 WW2 355–390 D370S) that forms a homodimer (Fig. 7). The values in the 15N NOE profile of the point mutant were slightly larger than those of the wild-type and C-terminally extended constructs, suggesting that the fast motion dynamics of the point mutant are more restricted upon dimerization than those of the monomers. The heteronuclear NOE values for the residues between the third β-strand and the α-helix, as well as those in the α-helix, were comparable to those in the β-strands, suggesting that the α-helix is stably formed, and lacks flapping motions. The longitudinal relaxation (R1) values of the wild type were larger than those of the longer construct and the mutant. This R1 trait is consistent with the size of each construct. There are obvious differences in the transverse relaxation (R2) profiles between the three constructs. The exposed residues on the β-sheet of MAGI1 WW2 355–390 have large values, ∼30–35 s−1, while those of MAGI1 WW2 355–401 have somewhat smaller values, ∼20–30 s−1. This suggests that these residues on the peptide binding face undergo chemical exchange. These WW domains may transiently form the β-clam-like homodimers using their peptide binding faces as an interface, but they are unstable due to the negative charge repulsion between E366 and D370. Such a process is also reflected in the fast motion dynamics, as a significant difference in the heteronuclear NOE values around Asp370 between the dimeric and monomeric constructs. This dynamic process is consistent with the results from the present analytical ultracentrifugation study that MAGI1 WW2 355–390 exists in a monomer–dimer equilibrium, with a Ka of 1.3 × 102 M−1 (Fig. 2). In contrast, MAGI1 WW2 355–390 D370S showed significantly smaller R2 values for these residues, again highlighting the more restricted dynamics of the homodimer.
Figure 7.

The 15N spin relaxation profiles of MAGI1 WW2 355–390 (black), MAGI1 WW2 355–401 (red), and MAGI1 WW2 355–390 D370S (green). The heteronuclear NOE (top), longitudinal (R1; middle), and transverse (R2; bottom) relaxation profiles are shown with error bars. (Cyan arrows) β-strands, (red bar) the α-helix of the longer construct, MAGI1 WW2 355–401.
Stability of the MAGI1 WW2 variants
We subsequently carried out a circular dichroism (CD) study to examine the thermal stabilities of MAGI1 WW2 355–390, MAGI1 WW2 355–401, and the D370S mutant (Fig. 7; Table 2). The far-UV CD spectrum of MAGI1 WW2 355–390, recorded at 20°C, showed a characteristic peak ∼230 nm, which was similarly observed for the D370S mutant (Fig. 8A). In contrast, the peak was absent in the spectrum of MAGI1 WW2 355–401. The CD spectrum of this long construct was similar to those typically observed for denatured proteins upon raising the temperature to 95°C, with the largest ellipticity difference around 217 nm. Therefore, thermal denaturation curves were obtained by monitoring the molar ellipticity at 232 nm for MAGI1 WW2 355–390 and the D370S mutant, and at 217 nm for MAGI1 WW2 355–401, with a 60-s equilibration period (Fig. 8B). The extension of the equilibration period to 90 sec did not affect the thermal denaturation profile of MAGI1 WW2 355–390 (data not shown). The melting temperature of MAGI1 WW2 355–390 was comparable to those reported for other WW domains (Koepf et al. 1999; Jäger et al. 2001; Nguyen et al. 2003). With respect to MAGI1 WW2 355–390, the longer construct showed higher stability, by 1.0 kJ/mol at 60°C (ΔTm = 13.1°), indicating that the extension of the C-terminal α-helix stabilizes the global fold of the WW domain. As expected from the dimeric nature of the D370S mutant, this mutant showed an apparent increase in stability, in a protein concentration-dependent manner. The thermal transition of the D370S mutant was steeper than those of MAGI1 WW2 355–390 and the C-terminal extended construct (Fig. 8B). This reflects the fact that the dimeric WW domain has larger surface exposure upon denaturation than the WW domain monomers, as the peptide-binding face of the β-sheet is buried in the interface.
Table 2.
Thermal stability summary of MAGI1 WW2 355–390 and its variants

Figure 8.

(A) Far-UV CD spectra of MAGI1 WW2 355–390 (black), MAGI1 WW2 355–401 (red), and MAGI1 WW2 355–390 D370S (green), recorded at 20°C. (B) Thermal denaturation profiles of MAGI1 WW2 355–390 (black), MAGI1 WW2 355–401 (red), and those of the D370S mutant, recorded with 55 μM (green) and 5 μM (blue) protein concentrations. The changes in ellipticity at 232 nm for MAGI1 WW2 355–390 and MAGI1 WW2 355–390 D370S, and that at 217 nm for MAGI1 WW2 355–401, were monitored and analyzed as a function of temperature.
Discussion
The WW domain is a well-known monomeric protein-binding module found in species ranging from yeast to mammals (Sudol et al. 1995; Sudol 1996). The discovery of SAV1 WW2 dimerization in our previous study (Ohnishi et al. 2007) suggested the possibility that more dimeric WW domains exist. The sequence analysis highlighted four WW domain entries from human origin in the UniProt database that have the sequence properties associated with homodimer formation (Fig. 1). With respect to the whole genome sequences predicted in Pfam (ver. 21.0), 37 entries were classified within the same subgroup as SAV1 WW2/MAGI1 WW2. The present study revealed the monomeric nature of MAGI1 WW2, which is critically controlled by the electrostatic repulsion of the acidic residues. These acidic residues are conserved among 32 of the 37 entries, suggesting that they probably exist as monomers. The rest of the entries (i.e., sp_SAV1_HUMAN, sp_SAV1_MOUSE, tr_Q28HC4_XENTR, tr_Q66IZ1_XENLA, tr_Q66HX5_DANRE) are all orthologs of mSAV1 WW2, and they probably function as homodimers.
SAV1 reportedly interacts with a serine/threonine kinase (Tapon et al. 2002; Udan et al. 2003), while MAGI1 itself is part of a kinase. SAV1 and MAGI1 both have tandem WW domains in common. Their second WW domains share significant sequence similarity (Fig. 1), which resulted in the high similarity in their global folds (Fig. 4A,B). In contrast, their following C-terminal segments have very low sequence similarity (identity = 0%, similarity = 27%). Indeed, we made a longer SAV1 WW2 construct with a C-terminal extension (P231–P277), and recorded its 1H-15N HSQC spectrum (data not shown). These resonances from the extended residues in the C-terminal region fall into the “random-coil region,” suggesting that this extra part is unstructured. Thus, despite the significant similarity in the amino acid sequence and the global fold, SAV1 WW2 and MAGI1 WW2 are stabilized by different mechanisms: the former by the dimerization, and the latter by the C-terminal α-helix extension. Taken together, these observations suggest that SAV1 and MAGI1 are evolutionarily distantly related.
In contrast to the significant difference in the midpoint denaturation temperatures (Fig. 8B), there are few structural differences, particularly in the canonical WW domain fold moiety (Leu361–Pro390), between MAGI1 WW2 355–390 and MAGI1 WW2 355–401. The α-helix is formed by Pro390–Gln399 in the longer construct, and its amino acid residues (Pro390, Val391, Leu392, and potentially Glu393) interact with the WW domain hydrophobic core, including Leu361 and Trp365 (Fig. 9A). Moreover, it appears that the formation of this α-helix induces the interaction between the N-terminal linker region and the WW domain main body, probably due to steric hindrance, as shown in Figure 9, B and C. The contacts between Ser357 and Lys367, and between Glu358 and Lys367 (Fig. 9A), are supported by the unambiguous NOE peaks in the longer construct (e.g., Ser357 Hα − Lys367 Hδ*, Hε*; Glu358 HN, Hα, Hγ *− Lys367 Hβ*), which were not observed in MAGI1 WW2 355–390. In addition, a series of residues in this N-terminal linker region in MAGI1 WW2 355–401 have slightly higher heteronuclear NOE values, in comparison to the shorter constructs (Fig. 7). Taken together, these results suggest that the packing of the N-terminal linker segment to the main body of the WW domain, which is led by the C-terminal α-helix formation, contributes to the stabilization of the global fold of MAGI1 WW2. Similarly, significant stabilization facilitated by the interaction between the N-terminal linker region and the main body of the WW domain was reported for the human Pin1 WW domain (Jäger et al. 2007).
Figure 9.

(A) Close-up view highlighting the interactions around the C-terminal α-helix in MAGI1 WW2 355–401. Key residues described in the main text are marked. (B) Structural comparison between MAGI1 WW2 355–390 (blue) and MAGI1 WW2 355–401 (red). Structures were superimposed using the backbone heavy atoms in the region between Leu361 and Pro390. (C) Comparison of the accessible surface areas of the residues between MAGI1 WW2 355–390 (black) and MAGI1 WW2 355–401 (red). (Cyan bars) β-strands; (red bar) the α-helix of the longer construct, MAGI1 WW2 355–401.
Wiesner and colleagues reported the structural characterization of the WW domain from the yeast splicing factor, Prp40, which has an extra α-helix at the C terminus (Wiesner et al. 2002). This WW domain is also part of a tandem WW domain, but it is the first domain, and the α-helix serves as the linker of the WW domain pair. The amino acid sequences of the α-helix region and the β-sheet region of this first WW domain are unlike those in MAGI1 WW2. In addition, the topology of the helix with respect to the WW domain main frame was distinct between these WW domains. Thus, it is unlikely that these WW domains are evolutionarily related.
The WW domain recognizes proline-rich amino acid sequences, using conserved aromatic and hydrophobic residues on the β-sheet. Typically, the WW domains are classified into four types based on the amino acid sequence pattern of the proline-rich ligand, which can be predicted from the pattern of the surface-exposed residues used for the peptide recognition (Kay et al. 2000; Kato et al. 2002, 2004). However, SAV1 WW2 has an atypical pattern for the conserved peptide binding residues, and it buries them within the homodimer interface (Ohnishi et al. 2007). MAGI1 WW2 adopts a very similar conformation to that of SAV1 WW2, but it exists as a monomer, and the peptide-binding face is exposed to the solvent. Its pattern of the critical residues for peptide recognition is quite unique, and thus the canonical classification cannot be applied. Exploring the sequence pattern that is preferentially recognized by MAGI1 WW2 is intriguing, but it is beyond the scope of this study, which has focused on the stabilization mechanisms of the WW domain. Selection of peptide ligands is underway in our group.
Materials and Methods
Plasmids
The DNA encoding the human MAGI1 gene was subcloned by PCR from the human cDNA clone locus NM_004742 (TrueClone Collection, OriGene, Cat No. SC117150). The regions between L355–P390 and L355–E401 were cloned into the expression vector pCR2.1 (Invitrogen), as fusions with an N-terminal histidine affinity tag and a tobacco etch virus (TEV) protease cleavage site. The actual constructs contain seven extra residues (GSSGSSG) after the TEV cleavage site that are derived from the PCR linker regions.
Proteins
The DNA fragments were incorporated into plasmids and expressed in the cell-free system using 13C/15N-labeled amino acids, with subsequent protein purification using chelating chromatography, as described previously (Tochio et al. 2006). The purified protein was concentrated to 0.6 mM in 20 mM d 11-Tris-HCl buffer (pH 7.0) containing 100 mM NaCl, 1 mM 1,4-DL-dithiothreitol-d 10 (d 10-DTT), 10% 2H2O, and 0.02% NaN3.
Analytical centrifugation
Sedimentation equilibrium experiments were performed using a Beckman Optima XL-I centrifuge. Experiments were carried out with 100-μL samples, at 0.6, 0.3, and 0.15 mg/mL total protein concentrations, using six channel centerpieces. An eight-position rotor (An-50 Ti) was rotated for 16 h at 34,000 rpm, 36,000 rpm, and 38,000 rpm for MAGI1 WW2 355-390, and 16,000 rpm, 18,000 rpm, and 20,000 rpm for MAGI1 WW2 355–390 D370S. Data were collected after 12 h, 14 h, and 16 h at each speed, by monitoring the UV absorbance at 280 nm. The protein samples were then completely sedimented at 42,000 rpm for 6 h, to generate the baseline. Data were analyzed under the assumption of the monodisperse protein system to determine the apparent molecular weight of the protein using the following relationship (McRorie and Voelker 1993):
where cr0, ω, R, T, M, 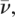 , and ρ are the concentration of the monomer at the reference radius r
0, angular velocity, gas constant, temperature, monomer molecular weight, partial specific volume of the protein, and solvent density, respectively. The partial specific volumes of hMAGI1 WW2 355–390 and hMAGI1 WW2 355–390 D370S were estimated from their amino acid sequences as 0.7129 and 0.7140 mL/g, respectively. Concentration distribution curves were fitted with a solvent density of 1.00293 g/mL, using the XL-A/XL-I Data Analysis software, Ver. 6.03. We also analyzed the sedimentation equilibrium data under the assumption of the monomer–dimer equilibrium system to obtain the association constant, Ka, using the following relationship with the theoretical molecular weight values for the monomer:
, and ρ are the concentration of the monomer at the reference radius r
0, angular velocity, gas constant, temperature, monomer molecular weight, partial specific volume of the protein, and solvent density, respectively. The partial specific volumes of hMAGI1 WW2 355–390 and hMAGI1 WW2 355–390 D370S were estimated from their amino acid sequences as 0.7129 and 0.7140 mL/g, respectively. Concentration distribution curves were fitted with a solvent density of 1.00293 g/mL, using the XL-A/XL-I Data Analysis software, Ver. 6.03. We also analyzed the sedimentation equilibrium data under the assumption of the monomer–dimer equilibrium system to obtain the association constant, Ka, using the following relationship with the theoretical molecular weight values for the monomer:
 |
NMR spectroscopy
All spectra were recorded on Bruker Avance 600 and 800 spectrometers at 298 K. Resonance assignments were carried out using a conventional set of triple resonance spectra, as described previously (Tochio et al. 2006). Interproton distance restraints were obtained from 15N- and 13C-edited NOESY spectra recorded with a mixing time of 120 ms. All spectra were processed using NMRPipe (Delaglio et al. 1995), and the programs NMRView (Johnson and Blevins 1994) and KUJIRA (Kobayashi et al. 2007) were employed for optimal visualization and spectral analyses.
Structural calculation
Automated NOE cross-peak assignments (Herrmann et al. 2002) and structure calculations with torsion angle dynamics (Güntert et al. 1997) were performed using the program CYANA 2.0 (Güntert 2004). Dihedral angle restraints were obtained from TALOS (Cornilescu et al. 1999) and were applied during the structure calculations. The 20 conformers with the lowest CYANA target function value out of the 100 calculated conformers were selected.
Spin relaxation study
All spectra were recorded on a Bruker Avance 600 spectrometer using 1.0 mM 15N-labeled samples of MAGI1 WW2 355–390, MAGI1 WW2 355–401, and MAGI1 WW2 355–390 D370S. Heteronuclear 15N NOEs as well as longitudinal (R1) and transverse (R2) 15N relaxation rates were measured using standard two-dimensional methods (Farrow et al. 1994). The relaxation delays were set to 10, 70, 150, 250, 370, 530, 760, 1150, 1510, and 2010 ms for R1 measurements and 14.4, 28.8, 43.2, 57.6, 72.0, 100.8, 144.0, 187.2, 230.4, and 288.0 ms for R2 measurements. The heteronuclear NOE experiments were run twice, with and without proton saturation during the recovery delay.
Circular dichroism
Circular dichroism (CD) experiments were performed using a JASCO J-820 spectrometer equipped with a Peltier temperature-control system. Far-UV spectra were recorded using 35–85 μM samples of MAGI1 WW2 355–390 and its variants in a 1-mm path length quartz cuvette, and 5 μM MAGI1 WW2 355–390 D370S in a 1-cm path length quartz cuvette. Thermal denaturation was monitored at 232 nm for MAGI1 WW2 355–390 and MAGI1 WW2 355–390 D370S, and at 217 nm for MAGI1 WW2 355–401. The temperature was raised from 20°C to 90°C in 0.2° intervals, with a 60-s equilibration period. There was no visible evidence of precipitation. The thermal denaturation curves were fitted with Equations 3 and 4:
where θobs is the observed ellipticity at temperature T, θN and mN are the intercept and the slope of the pre-transition region, and θU and mU are the intercept and the slope of the post-transition region, respectively. ΔCp is the heat capacity and ΔHm is the unfolding enthalpy at the mid-transition point, T m. The fraction of unfolded protein (α) was calculated using Equation 5:
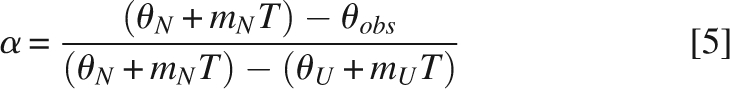 |
and the ΔGu at 60°C (333.15 K) was calculated using Equation 6:
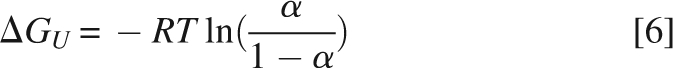 |
Accession codes
The coordinates for MAGI1 WW2 355–390 and MAGI1 355–401 have been deposited in the Protein Data Bank with the accession codes 2ZAJ and 2YSE, respectively.
Acknowledgments
We thank M. Sato, Y. Ide, A. Hiyoshi, and N. Iwasaki for the sample preparation. We thank A. Hasegawa for helpful discussions. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) of the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Reprint requests to: Shigeyuki Yokoyama, Systems and Structural Biology Center, RIKEN, 1-7-22 Suehiro-cho, Tsurumi, Yokohama 230-0045, Japan; e-mail: yokoyama@biochem.s.u-tokyo.ac.jp; fax: 81-45-503-9195.
Abbreviations: SAV1 WW2, the second WW domain of Salvador protein 1; MAGI1 WW2, the second WW domain of membrane associated guanylate kinase, WW and PDZ domain containing 1.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.035329.108.
References
- Ayed, A., Mulder, F.A., Yi, G.S., Lu, Y., Kay, L.E., Arrowsmith, C.H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001;8:756–760. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- Güntert, P., Mumenthaler, C., Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Herrmann, T., Guntert, P., Wüthrich, K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- Jäger, M., Nguyen, H., Crane, J.C., Kelly, J.W., Gruebele, M. The folding mechanism of a β-sheet: The WW domain. J. Mol. Biol. 2001;311:373–393. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- Jäger, M., Nguyen, H., Dendle, M., Gruebele, M., Kelly, J.W. Influence of hPin1 WW N-terminal domain boundaries on function, protein stability, and folding. Protein Sci. 2007;16:1495–1501. doi: 10.1110/ps.072775507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A., Blevins, R.A. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kato, Y., Ito, M., Kawai, K., Nagata, K., Tanokura, M. Determinants of ligand specificity in groups I and IV WW domains as studied by surface plasmon resonance and model building. J. Biol. Chem. 2002;277:10173–10177. doi: 10.1074/jbc.M110490200. [DOI] [PubMed] [Google Scholar]
- Kato, Y., Nagata, K., Takahashi, M., Lian, L., Herrero, J.J., Sudol, M., Tanokura, M. Common mechanism of ligand recognition by group II/III WW domains: Redefining their functional classification. J. Biol. Chem. 2004;279:31833–31841. doi: 10.1074/jbc.M404719200. [DOI] [PubMed] [Google Scholar]
- Kay, B.K., Williamson, M.P., Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- Kigawa, T., Yabuki, T., Yoshida, Y., Tsutsui, M., Ito, Y., Shibata, T., Yokoyama, S. Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett. 1999;442:15–19. doi: 10.1016/s0014-5793(98)01620-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N., Iwahara, J., Koshiba, S., Tomizawa, T., Tochio, N., Güntert, P., Kigawa, T., Yokoyama, S. KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J. Biomol. NMR. 2007;39:31–52. doi: 10.1007/s10858-007-9175-5. [DOI] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Sudol, M., Kelly, J.W. WW: An isolated three-stranded antiparallel β-sheet domain that unfolds and refolds reversibly; evidence for a structured hydrophobic cluster in urea and GdnHCl and a disordered thermal unfolded state. Protein Sci. 1999;8:841–853. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRorie, D.K., Voelker, P.J. Self-associating systems in the analytical ultracentrifuge. Beckman; Fullerton, CA: 1993. [Google Scholar]
- Nguyen, H., Jäger, M., Moretto, A., Gruebele, M., Kelly, J.W. Tuning the free-energy landscape of a WW domain by temperature, mutation, and truncation. Proc. Natl. Acad. Sci. 2003;100:3948–3953. doi: 10.1073/pnas.0538054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi, S., Güntert, P., Koshiba, S., Tomizawa, T., Akasaka, R., Tochio, N., Sato, M., Inoue, M., Harada, T., Watanabe, S., et al. Solution structure of an atypical WW domain in a novel β-clam-like dimeric form. FEBS Lett. 2007;581:462–468. doi: 10.1016/j.febslet.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Sudol, M. Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- Sudol, M., Chen, H.I., Bougeret, C., Einbond, A., Bork, P. Characterization of a novel protein-binding module—the WW domain. FEBS Lett. 1995;369:67–71. doi: 10.1016/0014-5793(95)00550-s. [DOI] [PubMed] [Google Scholar]
- Tapon, N., Harvey, K.F., Bell, D.W., Wahrer, D.C., Schiripo, T.A., Haber, D.A., Hariharan, I.K. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110:467–478. doi: 10.1016/s0092-8674(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochio, N., Umehara, T., Koshiba, S., Inoue, M., Yabuki, T., Aoki, M., Seki, E., Watanabe, S., Tomo, Y., Hanada, M., et al. Solution structure of the SWIRM domain of human histone demethylase LSD1. Structure. 2006;14:457–468. doi: 10.1016/j.str.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Udan, R.S., Kango-Singh, M., Nolo, R., Tao, C., Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- Wiesner, S., Stier, G., Sattler, M., Macias, M.J. Solution structure and ligand recognition of the WW domain pair of the yeast splicing factor Prp40. J. Mol. Biol. 2002;324:807–822. doi: 10.1016/s0022-2836(02)01145-2. [DOI] [PubMed] [Google Scholar]
- Yokoyama, S., Hirota, H., Kigawa, T., Yabuki, T., Shirouzu, M., Terada, T., Ito, Y., Matsuo, Y., Kuroda, Y., Nishimura, Y., et al. Structural genomics projects in Japan. Nat. Struct. Biol. 2000;7 Suppl:943–945. doi: 10.1038/80712. [DOI] [PubMed] [Google Scholar]