

Abstract
Lopinavir (LPV) is a second-generation HIV protease inhibitor (PI) designed to overcome resistance development in patients undergoing long-term antiviral therapy. The mutation of isoleucine at position 47 of the HIV protease (PR) to alanine is associated with a high level of resistance to LPV. In this study, we show that recombinant PR containing a single I47A substitution has the inhibition constant (Ki) value for lopinavir by two orders of magnitude higher than for the wild-type PR. The addition of the I47A substitution to the background of a multiply mutated PR species from an AIDS patient showed a three-order-of-magnitude increase in Ki in vitro relative to the patient PR without the I47A mutation. The crystal structure of I47A PR in complex with LPV showed the loss of van der Waals interactions in the S2/S2′ subsites. This is caused by the loss of three side-chain methyl groups due to the I47A substitution and by structural changes in the A47 main chain that lead to structural changes in the flap antiparallel β-strand. Furthermore, we analyzed possible interaction of the I47A mutation with secondary mutations V32I and I54V. We show that both mutations in combination with I47A synergistically increase the relative resistance to LPV in vitro. The crystal structure of the I47A/I54V PR double mutant in complex with LPV shows that the I54V mutation leads to a compaction of the flap, and molecular modeling suggests that the introduction of the I54V mutation indirectly affects the strain of the bound inhibitor in the PR binding cleft.
Keywords: HIV protease inhibitors, antiviral resistance development, X-ray structure, molecular recognition, enzyme kinetics
HIV-1 protease (PR) plays an essential role in the viral life cycle by cleaving the polyprotein precursors Gag and Gag-Pol into functional proteins, thus allowing the maturation of progeny viral particles (Peng et al. 1989). Inhibition of PR suppresses HIV viral replication (Kohl et al. 1988), and several protease inhibitors (PIs) have been recognized as highly potent drugs able to prolong the life expectancy of HIV-positive patients (Mastrolorenzo et al. 2006; De Clercq 2007). However, ongoing viral replication in the presence of a PI leads to the accumulation of mutations in the PR coding region and to the development of PI resistance and consequently to treatment failure (Yin et al. 2006). For the majority of PIs, there are one or more specific mutations in the PR coding region that are known to confer resistance, such as mutations G48V and L90M for saquinavir (Jacobsen et al. 1996), V82A for ritonavir (Schmit et al. 1996), and D30N and N88D for nelfinavir (Patick et al. 1998). These primary mutations are sometimes associated with compensatory mutations in the vicinity of Gag processing sites; these compensatory mutations improve the ability of the mutated PR to bind and cleave its native substrate (Doyon et al. 1996; Mammano et al. 1998). Recently, a novel mechanism of resistance involving primary mutations in Gag without any mutations in the PR coding region has been identified (Nijhuis et al. 2007). A detailed understanding of the mechanism of resistance development for individual commercially available PIs might lead to the design of a new generation of PIs capable of inhibiting even the highly resistant PR species from AIDS patients (Prejdová et al. 2004; Surleraux et al. 2005; for review, see De Clercq 2007).
Lopinavir (LPV; coadministered with ritonavir, LPV/r) was introduced into clinical practice in late 2000 as a highly active, second-generation PI designed to inhibit PR species with the common mutation V82A (Sham et al. 1998). Now it is a PI of the first choice for initial antiretroviral regimen (Hammer et al. 2006).
Until recently, no specific mutation or combination of mutations had been associated with decreased sensitivity toward lopinavir. Rather, the accumulation of mutations occurring at PR positions 10, 20, 24, 46, 53, 54, 63, 71, 82, 84, and 90 was believed to be associated with lopinavir resistance (“lopinavir mutation score”) (Kempf et al. 2001).
There are recent reports associating lopinavir resistance with a single mutation, I47A, in HIV-1 (Carrillo et al. 1998; de Mendoza et al. 2006) as well as HIV-2 (Masse et al. 2007). The selection of this mutation seems to be a two-step process in HIV-1 (I47-I47V-I47A) (Mo et al. 2005). The prevalence of the I47A substitution in HIV-positive patients is very low and is strongly associated with prolonged treatment with LPV/r (Kagan et al. 2005). Molecular modeling and energy calculations suggest that loss of van der Waals interactions between the lopinavir P2′ phenoxyacetyl moiety and Ala 47 of the mutant PR are responsible for the decreased affinity of LPV toward I47A HIV PR. In vitro analysis of the relative replicative capacity of HIV-1 and HIV-2 strains with the I47A mutation in the PR showed a substantial reduction of susceptibility to LPV, accompanied by hypersusceptibility to atazanavir and saquinavir (Masse et al. 2007). This is probably due to a tighter packing of the PR flaps (Friend et al. 2004; Kagan et al. 2005).
In order to analyze the molecular mechanism of resistance development to LPV on the molecular level, we prepared pure recombinant HIV PRs with the I47A mutation alone or in the background of other LPV resistance-affecting mutations found in a patient sample (Fig. 1), and we analyzed the recombinant proteins in terms of enzymatic activity and 3D structure. We also searched for other possible mutations with a synergic effect on LPV resistance, namely, V32I and I54V, and analyzed the corresponding HIV PRs on the molecular level. A detailed characterization of the interaction of LPV with HIV PR mutant species helps to explain the molecular mechanism of resistance development and is applicable to the design of PIs capable of potent inhibition of resistant HIV-PR species.
Figure 1.

Model of HIV-1 PR with highlighted resistance-conferring mutations studied in this work. Mutations in the PR1 isolated from the patient are colored in yellow; color coding for other mutations is: I47A (green), V32I (brown), and I54V (brown stripes).
Results
Recombinant proteins and kinetic analyses
We isolated a highly resistant HIV-1 PR species from a Czech patient treated by indinavir, lopinavir, amprenavir, and saquinavir for a prolonged period of time. This patient's antiviral chemotherapy failed as documented by increasing viral load and decreasing CD4 count (Supplemental Fig. S1). The HIV PR-coding region amplified from this patient revealed nine mutations considered to contribute to lopinavir resistance (L10I, L24I, L33F, M46L, I54V, L63P, A71V, V82A, and I84V). With the aim of evaluating the putative contribution of the I47A mutation to LPV resistance, we used the PR species derived from the patient DNA (PR1) as a framework and introduced the I47A substitution into a PR variant designated PR2. Furthermore, we introduced the I47A mutation alone and in combination with V32I, a mutation reported to occur along with I47A in AIDS patients with failing lopinavir therapy, into the wild-type PR sequence (generating PR3 and PR5, respectively) as a control. We also generated the double mutant I47A/I54V (PR4) in order to analyze our hypothesis that the substitution I54V, which is present in the patient-derived PR1 sequence, might synergistically influence lopinavir binding in combination with the I47A mutation (vide infra).
All PR variants (summarized in Table 1) were expressed in Escherichia coli, refolded from inclusion bodies, purified, and enzymatically characterized. Active-site titration revealed that the folding efficiency, expressed as a fraction of active enzyme from the total protein, varied between individual mutants (wt PR–80%, PR1–66%, PR2–39%, PR3–91%, PR4–32%, PR5–70%). The Km and k cat values were determined by spectrophotometric assay using a chromogenic peptide substrate derived from the CA-p2 cleavage site in the HIV Gag polyprotein (Table 1). The PR1 variant exhibited an approximately fourfold decrease in catalytic efficiency compared to the wild type. Notably, just 23% of wild-type activity is apparently sufficient to support maturation of the virus in vivo, since this variant was isolated from a replicating viral species. The introduction of the I47A mutation into the background of this patient-derived sequence leads to a dramatic effect on the enzymatic activity of PR2, resulting in only 4% of the wild-type catalytic efficiency for the PR2 variant. The decrease in catalytic efficiency is mainly due to a remarkable increase in Km. Proteases PR3, PR4, and PR5 also exhibited rather low proteolytic activities, 12%, 17%, and 11%, respectively, of the wild-type PR catalytic efficiency. While the introduction of V32I as the second substitution into the I47A-containing PR3 led to an improvement in the substrate turnover rate and simultaneously to a decrease in substrate binding affinity (Table 1, PR5), the introduction of I54V to the same background did not lead to a significant change in either the Km or k cat value (Table 1, PR4).
Table 1.
Enzyme characteristics (Km, k cat) and catalytic efficiencies (k cat/Km) determined by spectrophotometric assay at the pH optimum of the protease (pH 4.7)

Inhibition studies
The inhibition constants (Ki) for lopinavir, ritonavir, saquinavir, amprenavir, indinavir, atazanavir, and brecanavir were determined for a panel of HIV PRs (Table 2). LPV inhibits the wt PR with a Ki of 0.018 nM, and the Ki of LPV for PR1 is nearly 25-fold greater. The introduction of the I47A mutation into the patient-derived PR1 sequence (PR2) has a dramatic effect on Ki. The Ki of LPV for PR2 is three orders of magnitude higher than the Ki of LPV for PR1. The I47A mutation alone (PR3) also revealed a Ki value for LPV that is significantly higher than the wild-type value (two orders of magnitude higher). The introduction of I54V in addition to I47A leads to a further, approximately sevenfold increase in relative Ki values for LPV, RTV, and SQV, while the influence of the V32I mutation is less pronounced (PR4 and PR5 in Table 2). APV and IDV follow a pattern similar to that of LPV. BCV inhibits PRs 4 and 5 (I47A/I54V and V32I/I47A, respectively) with a comparable potency as PR3 (I47A only).
Table 2.
Ki values [nM] for the inhibition of PR mutants by clinically available inhibitors lopinavir, ritonavir, saquinavir, amprenavir, indinavir, atazanavir, and brecanavir

We observed the opposite trend for saquinavir, which exhibited a high Ki value for PR1 (180 ± 15 nM), whereas the introduction of I47A (PR2) decreased the inhibition constant almost eightfold.
In order to compare the relative selective advantage of a given PR mutant over another one in the presence of an inhibitor, the term “vitality,” v, has been introduced as a measure of resistance (Gulnik et al. 1995), defined as:
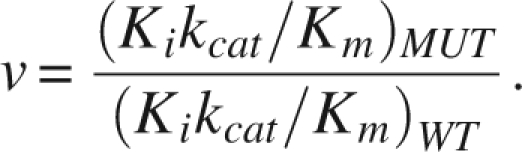 |
Vitality values were calculated for each mutant with the corresponding inhibitor and compared to the values for the wild-type PR as a reference enzyme (Fig. 2). Protease PR1, which harbors mutations that are considered to be relevant for lopinavir resistance (lopinavir mutation score), does not have a high vitality value for LPV. However, the introduction of the I47A mutation into the background of PR1 (PR2) caused a dramatic increase in the vitality value, which indicates high-level resistance to lopinavir. Introduction of the I47A mutation alone into the wt sequence (PR3) has only a small effect on vitality. On the other hand, the I47A/I54V double mutant (PR4) has an approximately 10× higher vitality value than PR3, indicating high-level resistance. The introduction of compensatory mutation V32I (PR5, V32I/I47A) led to a less dramatic twofold increase in vitality (compared to PR3).
Figure 2.

Vitality values for all tested proteases with lopinavir, amprenavir, ritonavir, indinavir, saquinavir, atazanavir, and brecanavir. The PRs ordered along the Y-axis were changed for optimal visibility of the values.
While our data support the key role of the I47A mutation in LPV resistance, it seems that this mutation also sensitizes the PR to the binding of saquinavir, since the introduction of the I47A mutation into PR1 leads to a 50-fold drop in vitality (Fig. 2).
Crystal structures of HIV protease mutants in complex with lopinavir
Crystal structures of two PR variants, PR3 and PR4, were determined in complex with lopinavir at 2.8 Å and 2.3 Å resolution, respectively. Our primary aim was to investigate the potential structural differences caused by the I47A and I54V mutations that could explain the observed enzymatic and inhibition data and provide structural information essential for computational modeling studies. The X-ray data collection statistics and model refinement statistics are given in Supplemental Table S1. Both mutant complexes crystallized in the P61 space group with one PR dimer per asymmetric unit. The structures were refined with two inhibitor molecules per dimer bound in opposite orientations with occupancy 0.5.
Overall, the two crystal structures are very similar; they can be superimposed with root-mean-square deviations (RMSDs) of 0.33 Å for main-chain atoms and 0.18 Å for Cα atoms. This value is within the range observed for structures of identical proteins (Betts and Sternberg 1999). When compared to the structure of the wild-type protease in complex with lopinavir (PDB code 1MUI) (Stoll et al. 2002), the main-chain atoms of the mutant PRs can be superimposed with the wild-type enzyme with RMSDs of 0.81 Å (PR3-LPV) and 0.84 Å (PR4-LPV). The largest structural differences are observed in the vicinity of the substituted residues. Both I47 and I54 are located in the antiparallel β-sheet of the protease flap with side chains pointing toward the enzyme substrate-binding site. The substitution of two isoleucines with alanine and valine not only increases the volume of the active site-binding cavity, but also causes structural alteration of the flap region. The structural shift of the α-carbon of residue 47 is 0.96 Å for PR3 and 1.26 Å for PR4 relative to the wild type. The structural rearrangements can be qualitatively described as a narrowing of the flap antiparallel β-strand (Fig. 3). In the wild-type PR, the side-chain atoms of I47 and I54 are in close contact, and mutations I47A and I54V allow closer interactions between main chains in the antiparallel strands (cf. the hydrogen-bond lengths in Fig. 3B–D).
Figure 3.

Top view of flap region showing structural changes induced by I47A and I54V mutations. (A) Superimposition of the main-chain atoms of wild type (wt), PR3, and PR4. Hydrogen-bond distances in antiparallel β-sheet in the structure of (B) wild-type PR (pdb code 1MUI); (C) I47A PR3; and (D) I47A/I54V PR4. (Red) Oxygen atoms; (blue) nitrogen atoms. The carbon atoms are (cyan) wt (1MUI), (green) PR3 (I47A), and (magenta) PR4 (I47A/I54V).
Mutation of I47 to alanine results in a loss of three methyl groups in each PR monomer. As a result, numerous vdW contacts to the P2 and P2′ segments of LPV are lost or weakened (see Fig. 4A,B). The position of LPV in PR3 and PR4 remains almost unaltered compared to the wild-type complex structure. The only structural change observed is the position of the P2′ phenoxyacetyl moiety of LPV. The position of the P2′ segment in the mutant complexes is shifted by ∼1.4 Å compared to its position in the wt complex (Fig. 4). In contrast, the position of the P2 segment of LPV remains unaltered in our PR3 and PR4 complex structures compared to the wild-type complex.
Figure 4.

Van der Waals interactions between PR and LPV lost due to the I47A mutation. The distances are represented as dashed lines with numbers in angstroms. (A) Wild-type PR (1MUI) complexed with LPV. Segmentation of LPV into P2, P1, P1′, and P2′ subsites is depicted. (B) PR3 (I47A) complexed with LPV. (Thin gray lines) Superimposition of LPV as bound to the wild-type PR.
Another reduction in PR–LPV interactions emerges in the PR4 double mutant as a result of structural change in the flap region (Fig. 3). The weakening of the overall interactions between PR4 and LPV is demonstrated by the increased atomic displacement (ADP) values for the LPV atoms in the mutant complex compared to the wild type. The greatest difference is observed for the P1′ side chain (Supplemental Fig. S2).
Molecular modeling and computation
To interpret the deleterious effect of the I47A mutation on PR inhibition by LPV (Table 2) at the atomic level, we performed molecular modeling and calculated the PR–LPV interaction energies in four complexes of PR variants (wt, PR2, PR3, and PR4) with LPV. The comparison of the two models of protonation of catalytic aspartates (see Supplemental material) showed that more favorable interaction energies were consistently obtained with the protonated inner carboxylate oxygen of Asp 25. This model, consistent with several recent experimental studies (Brynda et al. 2004a; Liu et al. 2005; Kovalevsky et al. 2007) was therefore used in further analyses.
The calculated total interaction free energies between LPV and the four PR variants show a clear decrease in the series wt > PR3 > PR4 > PR2 (Table 3A). Quantitatively, the energy differences between wt PR and PR3, PR4, and PR2 are 1.9, 3.0, and 8.1 kcal/mol, respectively. These values agree well with the Gibbs free energy differences of 2.5, 3.6, and 5.7 kcal/mol obtained from the experimental inhibition constants from Table 2 using the equation ΔΔG (MUT-WT) = RT ln(Ki MUT/Ki WT). We can therefore be confident about the decomposition of the total interaction energies.
Table 3.
Interaction energies (in kcal/mol) between LPV and four PR variants (WT, PR3, PR4, and PR2) separated into contributions from PR subsites S2–S2′

To understand the basis of the energetic losses in the PR–LPV interaction upon mutations in the PR, we decomposed the total interaction energies into contributions from the S2–S2′ subsites of the PR. Table 3A shows that the interactions of subsites S2/S2′ in wt with the P2/P2′ segments of LPV are the strongest, while subsites S1 and S1′contribute more weakly to the total interaction. In the mutated PRs, subsites S2, S1′, and S2′ lose binding affinity to LPV, with the exception of S1′ in PR2. In contrast, interactions in the S1 pocket become stronger in the mutated PRs compared to wt (Table 3A; Supplemental Fig. S3).
In addition to the contributions of PR residues toward LPV binding, the strain of the inhibitor in the PR cavity also affects the energetics of binding. Table 3B shows that the calculated strain of LPV in the four PR molecules disfavors binding by 7–8.5 kcal/mol. More insight can be obtained by decomposition to contributions by segments of LPV: About half of the strain energy can be attributed to the P1′ moiety. This finding corresponds to the increase in the ADP factors of the P1′ segment of LPV observed for the PR4 crystal structure (Supplemental Fig. S2).
For more detailed insight into the relative importance of individual PR residues in the PR subsites to lopinavir binding, we further decomposed the interaction energies into contributions by individual PR residues. The strongest PR–LPV interaction pairs are Asp 29–P2 (−6.3 kcal/mol) and Asp 25–P1′ (−4.5 kcal/mol) followed by residues in three clusters in both PR chains: residues 25–32 in the active site, residues 47–50 in the flap, and residues in the 80s loop (Supplemental Fig. S3). The two above mentioned hydrogen-bonded interaction pairs were previously identified by X-ray crystallography (Stoll et al. 2002) and quantum chemical calculations (Zhang and Zhang 2005) to be important determinants of the wt-LPV binding affinity.
The changes in residue contributions to the binding energetics upon mutations are small overall, with the notable exception of Ile 47/47′ → Ala exchange (1.9/1.5 kcal/mol decrease) (Supplemental Fig. S3). This large drop in interaction energy corresponds to the decreased van der Waals interactions with the P2/P2′ segments of LPV observed in the crystal structure (Fig. 4).
The deleterious effect of the I47A mutation is alleviated by several compensatory interactions in the mutated PRs (Supplemental Fig. S3). The most important ones (energy difference compared to wt >0.5 kcal/mol) are mediated by residues Asp 30, Gly 49, and the flap water (Wat) in the S2 subsite; V82A and I84V/I84′V substitutions in the S1/S1′ pockets; and Gly48′ in the S2′ subsite. With the exception of Asp 30, these residues bind more strongly to LPV owing to more favorable van der Waals interactions in the mutated PRs. There are also several compensatory interactions observed specifically in the PR2 complex; the most important ones originate from residues Leu 23, Gly 27/27′, and Ile 50′ in the S1/S1′ subsites and are also of van der Waals character.
Unlike the clear-cut effect of the I47A mutation on LPV binding, the I54V substitution by itself does not have an impact from an energetic point of view. However, the I54V mutation in cooperation with I47A does affect the structure of the flaps, as observed in the crystal structures (Fig. 3). In terms of energy, the strain of the P1′ segment of LPV increases in PR4, which harbors the double mutation I47A/I54V (Table 3B).
Discussion
In this study, we set out to elucidate the structural features of HIV-1 PR responsible for resistance to lopinavir, a second-generation protease inhibitor often used for salvage therapy in patients who have experienced numerous previous treatment regimens (Loutfly and Walmsley 2002; Václavíková et al. 2005). We used a PR variant (designated PR1) amplified from a patient under prolonged treatment with lopinavir as a framework to investigate the structural background of the influence of the I47A mutation on lopinavir resistance. While PR1 showed a significant increase in Ki values for saquinavir, ritonavir, indinavir, and brecanavir (relative to the values for wt PR), it was still rather effectively inhibited by lopinavir.
The introduction of the I47A mutation into the PR1 sequence leads to a dramatic, 90-fold increase in the vitality value for LPV. Strikingly, this effect was rather specific for LPV, since the relative vitality gain for the other inhibitors tested was much less pronounced (see Fig. 2). It should be noted that the vitality value incorporates the ability of the mutated PR to cleave its substrate; the introduction of the I47A mutation led to a dramatic sixfold decrease in the specific activity of the enzyme, mainly owing to an ∼10-fold increase in Km for the chromogenic peptide substrate used. While the overall decrease in specific activity of this mutant is significant, it could be argued that the ability to bind the substrate is less relevant in the crowded environment of the budding virus particle than the turnover number. PR species with a similar defect in substrate binding (Km increase) showed replicative capacity in vitro comparable to that of the wild-type virus (Konvalinka et al. 1995; Rose et al. 1995). On the other hand, the replicative capacity of a PR variant including I47A/V32I mutations was shown to be only 2.9% that of the wild-type virus (Friend et al. 2004), which is a very low value for a virus to be selected in vivo.
The rather unexpected influence of a single mutation on LPV and substrate binding led us to analyze the contribution of the isolated I47A mutation (without any other mutations, PR3) to enzyme activity and inhibitor susceptibility. PR3 (Tables 1 and 2) shows decreased substrate turnover and, surprisingly, similar substrate binding compared to the wt PR. This single mutation leads to an ∼60-fold increase in the relative Ki value of LPV and also significant increases in relative Ki values for indinavir, amprenavir, and brecanavir. The X-ray structure analysis of the PR3–LPV complex provides an explanation for the decreased binding: The loss of three interacting methyl groups leads to spatial enlargement of the S2 and S2′ pockets of the PR3-binding site and subsequently to loss of numerous van der Waals contacts with the S2 and S2′ pockets of the PR3-binding site (see Supplemental Fig. S3). In particular, we confirm the important role of the interaction of the side-chain terminal Cδ methyl group of the I47 residue with the ring atoms of the P2 phenoxyacetyl moiety of lopinavir (Kagan et al. 2005). The decomposition of interaction energies showed that the S2/S2′ pockets mediate the strongest wt PR–LPV interaction, most importantly with the Asp 29 and Ile 47 residues of PR. Another important interaction occurs between Asp 25 and P1′.
Although the isolated I47A mutation brings about a significant increase in the relative Ki value for LPV (PR3 in Table 2), it is still ∼240× lower than the Ki obtained for PR2, which harbors nine mutations from the lopinavir mutation score in addition to the I47A mutation. Clearly, some other amino acid mutations in the PR2 sequence contribute synergistically to the overall relative inhibition and vitality (Clemente et al. 2004). Specifically, the V32I mutation has been reported to compensate for the loss of S2/S2′ interactions in the active-site cleft (Parkin et al. 2003). The covariation frequency of I47V and V32I is the second highest among resistance conferring mutations after D30N + N88D (Kagan et al. 2006). The annual frequency of I47V/A + V32I increased sharply after lopinavir was introduced in late 2000 (Supplemental Table S2).
In a structural model based on the X-ray structure of PR4 (data not shown), we observed that the additional methyl group present due to the V32I mutation can partially replace the methyl groups missing because of the I47A substitution. This effect does not play a major role in interactions with the P2 and P2′ moieties of LPV but could help to stabilize the flaps (Kagan et al. 2005).
V32 is not the only amino acid that potentially contributes to the interaction with the side chain of the amino acid in position 47. As an example of a potential synergistic cooperation effect, we chose the I54V mutation, which is located in the vicinity of the side chain of the amino acid residue in position 47. The introduction of the I54V mutation together with the I47A mutation increased the relative Ki value of LPV for the corresponding mutated HIV PR (PR4) sevenfold, without compromising the ability of the mutated enzyme to cleave its substrate. This amino acid exchange also led to a significant increase in relative Ki values (and vitalities) for brecanavir, amprenavir, and indinavir. The X-ray structure of the complex of I47A/I54V PR (PR4) with LPV shows that the introduction of I54V leads to the compaction of the distance within the antiparallel β-sheets of the flaps accompanied by shortening of main-chain H-bonds (cf. Fig. 3). Concerning the binding of LPV, a comparison of the structural atomic displacement parameters (ADPs) of both X-ray structures shows an increase in ADPs in the P1′ position of the inhibitor, suggesting a weaker interaction of the inhibitor with the mutated PR. The structure of I54V HIV PR with a peptide substrate in the active site at 1.5 Å resolution was recently published (Kovalevsky et al. 2007). This high-resolution structure revealed a cascade of subtle structural changes caused by the I54V substitution with the largest shifts in residues I50 and I50′. The single I54V substitution does not cause structural changes (compaction) of flaps as we observe in our I47A/I54V complex structure. The flap compaction is probably triggered by the I47A mutation, and this structural change is only intensified by the addition of the I54V substitution.
It cannot be ruled out that additional factors, not observable in the X-ray structure, might be responsible for the differences in binding. Since the mutations are localized at the flap hinge, it is tempting to speculate that the flap dynamics are affected, which might provide an additional explanation for the resistant phenotype observed. The experimental verification of these speculations would require time-resolved NMR analysis of the inhibitor/mutated PR complexes.
Based on a molecular model, Kagan et al. (2005) suggest that the I54 side chain is tightly packed between the Cγ atoms of I47 and V56 in the same PR subunit and of I50 in the second subunit. They claim that these interactions of I54 stabilize the closed conformation of the PR flaps, which may be important for the enzymatic activity of the protease. Modeling studies performed by Kagan et al. showed that mutation of I54 to Leu or Met in a heavily mutated background is likely to be sterically unfavorable, since the longer side chains would not fit into the space available between the side chains of I32, A47, and I50 (Kagan et al. 2005). In order to estimate the clinical relevance of this potential synergistic mutation, we surveyed a mutation database constructed from more than 195,000 clinical HIV-1 sequences submitted for genotypic analysis between 2001 and September 2007. Remarkably, only two sequences were found to carry I47A together with I54V. In fact, the mutations I47A and I54V, L or M seem to be mutually exclusive. In contrast, I47V readily co-occurs with I54/V, M and L (cf. Supplemental material). The structural reason for this apparent mutual exclusivity is unclear. The enzymological data suggest that the combination I47A/I54V yields PR species with very similar kinetic properties as I47A/V32I. In fact, the vitality profile of I47A/I54V (PR4) seems to be more favorable than that of I47A/V32I PR (PR5). It remains to be seen whether or not the prevalence of these two mutations will increase in the future.
Materials and Methods
Patients
Within a long-term epidemiological study, HIV-positive patients receiving highly active antiretroviral therapy (HAART) at the University Clinic Bulovka in Prague have been closely monitored for the presence of resistant HIV species (Václavíková et al. 2005). All patients provided informed consent. The selection of the sample for this study (protease PR1) (Table 1) was based on the mutation outcome in the PR coding region and clinical markers suggesting resistance development (data on the patient's treatment are summarized in the Supplemental Fig. S1). The HIV-1 RNA copy number was determined by quantitative RT-PCR (Amplicor HIV-1 Monitor version 1.5; Roche Diagnostics), and the CD4+ cell count was determined by FACS analysis using CaliburE2771 as described (Václavíková et al. 2005). The viral species analyzed belongs to the B subtype of HIV-1.
The PR coding region from HIV-1 was isolated and amplified from patient plasma by RT-PCR as published previously (Prejdová et al. 2004; Václavíková et al. 2005).
Generation of recombinant PRs with PI resistance mutations
The RT-PCR product was used as a template for amplification of the PR coding region (corresponds to PR1) with the forward primer 5′- ATCCTTTCATATGCCTCAGATCACTTTTG-3′, which is specific to the 5′-end of the PR coding region and includes an NdeI cleavage site, and the reverse primer 5′-TTGAATTCGATATCATTAAAAATTTAAAGTGCAGCC-3′, which includes a recognition site for EcoRI. PCR was performed with the following program: a 5-min hold at 95°C and then 34 cycles of 30 s at 95°C, 30 s at 55°C, and 1 min at 72°C. The PCR product was ligated into the expression vector pET24a (Novagen). Mutagenesis reactions were performed according the procedure from the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
The recombinant PRs prepared for this study are summarized in Table 1. The wild-type HIV-1 PR (pNL 4-3) served as a template for constructing PR variant PR3, which harbors only the I47A mutation and no other mutations. The following primers were used: 5′-GGAAACCAAAAATGGCAGGGGGAATTGGAGG-3′ and 5′-CCTCCAATTCCCCCTGCCATTTTTGGTTTCC-3′. The product of this PCR reaction was used as a template for the preparation of the variant PR4, which harbors the double mutation I47A/I54V, using primers 5′-GGAATTGGAGGTTTTGTCAAAGTAAGACAGTATG-3′ and 5′-CATACTGTCTTACTTTGACAAAACCTCCAATTCC-3′. To introduce the V32I substitution, the following primers were used: 5′-CAGGAGCAGATGATACAATATTAGAAGAAATG-3′ and 5′-CATTTCTTCTAATATTGTATCATCTGCTCCTG-3′. In order to introduce the I47A mutation into protease PR1, which was amplified from an HIV-1-positive patient under prolonged therapy with PIs, the following primers were used: 5′-GGAAACCCAAACTGGCAGGGGGAATTGGAGG-3′ and 5′-CCTCCAATTCCCCCTGCCAGTTTTGGTTTCC-3′. The sequences of the products were verified after each mutagenesis reaction by dideoxynucleotide sequencing.
Protein expression and purification
The recombinant proteases were overexpressed in E. coli BL21 (DE3) RIL (Novagen). Protein expression and isolation of inclusion bodies were carried out as previously described (Konvalinka et al. 1997; Weber et al. 2002). Inclusion bodies were solubilized in 67% (v/v) acetic acid and refolded by dilution into a 25-fold excess of water, followed by overnight dialysis at 4°C against water and then against 50 mM MES (pH 5.8), 10% (v/v) glycerol, 1 mM EDTA, and 0.05% (v/v) 2-mercaptoethanol (Stríšovšký et al. 2000). The proteases were purified by cation exchange chromatography using MonoS FPLC (Pharmacia). The enzymes were stored at −70°C until further use.
Activity and inhibition assay
Enzyme kinetic parameters (Km and k cat) and inhibition constants (Ki) were determined by spectrophotometric assay with the chromogenic peptide substrate KARVNle*NphEANle-NH2 as previously described (Richards et al. 1990; Kožíšek et al. 2004). Typically, 8 pmol of PR was added to 1 mL of 0.1 M sodium acetate buffer (pH 4.7), 0.3 M NaCl, and 4 mM EDTA, containing substrate at a concentration near the Km of the enzyme and various concentrations of an inhibitor dissolved in DMSO. The final concentrations of DMSO were kept below 2.5% (v/v). Substrate hydrolysis was followed as a decrease of absorbance at 305 nm using a Unicam UV 500 spectrophotometer (ThermoSpectronic). The data were analyzed using the equation for competitive inhibition according to Williams and Morrison (1979).
Crystallization and data collection
The PR–inhibitor complexes were prepared by mixing the enzyme with a fivefold molar excess of lopinavir dissolved in DMSO and concentrated up to 3–5 mg/mL by ultrafiltration using Microcon-10 filters (Millipore). Crystals were grown by the hanging-drop vapor diffusion technique at 19°C. The crystallization drops consisted of 2 μL of PR–lopinavir complex and 1 μL of reservoir solution. The reservoir solution for the PR3–LPV complex contained 0.5 M NaCl and 0.1 M MES (pH 6.0); the conditions were 0.7 M NaCl and 0.1 M MES (pH 6.0) for crystallization of the PR4–LPV complex.
For diffraction measurement at 120K, crystals were soaked in the corresponding reservoir solution supplemented with 30% glycerol (v/v) and cryo-cooled in liquid nitrogen. X-ray diffraction data were collected on a Mar345 image plate system using a Nonius FR591 rotating anode generator. All data were integrated and reduced using MOSFLM (Leslie 1999) and scaled using SCALA (Evans 1993) from the CCP4 suite (Baily 1994). Crystal parameters and data collection statistics are summarized in Supplemental Table S1.
Structure refinement and analysis
The hexagonal crystals obtained were isomorphous with all other P61 crystals of HIV-1 protease complexes, and the structure determination was performed by the difference-Fourier method using protease coordinates from PDB structure 1U8G (Brynda et al. 2004b) as a starting model. Initial rigid-body refinement and subsequent restrained refinement cycles were performed with the program REFMAC 5.1.24 (Murshudov et al. 1997) from the CCP4 package (Baily 1994). For manual model rebuilding and inhibitor building, the program Coot (Emsley and Cowtan 2004) was used. Final TLS refinement (Winn et al. 2003) was done with TLS groups corresponding to HIV PR subdomains (Rose et al. 1998). Atomic coordinates and structure factors have been deposited into the Protein Data Bank (PDB) with accession codes 2QHC and 2Z54 for PR3 and PR4, respectively.
Molecular modeling
Interaction energy calculations were performed for four HIV-1 protease variants (wild-type, PR3, PR4, and PR2) (see Table 1) complexed with LPV. The starting coordinates were obtained from X-ray crystallography (wt, PDB code 1MUI) (Stoll et al. 2002) (PR3 and PR4) (this study) and molecular modeling (PR2). To derive the latter, the wt amino acids (Lys 20 and Phe 53) were modeled into the X-ray structure of PR1-K20R-F53L (data not shown). Details regarding the preparation of structures for calculations are presented in the Supplemental material.
The geometries of the complexes were relaxed in a vacuum prior to calculating the interaction energies. These were computed with the molecular mechanics-generalized Born/surface area (MM-GBSA) methodology (Massova and Kollman 2000) using the MM-PBSA and SANDER modules of AMBER 8 (Case et al. 2004). All water molecules except the flap water (Wat) were stripped, and Wat was included as part of the PR. The total PR–LPV interaction energies were decomposed to contributions from the S2–S2′ subsites and to pairwise PR residue-LPV segment contributions.
The strain in the LPV molecule was obtained as the MM-GBSA energy difference between the LPV conformation in the PR complex and that of the free inhibitor in solution. The latter was obtained by minimizing the LPV molecule for 10,000 cycles in the implicit generalized Born model of solvation.
Electronic supplemental material
The Supplemental material contains X-ray data collection and model refinement statistics for structures of mutants PR3 and PR4 (Supplemental Table S1); the frequencies of mutations I47V/A and I54V/L/M among HIV-positive patients (Supplemental Table S2); virologic and immunologic response of the HIV-positive patient who provided the sample for the isolation of PR1 correlated to his PI-therapy (Supplemental Fig. S1); a bar chart showing the ADPs of LPV atoms in complexes with PR3 and PR4 (Supplemental Fig. S2); and a graph showing the distribution of the PR residue interaction energies with LPV segments in the S2–S2′ subsites of individual PR mutants.
Acknowledgments
This work was supported by grant NR-8571-3 from the Ministry of Health Care of the Czech Republic, a grant from the Ministry of Education (MSMT) of the Czech Republic within Programme 1M0508 “Research Centre for new Antivirals and Antineoplastics,” by Research Plan AVOZ40550506 and AVOZ50520514 from the Academy of Sciences of the Czech Republic, and by donations from Abbott Laboratories and Gilead Sciences. The authors thank Hillary Hoffman for critical proofreading of the manuscript and many valuable comments.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Jan Konvalinka, Gilead Sciences and IOCB Research Center, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo n. 2, 166 10, Prague 6, Czech Republic; e-mail: konval@uochb.cas.cz; fax: 420-220183578.
Abbreviations: ADP, atomic displacement parameter; APV, amprenavir; ATV, atazanavir; BCV, brecanavir; ESP, electrostatic potential; HAART, highly active antiretroviral therapy; HIV-1, human immunodeficiency virus type 1; IDV, indinavir; k cat, catalytic rate constant; Ki, inhibition constant; Km, Michaelis constant; LPV, lopinavir; LPV/r, lopinavir coadministered with ritonavir; mut, mutant; Nle, norleucine; Nph, p-nitrophenylalanine; PI, protease inhibitor; PR, protease; RTV, ritonavir; SQV, saquinavir; Wat, flap water; wt, wild type.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.036079.108.
References
- Baily, S. The CCP4 Suite—programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Betts, M.J., Sternberg, M.J. An analysis of conformational changes on protein–protein association: Implications for predictive docking. Protein Eng. 1999;12:271–283. doi: 10.1093/protein/12.4.271. [DOI] [PubMed] [Google Scholar]
- Brynda, J., Řezáčová, P., Fábry, M., Hořejší, M., Štouráčová, R., Sedláček, J., Souček, M., Hradílek, M., Lepšík, M., Konvalinka, J. A phenylnorstatine inhibitor binding to HIV-1 protease: Geometry, protonation, and subsite-pocket interactions analyzed at atomic resolution. J. Med. Chem. 2004a;47:2030–2036. doi: 10.1021/jm031105q. [DOI] [PubMed] [Google Scholar]
- Brynda, J., Řezáčová, P., Fábry, M., Hořejší, M., Štouráčová, R., Souček, M., Hradílek, M., Konvalinka, J., Sedláček, J. Inhibitor binding at the protein interface in crystals of a HIV-1 protease complex. Acta Crystallogr. D Biol. Crystallogr. 2004b;60:1943–1948. doi: 10.1107/S0907444904021572. [DOI] [PubMed] [Google Scholar]
- Carrillo, A., Stewart, K.D., Sham, H.L., Norbeck, D.W., Kohlbrenner, W.E., Leonard, J.M., Kempf, D.J., Molla, A. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 1998;72:7532–7541. doi: 10.1128/jvi.72.9.7532-7541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A., Darden, T.A., Cheatham T.E., III, Simmerling, C.L., Wang, J., Duke, R.E., Luo, R., Merz, K.M., Wang, B., Pearlman, D.A., et al. AMBER 8. University of California; San Francisco, CA: 2004. [Google Scholar]
- Clemente, J.C., Mosse, R.E., Hemrajani, R., Whitford, L.R., Govindasamy, L., Reutzel, R., McKenna, R., Agbandje-McKenna, M., Goodenow, M.M., Dunn, B.M. Comparing the accumulation of active- and nonactive-site mutations in the HIV-1 protease. Biochemistry. 2004;43:12141–12151. doi: 10.1021/bi049459m. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. Anti-HIV drugs. Verh. K. Acad. Geneeskd. Belg. 2007;69:81–104. [PubMed] [Google Scholar]
- de Mendoza, C., Valer, L., Bacheler, L., Pattery, T., Corral, A., Soriano, V. Prevalence of the HIV-1 protease mutation I47A in clinical practice and association with lopinavir resistance. AIDS. 2006;20:1071–1074. doi: 10.1097/01.aids.0000222084.44411.cc. [DOI] [PubMed] [Google Scholar]
- Doyon, L., Croteau, G., Thibeault, D., Poulin, F., Pilote, L., Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996;70:3763–3769. doi: 10.1128/jvi.70.6.3763-3769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley, P., Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans, P.R. Data reduction. In: Helliwell J.R., et al., editors. Proceedings of CCP4 Study Weekend, on Data Collection and Processing; Warrington, UK: Daresbury Laboratory; 1993. pp. 114–122. [Google Scholar]
- Friend, J., Parkin, N., Lieger, T., Martin, J.N., Deeks, S.G. Isolated lopinavir resistance after virological rebound of a ritonavir/lopinavir-based regimen. AIDS. 2004;18:1965–1970. doi: 10.1097/00002030-200409240-00016. [DOI] [PubMed] [Google Scholar]
- Gulnik, S.V., Suvorov, L.I., Liu, B., Yu, B., Anderson, B., Mitsuya, H., Erickson, J.W. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- Hammer, S.M., Saag, M.S., Schechter, M., Montaner, J.S., Schooley, R.T., Jacobsen, D.M., Thompson, M.A., Carpenter, C.C., Fischl, M.A., Gazzard, B.G., et al. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-USA panel. Top. HIV Med. 2006;14:827–843. [PubMed] [Google Scholar]
- Jacobsen, H., Hänggi, M., Ott, M., Duncan, I.B., Owen, S., Andreoni, M., Vella, S., Mous, J. In vivo resistance to a human immunodeficiency virus type 1 proteinase inhibitor: Mutations, kinetics, and frequencies. J. Infect. Dis. 1996;173:1379–1387. doi: 10.1093/infdis/173.6.1379. [DOI] [PubMed] [Google Scholar]
- Kagan, R.M., Shenderovich, M.D., Heseltine, P.N.R., Ramnarayan, K. Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 2005;14:1870–1878. doi: 10.1110/ps.051347405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan, R.M., Cheung, P.K., Huang, T.K., Lewinski, M.A. Increasing prevalence of HIV-1 protease inhibitor-associated mutations correlates with long-term non-suppressive protease inhibitor treatment. Antiviral Res. 2006;71:42–52. doi: 10.1016/j.antiviral.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Kempf, D.J., Isaacson, J.D., King, M.S., Brun, S.C., Xu, Y., Real, K., Berstein, B.M., Japour, A.J., Sun, E., Rode, R.A. Identification of genotypic changes in human immunodeficiency virus protease that correlate with reduced susceptibility to the protease inhibitor lopinavir among viral isolates from protease inhibitor-experienced patients. J. Virol. 2001;75:7462–7469. doi: 10.1128/JVI.75.16.7462-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, N.E., Emini, E.A., Schleif, W.A., Davis, L.J., Heimbach, J.C., Dixon, R.A., Scolnick, E.M., Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konvalinka, J., Litterst, M.A., Welker, R., Kottler, H., Rippmann, F., Heuser, A.M., Kräusslich, H.G. An active-site mutation in the human immunodeficiency virus type 1 proteinase (PR) causes reduced PR activity and loss of PR-mediated cytotoxicity without apparent effect on virus maturation and infectivity. J. Virol. 1995;69:7180–7186. doi: 10.1128/jvi.69.11.7180-7186.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konvalinka, J., Litera, J., Weber, J., Vondrášek, J., Hradílek, M., Souček, M., Pichová, I., Majer, P., Štrop, P., Sedláček, J., et al. Configurations of diastereomeric hydroxyethylene isosteres strongly affect biological activities of a series of specific inhibitors of human-immunodeficiency-virus proteinase. Eur. J. Biochem. 1997;250:559–566. doi: 10.1111/j.1432-1033.1997.0559a.x. [DOI] [PubMed] [Google Scholar]
- Kovalevsky, A.Y., Chumanevich, A.A., Liu, F., Louis, J.M., Weber, I.T. Caught in the act: The 1.5 Å resolution crystal structures of the HIV-1 protease and the I54V mutant reveal a tetrahedral reaction intermediate. Biochemistry. 2007;46:14854–14864. doi: 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kožíšek, M., Prejdová, J., Souček, M., Machala, L., Staňková, M., Linka, M., Brůčková, M., Konvalinka, J. Characterisation of mutated proteinases derived from HIV-positive patients: Enzyme activity, vitality and inhibition. Collect. Czech. Chem. Commun. 2004;69:703–714. [Google Scholar]
- Leslie, A.G. Integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- Liu, F., Boross, P.I., Wang, Y.F., Tozser, J., Louis, J.M., Harrison, R.W., Weber, I.T. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J. Mol. Biol. 2005;354:789–800. doi: 10.1016/j.jmb.2005.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutfly, M.R., Walmsley, S.L. Salvage antiretroviral therapy in HIV infection. Expert Opin. Pharmacother. 2002;3:81–90. doi: 10.1517/14656566.3.2.81. [DOI] [PubMed] [Google Scholar]
- Mammano, F., Petit, C., Clavel, F. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: Phenotypic analysis of protease and gag coevolution in protease inhibitor-treated patients. J. Virol. 1998;72:7632–7637. doi: 10.1128/jvi.72.9.7632-7637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse, S., Lu, X., Dekhtyar, T., Lu, L., Koev, G., Gao, F., Mo, H., Kempf, D.J., Bernstein, B., Hanna, G.J., et al. In vitro selection and characterization of human immunodeficiency virus type 2 with decreased susceptibility to lopinavir. Antimicrob. Agents Chemother. 2007;51:3075–3080. doi: 10.1128/AAC.00146-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova, I., Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. [Google Scholar]
- Mastrolorenzo, A., Rusconi, S., Scozzafava, A., Supuran, C.T. Inhibitors of HIV-1 protease: 10 years after. Expert Opin. Ther. Pat. 2006;16:1067–1091. [Google Scholar]
- Mo, H.M., King, S., King, K., Molla, A., Brun, S., Kempf, D.J. Selection of resistance in protease inhibitor-experienced, human immunodeficiency virus type 1-infected subjects failing lopinavir-and ritonavir-based therapy: Mutation patterns and baseline correlates. J. Virol. 2005;79:3329–3338. doi: 10.1128/JVI.79.6.3329-3338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., Dodson, E.J. Refinment of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nijhuis, M., van Maarseveen, N.M., Lastere, S., Schipper, P., Coakley, E., Glass, B., Rovenská, M., de Jong, D., Chappey, C., Goedegebuure, I.W., et al. A novel substrate based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007;4:152–163. doi: 10.1371/journal.pmed.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin, N.T., Chappey, C., Petropoulos, Ch.J. Improving lopinavir genotype algorithm through phenotype correlations: Novel mutation patterns and amprenavir cross-resistance. AIDS. 2003;17:955–961. doi: 10.1097/00002030-200305020-00003. [DOI] [PubMed] [Google Scholar]
- Patick, A.K., Duran, M., Cao, Y., Shugarts, D., Keller, M.R., Mazabel, E., Knowles, M., Chapman, S., Kuritzkes, D.R., Markowitz, M. Genotypic and phenotypic characterization of human immunodeficiency virus type 1 variants isolated from patients treated with the protease inhibitor nelfinavir. Antimicrob. Agents Chemother. 1998;42:2637–2644. doi: 10.1128/aac.42.10.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, C., Ho, B.K., Chang, N.T. Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J. Virol. 1989;63:2550–2556. doi: 10.1128/jvi.63.6.2550-2556.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prejdová, J., Souček, M., Konvalinka, J. Determining and overcoming resistance to HIV protease inhibitors. Curr. Drug Targets Infect. Disord. 2004;4:137–152. doi: 10.2174/1568005043340984. [DOI] [PubMed] [Google Scholar]
- Richards, A.D., Phylip, L.H., Farmerie, W.G., Scarborough, P.E., Alvarez, A., Dunn, B.M., Hirel, P.H., Konvalinka, J., Štrop, P., Pavlíčková, L., et al. Sensitive, soluble chromogenic substrates for HIV-1 proteinase. J. Biol. Chem. 1990;265:7733–7736. [PubMed] [Google Scholar]
- Rose, J.R., Babe, L.M., Craik, C.S. Defining the level of human-immunodeficiency-virus type-1 (HIV-1) protease activity required for HIV-1 particle maturation and infectivity. J. Virol. 1995;69:2751–2758. doi: 10.1128/jvi.69.5.2751-2758.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, R.B., Craik, C.S., Stroud, R.M. Domain flexibility in retroviral proteases: Structural implications for drug resistant mutations. Biochemistry. 1998;37:2607–2621. doi: 10.1021/bi9716074. [DOI] [PubMed] [Google Scholar]
- Schmit, J.C., Ruiz, L., Clotet, B., Raventos, A., Tor, J., Leonard, J., Desmyter, J., De Clercq, E., Vandamme, A.M. Resistance-related mutations in the HIV-1 protease gene of patients treated for 1 year with the protease inhibitor ritonavir (ABT-538) AIDS. 1996;10:995–999. doi: 10.1097/00002030-199610090-00010. [DOI] [PubMed] [Google Scholar]
- Sham, H.L., Kempf, D.J., Molla, A., Kennan, C.M., Gondi, N.K., Chin-Ming, Ch., Kati, W., Steward, K., Lal, R., Hsu, A., et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998;42:3218–3224. doi: 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll, V., Qin, W., Stewart, K.D., Jakob, C., Park, C., Walter, K., Simmer, R.L., Helfrich, R., Bussiere, D., Kao, J., et al. X-ray crystallographic structure of ABT-378 (lopinavir) bound to HIV-1 protease. Bioorg. Med. Chem. 2002;10:2803–2806. doi: 10.1016/s0968-0896(02)00051-2. [DOI] [PubMed] [Google Scholar]
- Stříšovský, K., Tessmer, U., Langner, J., Konvalinka, J., Kräusslich, H.G. Systematic mutational analysis of the active-site threonine of HIV-1 proteinase: Re-thinking the “fireman's grip” hypothesis. Protein Sci. 2000;9:1631–1641. doi: 10.1110/ps.9.9.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surleraux, D.L., de Kock, H.A., Verschueren, W.G., Pille, G.M., Maes, L.J., Peeters, A., Vendeville, S., De, M.S., Azijn, H., Pauwels, R., et al. Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J. Med. Chem. 2005;48:1965–1973. doi: 10.1021/jm049454n. [DOI] [PubMed] [Google Scholar]
- Václavíková, J., Machala, L., Staňková, M., Linka, M., Brůčková, M., Vandasová, J., Konvalinka, J. Response of HIV positive patients to the long-term salvage therapy by lopinavir/ritonavir. J. Clin. Virol. 2005;33:319–323. doi: 10.1016/j.jcv.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Weber, J., Mesters, J.R., Lepšík, M., Prejdová, J., Švec, M., Šponarová, J., Mlčochová, P., Skalická, K., Stříšovský, K., Uhlíková, T., et al. Unusual binding mode of an HIV-1 protease inhibitor explains its potency against multi-drug-resistant virus strains. J. Mol. Biol. 2002;324:739–754. doi: 10.1016/s0022-2836(02)01139-7. [DOI] [PubMed] [Google Scholar]
- Williams, J.W., Morrison, J.F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- Winn, M.D., Murshudov, M.N., Papiz, M.Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Yin, P.D., Das, D., Mitsuya, H. Overcoming HIV drug resistance through rational drug design based on molecular, biochemical, and structural profiles of HIV resistance. Cell. Mol. Life Sci. 2006;63:1706–1724. doi: 10.1007/s00018-006-6009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.W., Zhang, J.Z.H. Full quantum mechanical study of binding of HIV-1 protease drugs. Int. J. Quantum Chem. 2005;103:246–257. [Google Scholar]