

Abstract
A null mutation was prepared in the mouse for CD18, the β2 subunit of leukocyte integrins. Homozygous CD18 null mice develop chronic dermatitis with extensive facial and submandibular erosions. The phenotype includes elevated neutrophil counts, increased immunoglobulin levels, lymphadenopathy, splenomegaly, and abundant plasma cells in skin, lymph nodes, gut, and kidney. Very few neutrophils were found in spontaneously occurring skin lesions or with an induced toxic dermatitis. Intravital microscopy in CD18 null mice revealed a lack of firm neutrophil attachment to venules in the cremaster muscle in response to N-formyl- methionyl-leucyl-phenylalanine. A severe defect in T cell proliferation was found in the CD18 null mice when T cell receptors were stimulated either by staphylococcal enterotoxin A or by major histocompatibility complex alloantigens demonstrating a greater role of CD11/CD18 integrins in T cell responses than previously documented. The null mice are useful for delineating the functions of CD18 in vivo.
Keywords: infection; integrin β2; leukocyte-adhesion deficiency syndrome; receptors, antigen, T cell; leukocytes
Leukocyte integrins play a major role in cell–cell and cell–matrix interactions (1, 2). The β2 integrins include LFA-1, macrophage antigen-1 (Mac-1),1 p150,95, and possibly a fourth form (3); each functions as heterodimeric molecules, with a separate α chain (CD11a, b, c, or d) and a common β chain (CD18). These leukocyte receptors are of importance in adhesion and signaling in a variety of physiological and pathological conditions.
Substantial information is available regarding the complex cellular function of CD18 based on studies using blocking antibodies and on analysis of genetic deficiencies in humans and in cattle (4–7). The human disorder is known as leukocyte adhesion deficiency type I (LAD I), and is caused by heterogeneous mutations in CD18. CD18-deficient patients suffer from recurrent microbial infections, leukocytosis, impaired wound healing, and lack of pus formation. A hypomorphic mutation in CD18 was generated in mice earlier by gene targeting, and homozygotes displayed 2–16% of normal levels of CD18 on leukocytes (8). This hypomorphic CD18-deficient mouse does not show spontaneous infections as occur in LAD I patients; however, when this mutation was backcrossed onto PL/J inbred strain, homozygous mice developed a dermatitis resembling human psoriasis (9). Although the hypomorphic mutation is valuable for assessing the effect of reduced expression of β2 integrins in chronic inflammatory processes, it is less valuable for functional evaluation of the role of CD18, since animals are not completely deficient for CD18. Human CD18 deficiency includes a range of severity that overlaps the phenotypes seen with the mouse hypomorphic and null mutations, with many severely affected and some mildly affected patients (7).
We report here the characterization of mice completely deficient in CD18 that were generated by homologous recombination in embryonic stem (ES) cells. In contrast to CD18 hypomorphic mutant mice (8), CD11a-deficient mice (10), and CD11b-deficient mice (11, 12), the CD18 null mice reveal a phenotype closely resembling LAD I patients, including spontaneous occurrence of mucocutaneous infections. These CD18 null mice provide the first murine model of severe human LAD I and reveal a prominent requirement for CD18 integrins in T cell activation by staphylococcal enterotoxin A or by MHC alloantigens.
Materials and Methods
Targeting Construct and Generation of Mutant Mice.
We have used a previously published (8) targeting construct with a ∼10-kb segment of the CD18 gene containing exon 2 and 3 and a neomycin-resistance gene (neo) cassette with a short version of the RNA polymerase II promoter and the bovine growth hormone polyadenylation signal; the neomycin cassette thus, disrupts the splice-acceptor site of exon 3 (8). The AB1 ES cell line was electroporated (13) with the construct after digestion with KpnI for use as a replacement vector. Digestion with HindIII was used to identify homologous recombinants on Southern blots hybridized with the 3′ flanking probe as indicated (Fig. 1 A). The mutation resulted in a ∼6.5-kb HindIII fragment compared with a 5.0-kb HindIII fragment for the wild-type genomic DNA. Embryonic stem cells confirmed by Southern blotting to carry the replacement mutation were used for germline transmission which was confirmed by Southern blot analysis of tail DNA using HindIII digestion and the 3′ flanking probe. Progeny containing the mutant CD18 allele were intercrossed resulting in CD18 homozygous mutant pups. The results presented are from mice of mixed 129/Sv and C57BL/6J background. For all experiments, homozygous progeny and wild-type littermates from heterozygous intercrosses were used. Sentinel mice housed in the same rooms at the barrier facility as the CD18 null mice repeatedly tested negative for common viruses.
Figure 1.

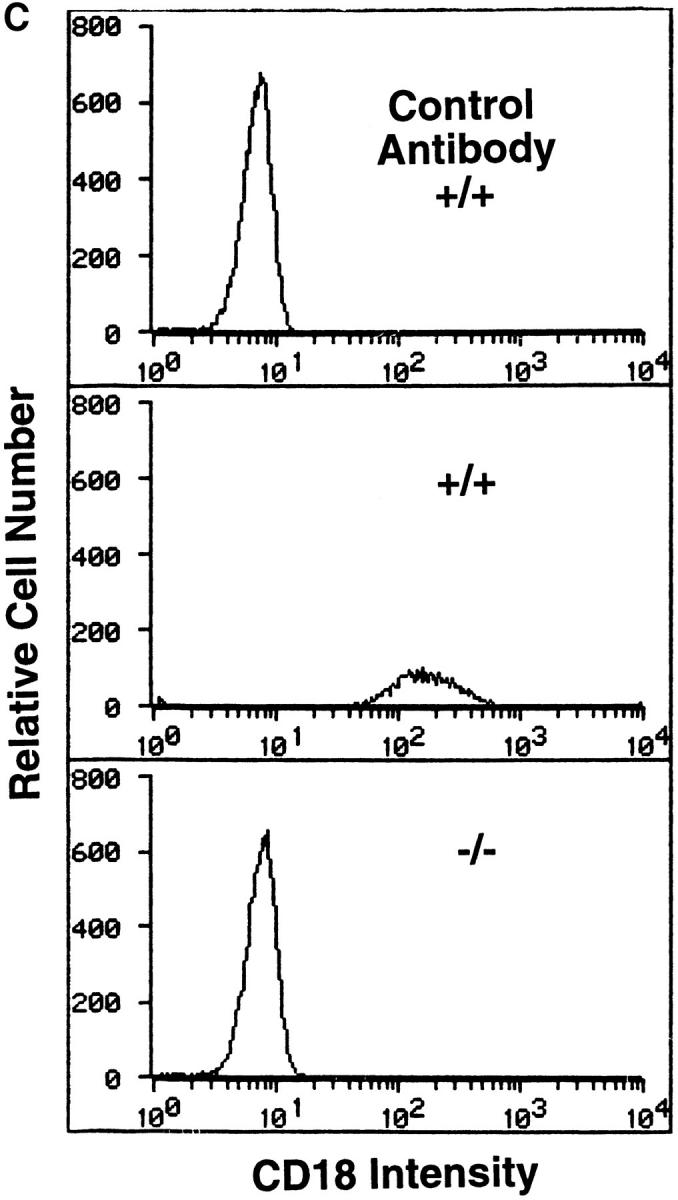

Preparation and analysis of the CD18 null mutation. A shows the genomic region to be targeted above with the targeted mutation below. DNA length is drawn to scale and the first three exons are indicated as solid rectangles. The location of the 3′ and 5′ flanking probes are shown as hatched boxes. Restriction enzyme sites are indicated as follows: E, EcoRI; H, HindIII; K, KpnI; and P, Pst. The targeting vector was linearized with KpnI, and a replacement mutation was obtained with insertion of a neomycin cassette at a PstI site that occurs precisely at the boundary of the intron and the 5′ end of exon 3. B shows a Southern blot with the 3′ flanking probe and genomic DNA isolated from the tails of wild-type (+/+), homozygous CD18 null (−/−), and heterozygous (+/−) mice. C presents the analysis of expression of CD18 on the surface of leukocytes from wild-type and homozygous CD18 null mice. FACS® analysis was performed as described in Materials and Methods. An isotype-matched control antibody was used (top). Expression of CD18 was quantitated by immunostaining with FITC-conjugated mAb C71/16.
Histopathology.
A total of 33 animals were examined representing three age groups, both sexes, and both homozygous and wild-type mice. Affected and control mice were killed for histological analysis and 32 different tissues were fixed in 10% (vol/ vol) neutral buffered formalin supplemented with zinc chloride. Paraffin-embedded sections were stained with hematoxylin-eosin.
Peripheral White Blood Cell Counts.
Mice were bled from the retroorbital sinus into EDTA-anticoagulated tubes while under anesthesia. Total white blood cell counts and differential leukocyte counts were determined using a Technicon H.1 analyzer (Miles Diagnostics, Elkhart, IN) calibrated for analysis of murine samples. Blood smears were prepared with Wright's stain.
Microbiological Analyses.
For bacterial culture, sterile swabs were moistened in phosphate-buffered saline containing 0.1% Triton X-100 and used to scrub the area for inoculation onto Columbia sheep blood agar plates. Swabs were also inoculated onto Mycosel agar and incubated at 25°C for 1 mo for the culture of fungi. Swabs also were inoculated on Löwenstein Jensen's agar and incubated at 30°C for 2 mo for isolation of mycobacteria.
Susceptibility to Streptoccus pneumoniae Infection.
Wild-type and CD18 null mice aged 8 wk were inoculated intraperitoneally with a single 0.1-ml dose (6–8 × 106 CFU/ml) of S. pneumoniae as described in detail elsewhere (14). Blood (10 μl) was obtained by nicking the tail vein using sterile technique for culture by inoculation on blood agar incubated at 35°C. The degree of bacteremia was characterized by the number of CFU on the blood culture plate. Statistical analyses were performed using Kaplan Meier and log rank analysis for bacterial load, presence or absence of bacteremia, and mortality.
Cell Staining and Flow Cytometry.
Peripheral blood was collected from homozygous mutants or littermate controls from the retroorbital sinus with EDTA anticoagulation. Expression of CD18 was determined by immunostaining with a fluorescein isothiocyanate-conjugated anti-CD18 mAb (C71/16; PharMingen, San Diego, CA). Also, 4 × 105 fresh splenocytes or thymocytes were stained with 1 μg of FITC-conjugated and/ or PE-conjugated CD3, CD4, and CD8-specific or isotype control antibodies for 20–30 min on ice and fixed with 1% paraformaldehyde. Cell surface phenotype was determined by immunofluorescent analysis with an EPICS Profile flow cytometer (Coulter Immunology, Hialeah, FL).
Toxic Dermatitis.
Six sex-matched mutant and wild-type animals were used at 7– 9 wk of age. The irritant response was initiated by applying 10 μl of 2% (vol/vol) dinitrofluorobenzene (DNFB; Sigma Chemical Co., St. Louis, MO) in olive oil to the dorsal and ventral aspects of the right ear. The resulting inflammatory response was monitored by microscopic examination at 5 h after DNFB application.
Intravital Microscopy.
Three CD18 null mice and three wild-type littermate controls were anesthetized with an i.p. injection of ketamine hydrochloride (100 mg/kg, Ketalar; Parke-Davis, Morris Plains, NY) after premedication with sodium pentobarbital (30 mg/kg, Nembutal; Abbott Laboratories, Chicago, IL,) and atropine (0.1 mg/kg, Elkins-Sinu, Cherry Hill, NJ) i.p. The trachea, left jugular vein, and left carotid artery were cannulated to facilitate respiration and allow injection of anesthetic, measurement of blood pressure, and blood sampling. Blood pressure was monitored continuously (model BPMT-2; Stemtech, Inc., South Natick, MA) and carotid blood samples were drawn at the beginning and end of the observation period. Mice were thermocontrolled using a small animal heating pad (model 50-7503; Harvard Apparatus, Inc., South Natick, MA) and received anesthetic as required to maintain blood pressure in the range 60–100 mm of Hg.
The cremaster muscle was prepared for intravital microscopy as described (15). During surgery (typically 10 min) and data collection, the cremaster was superfused with thermocontrolled (37°C) bicarbonate-buffer (132 mM NaCl, 4.7 mM KCl, 2 mM CaCl2, 1.2 mM MgCl2, and 18 mM NaHCO3 equilibrated with 5% CO2 in N2 to adjust pH to 7.35).
Microscopic observations were made up to 1 h after exteriorization of the cremaster using a (Carl Zeiss Inc., Thornwood, NY) intravital microscope with a saline immersion objective (SW40.0.75 numerical aperture). Venules with diameters between 20 and 40 μm were observed and recorded through a CCD camera system (model VE-1000CD; Dage-MTI, Inc., Michigan City, IN).
Glass micropipettes were drawn from standard borosilicate glass (outer diameter 1.0 mm; Stoelting, Wood Dale, IL) on a vertical micropipette puller (Stoelting) and beveled to a tip diameter of 7–10 μm using a micropipette grinder (model EG-40; Narishige, Sea Cliff, NY) with 0.3 μm abrasive foil (no. 6775; A.H. Thomas, Philadelphia, PA). The pipettes were filled with ∼15 μl of filtered (0.45 μm, Millex HV4; Millipore Corp., Bedford, MA) 10 μM fMLP (Sigma Chemical Co.) solution. An air-tubing and syringe system attached to the pipette holder served as a pressure reservoir controlled by a 10-ml syringe and allowed continuous, stable microinfusions. Venules were selected for injection on the basis of size (20–40 μm) and presence of baseline rolling with little or no firm adhesion of cells; these venules were recorded for 60 s before placement of the pipette. A pipette was placed in proximity (within 40 μm) to the observed vessel using a piezo-driven micromanipulator (model DC-3K; Märzhäuser-Wetzlar, Upper Saddle River, NJ) with the beveled tip penetrating the interstitial tissue. The vessel was recorded for 60 s in the presence of the pipette tip without infusion. No difference was seen between the number of firmly adhered cells before and after pipette placement (data not shown). Microinfusion of 10 μM fMLP which caused a slight swelling of the impaled interstitial tissue, was initiated by application of pressure to the air-tubing system and was maintained for 60 s. The vessel was recorded for a further 60 s after termination of microinfusion.
The number of firmly adhered leukocytes in the observed vessel was counted off-line at four time points; (a) after 60 s recording, (b) after 60 s + pipette, (c) after 60 s fMLP infusion, and (d) 60 s after termination of fMLP. Length and diameter of the observed vessel was measured using a commercial digital image processing system (Macintosh 7100 computer video capture board Scion Corporation, Frederick, MD). Results are presented as number of firmly adhered cells/100 μm of vessel length. Rolling leukocyte fluxes were also determined for the four observation periods by counting the number of leukocytes distinguishable from the blood stream passing a line perpendicular to the vessel axis. For this purpose, the video tapes were replayed at one half to one-fifth original speed. Counting visibly distinguishable cells is widely used for analysis of leukocyte rolling and is known to be accurate at blood flow velocities above 0.5 mm/s (16). Vessels with flow velocities below 0.5 mm/s were not studied as it is difficult to distinguish rolling cells from free flowing cells at lower flow rates. Total leukocyte flux through a given venule was estimated as a product of systemic leukocyte count and volumetric flow rate of blood through the venule (calculated using microvessel diameter and mean blood flow velocity assuming cylindrical geometry). Leukocyte rolling flux fraction is defined as the flux of rolling leukocytes as a percentage of total leukocyte flux and is thus independent of variation in systemic leukocyte counts.
In Vitro Adherence Assay for Neutrophils.
Neutrophils from mutant mice and wild-type littermates were isolated from bone marrow as described (12). A visual adherence assay was used for quantitating adhesion of murine neutrophils to KLH-coated glass, a surface to which neutrophils adhered only after stimulation (17). Briefly, neutrophils were preincubated at 37°C for 10 min and exposed to zymosan activated serum (ZAS) for an additional 10 min in the presence or absence of a saturating level of antibody (5 μg/ml) before injection into the adherence chambers. Cells were allowed to settle for 500 s and inverted for 500 s; the percent adherence was determined by counting 5–10 high power fields per experimental condition.
Assays of Luminal-enhanced Chemiluminescence.
The evolution of light from neutrophils undergoing iC3b-mediated phagocytosis after activation by opsonized zymosan particles was quantitated as described previously (18). Zymosan particles (ICN Pharmaceuticals, Irvine, CA) were opsonized in freshly obtained serum from healthy mice for 45 min at 37°C. Opsonized zymosan particles were then washed three times and resuspended in PBS to a final concentration of 0.5 mg/ml. Phagocytotic reaction mixtures contained 106 neutrophils with 10−8 M luminal. Chemiluminescence was quantitated after the addition of PBS or 0.5 μg/ml of opsonized zymosan particles to reaction mixture.
T Cell Proliferation Upon Stimulation with SEA or Mixed Lymphocyte Reaction.
Splenocytes were treated with low ionic strength media and Tris ammonium chloride to remove dead cells and red blood cells, respectively (19). 4 × 105 cells/well were plated in 96-well round bottom microtiter plates in a total volume of 200 μl in supplemented Dulbecco's Modified Eagle's medium (sDME) with 10% FCS (19). The cells were cultured alone or with SEA at 0.1 ng/ml to 20 μg/ml (Toxin Technology, Sarasota, FL) for 96 h and labeled with [3H]thymidine (DuPont-NEN, Boston, MA) for the last 5–6 h. They were processed for liquid scintillation counting (Betaplate, Wallace, Gaithersburg, MD) and the resulting data expressed as mean cpm ± SEM of triplicate cultures.
CD18 null and wild-type splenocytes (4 × 105 cells/well) were also stimulated in the MLR by increasing concentrations of allogeneic BALB/c (H-2d) or syngeneic CD18 null or wild-type control (H-2b) spleen cells that had been irradiated to 1,500 rads. MLR were labeled and harvested as above after 96 h culture in 10% sDME with 10 μM 2-mercaptoethanol (Sigma Chemical Co.), in round bottom microtiter plates. CD18 null and wild-type splenocytes were also stimulated with ionomycin (200 ng/ml) and phorbol 12-myristate 13-acetate (1 ng/ml; Sigma Chemical Co.), and were labeled and harvested at 24-h intervals through 120 h of culture.
Determination of Immunoglobulin and Cytokine Levels.
Serum immunoglobulin isotypes were determined by sandwich ELISA as previously described (20). Bioassays for IL-3, GM-CSF, and IL-6 were performed as described previously (21, 22).
Results
Generation of a CD18 Null Mutation in Mice.
To introduce a null mutation into the CD18 locus, a previously reported targeting construct (8) that was used to introduce a duplication resulting in a hypomorphic allele was now used to produce a replacement mutation (Fig. 1 A). The construct contains the neomycin-resistance cassette disrupting the 5′ boundary of exon 3, thus preventing any synthesis of normal CD18 protein. Using Southern analysis, recombinant colonies were found with a frequency of 1 in 125 among the G418-resistant clones. The mutation was transmitted to the germline, and heterozygous mutant mice were intercrossed. Homozygous mutant (hereafter referred to as CD18 null) mice were detected by Southern blotting (Fig. 1 B) at 3 wk of age at a lower than expected frequency ranging from 12 to 18% of 102 progeny tested over different intervals. However, when performing Southern blot analysis of 18-d-old embryos, an expected frequency of 25% was obtained, suggesting that some CD18 null offspring die perinatally. As a quantitative assessment of cell surface expression of CD18, FACS® analysis of leukocytes was performed (Fig. 1 C). Surface expression of CD18 was found on leukocytes from wild-type animals, but there was no detectable expression of CD18 on leukocytes from CD18 null mice.
The function of CD18 was assessed by measuring the ability of neutrophils to attach to KLH-coated glass and to generate an oxidative burst after exposure to chemoattractants; this method reveals a severe deficit in CD18-deficient human cells (17). There was almost no adhesion of neutrophils from wild-type or CD18 null mice without stimulation, but exposure of neutrophils to zymosan activated serum, which is rich in C5a, resulted in a substantial increase in adhesion of neutrophils derived from wild-type mice (Fig. 2 A). By contrast, no significant neutrophil adhesion was observed with cells from CD18 null mice. Neutrophil adhesion of wild-type mice was substantially abrogated by blocking antibodies against CD11b, whereas blocking antibodies against CD11a and an isotype-matched rat IgG did not affect neutrophil binding to KLH (Fig. 2 A). Similar results were obtained for phagocytosis associated oxidative burst that earlier had been shown to be Mac-1 (CD11b/ CD18) dependent (23). The evolution of light by neutrophils in association with iC3b mediated phagocytosis was quantitated by a luminal-enhanced chemiluminescence assay (23). Neutrophils isolated from wild-type mice exhibited substantial chemiluminescence when mixed with opsonized zymosan particles, whereas neutrophils from CD18 null mice did not exhibit significant activity (Fig. 2 B). The absence of surface expression of CD18, the lack of neutrophil adhesion, and the deficiency of oxidative burst are consistent with the complete disruption of the CD18 molecule.
Figure 2.


Analysis of neutrophil adherence and respiratory burst in wild-type and CD18 null mice. In A, adherence of murine neutrophils to a KLH-coated glass surface was measured as described in Materials and Methods. Isolated neutrophils were incubated with PBS or mAb for 30 min at room temperature: 10 μg/ml Ml/70 (anti-CD11b), 10 μg/ml KBA (anti-CD11a), or 10 μg/ml SFDR5 (Ig-matched control Ab). ZAS was added immediately before injecting the cell mixture into the adhesion chamber. Data are presented as mean ± SEM (n = 3); P <0.01 for wild-type (solid bars) versus CD18 null (open bars) in absence of antibodies. B depicts chemiluminescence of neutrophils from wild-type and CD18 null mice. Isolated neutrophils were mixed with 10−8 M luminal before the addition of PBS or 0.5 μg/ml of freshly opsonized zymosan particles (OpZ ). Result represents mean ± SEM (n = 3); P <0.01 for wild-type versus CD18 null with OpZ.
The Phenotype of Homozygous CD18 Null Mice Resembles Human LAD I.
At various times in managing the mouse colony, 10–40% of the newborn CD18 null animals died perinatally. The surviving CD18 mutant mice developed progressive perioral soft tissue swelling, facial alopecia, reddening of the skin, and extended facial and submandibular ulcerative dermatitis at ∼3 mo of age (Fig. 3, A and B). This phenotype was observed in 95% (48 of 51) of mice on a mixed 129/Sv and C57BL/6J background.
Figure 3.




Gross morphology and histology of CD18 null mice. A demonstrates alopecia in the neck and upper thorax with severe ulcerative dermatitis and perioral soft tissue swelling. B shows extended facial alopecia and erosions with conjunctivitis of the eye. C demonstrates ulcerative dermatitis with full thickness necrosis of the epidermis covered by an eosinophilic crust with entrapped inflammatory cells and bacterial colonies (arrows). There is adjacent epidermal hyperplasia. The dermal inflammatory infiltrate consists of lymphocytes, histiocytes, mast cells, occasional plasma cells, and few neutrophils. E, epidermis; D, dermis; and U, ulceration. Original magnification 250×. The spleen is shown in D with hyperplasia of the red pulp, multiple plasma cells (*), and occasional Russell bodies (arrows), the latter indicative of abnormal deposition of immunoglobulins within plasma cells. Original magnification 400×.
Histopathology of the skin revealed superficial erosions and ulcerations covered by an eosinophilic crust with entrapped necrotic inflammatory cells and bacterial colonies (Fig. 3 C). The dermis beneath and adjacent to the erosions revealed a predominantly mononuclear inflammatory cell infiltrate consisting of lymphocytes, histiocytes, and occasional perivascular plasma cells with far fewer neutrophils than would be expected in a wild-type animal at a site of bacterial infection, suggesting that neutrophil extravasation into the interstitial tissue is severely impaired (Fig. 3 C). Affected mice also displayed splenomegaly with a 2.5-fold increase in weight (data not shown) due to a myeloid hyperplasia within the red pulp; similar hyperplasia was present in the bone marrow of CD18 null mice. Occasional inflammatory lesions were noted in mutant mice in heart, salivary gland, liver, and intestine. No consistent pattern was noted, and these lesions were usually associated with concurrent cutaneous disease. In addition, a reactive lymphadenopathy predominantly involving nodes that drain the anatomical sites of skin erosions was observed. The major histological abnormality in these nodes was an accumulation of plasma cells, some revealing periodic acid-Schiff (PAS)-positive inclusions (Russell bodies) indicative of increased immunoglobulin deposition. At 2.5–3 mo of age, serum total IgG, IgG1, IgG2a, and IgG2b were increased 10–15-fold in CD18 null mice compared with wild-type littermates (Table 1). CD18 null mice showed significant but more modest increases in IgG3, IgM, and IgA. Plasma cell infiltration with Russell body formation was also observed in spleen (Fig. 3 D), kidney, small intestine, iliac lymph nodes, and the dermis and subcutaneous tissue of the skin from the dorsum of the head, shoulders, and the dorsum of the neck. Numbers of circulating leukocytes in CD18 null mice were increased up to sixfold compared with littermates (Table 2).
Table 1.
Ig Concentrations in the Sera of Wild-type and CD18 Null Mice
| Ig Class | Mean ± SD | |||
|---|---|---|---|---|
| Wild-type (n = 6) | CD18 null (n = 6) | |||
| μg/ml | ||||
| IgG total | 705 ± 312 | 18,162 ± 9,049 | ||
| IgG1 | 243 ± 164 | 4,568 ± 2,769 | ||
| IgG2a | 486 ± 440 | 5,322 ± 6,293 | ||
| IgG2b | 81 ± 42 | 1,380 ± 834 | ||
| IgG3 | 234 ± 45 | 397 ± 104 | ||
| IgM | 116 ± 6 | 242 ± 66 | ||
| IgA | 1,463 ± 149 | 2,310 ± 448 | ||
| IgE | 162 ± 197 | 335 ± 223 | ||
Table 2.
Leukocyte Counts in CD18 Null and Wild-type Mice
| Cells | Mean ± SD | |||
|---|---|---|---|---|
| Wild-type (n = 6) | CD18 null (n = 4) | |||
| × 103/μl | ||||
| Leukocytes | 7.9 ± 3.1 | 43.9 ± 8.2 | ||
| Neutrophils | 2.2 ± 1.5 | 24.0 ± 10.4 | ||
| Lymphocytes | 5.2 ± 3.1 | 18.0 ± 2.8 | ||
| Monocytes | 0.23 ± 0.08 | 0.87 ± 0.16 | ||
| Eosinophils | 0.14 ± 0.07 | 0.49 ± 0.17 | ||
| Basophils | 0.04 ± 0.02 | 0.15 ± 0.02 | ||
Increased circulating leukocyte counts were largely due to elevated numbers of circulating neutrophils, but lymphocyte and monocyte numbers were also increased. The neutrophils revealed hypersegmentation and appendix formation of the nuclei. Since IL-3 and GM-CSF have been shown to stimulate granulopoesis (21) and IL-6 to prevent apoptosis (24) and thus contribute to elevation of peripheral neutrophil counts, serum levels of these cytokines were studied by specific bioassays in mice >10 wk of age. Serum levels of IL-3 and IL-6 but not GM-CSF were ≥20-fold elevated in CD18 null mice; values for CD18 null versus wild-type mice, respectively, were as follows: IL-3 mean 279 pg/ml (range 60–550; n = 4) versus mean 52 pg/ml (range 30–160; n = 4) and IL-6 mean 715 pg/ml (range 562–1258, n = 6) versus mean 42 pg/ml (range <25–68, n = 6). Phenotyping of CD4, CD8, and CD3 was performed on T cells from spleen and thymus using FACS® analysis (not shown), and no significant differences were found between CD18 null and wild-type mice indicating that CD18 is not essential for T cell development.
Since LAD I patients have been reported to suffer from bacterial infection, and because of observed perioral and conjunctival lesions, oral and conjunctival cultures were obtained from 12 CD18 null and 4 wild-type mice. All 16 oral cultures reflected the coprophagic behavior of mice with various fecal contaminants. One of the four wild-type conjunctival cultures was positive for Staphylococcus xylosus, and the other three cultures were negative. All 12 conjunctival cultures from the CD18 null mice were positive with one to three organisms of commensal or fecal origin including Escherichia coli, Lactobacillus species, Enterococcus faecalis, Proteus mirabilis, Streptococcus, and Staphylococcus epidermidis. The number of organisms increased with the severity of conjunctivitis noted clinically. None of the fungal cultures from six CD18 null and six wild-type mice showed any growth. Conventional hematoxylin-eosin and Gram staining occasionally detected bacterial colonies on the crust of erosions, but bacteria were not detected invading the tissue beneath. Acid fast, PAS, and Grocott staining of skin sections failed to detect organisms below the surface.
Increased Mortality in CD18-deficient Mice Inoculated with S. pneumoniae.
Wild-type mice and CD18 null mice aged 8 wk were inoculated intraperitoneally with 6–8 × 106 CFU of S. pneumoniae. There was a 53% survival by day 10 in wild-type mice compared with no survival of CD18 mice (P = 0.003; Fig. 4). In wild-type mice, survivors cleared bacteremia so that all animals were either dead or free of bacteremia by day 10. In contrast, almost all blood cultures were positive in CD18 null mice until death. CD18 null mice consistently had a higher mean number of bacterial colony forming units/10 μl in their blood cultures at various times after inoculation with S. pneumoniae (167 ± 145 at 24 h, 181 ± 139 at 48 h, 177 ± 126 at 72 h, 159 ± 137 at 96 h, 157 ± 142 at 120 h, 52 ± 84 at 144 h, and 35 ± 99 at 192 h) compared with wild-type mice (134 ± 133 at 24 h, 58 ± 99 at 48 h, 61 ± 118 at 72 h, 24 ± 68 at 96 h, 2.3 ± 8.2 at 120 h, 9.0 ± 30 at 144 h, and 0.0 ± 192 h; mean ± SD); P value by log rank analysis <0.0001. The baseline leukocyte and neutrophil counts in the blood of CD18 null mice were approximately four times those of wild-type in these studies (compare to Table 1). In both groups, the leukocyte counts dropped substantially in the first 24 h after S. pneumoniae inoculation (54% decrease in controls, 66% decrease in CD18 mice) and then increased gradually. The neutrophil counts in the peritoneal fluid 48 h after intraperitoneal inoculation of S. pneumoniae, though not significant, were slightly increased in CD18 null mice compared with wild-type indicating that significant numbers of neutrophils can emigrate in the complete absence of CD18. Thus, CD18 null mice demonstrated a marked increase in susceptibility to death from bacteremia after inoculation with S. pneumoniae.
Figure 4.

Increased bacteremia and mortality in CD18 null mice inoculated with S. pneumoniae. Mice were injected intraperitoneally with S. pneumoniae as described in Materials and Methods. Significant increased mortality (P = 0.0003; solid bars) and increased bacteremia (hatched bars) in surviving mice (P = <0.0001) were found for CD18 null mice.
Neutrophil Emigration Is impaired in Toxic Dermatitis.
As reported earlier for bovine and human LAD (25–27), we detected extremely low numbers of neutrophils in the interstitial connective tissue beneath the spontaneously occurring skin erosions in CD18 null mice (Fig. 3 C). To study neutrophil extravasation in a more controlled fashion, we induced a toxic dermatitis by applying 2% DNFB onto the ear of mice and studied neutrophil extravasation at 5 h after application of the irritant. A severe toxic response was observed both in CD18 null mice and in littermate controls with spongiosis of the epidermis and edema formation within the dermis. However, although many neutrophils were detected in the interstitial connective tissue in wild-type mice (Fig. 5 A), virtually no neutrophils were seen within the dermis of CD18 null mice (Fig. 5 B). In CD18 null mice, neutrophils were seen within the lumen of dermal vessels.
Figure 5.
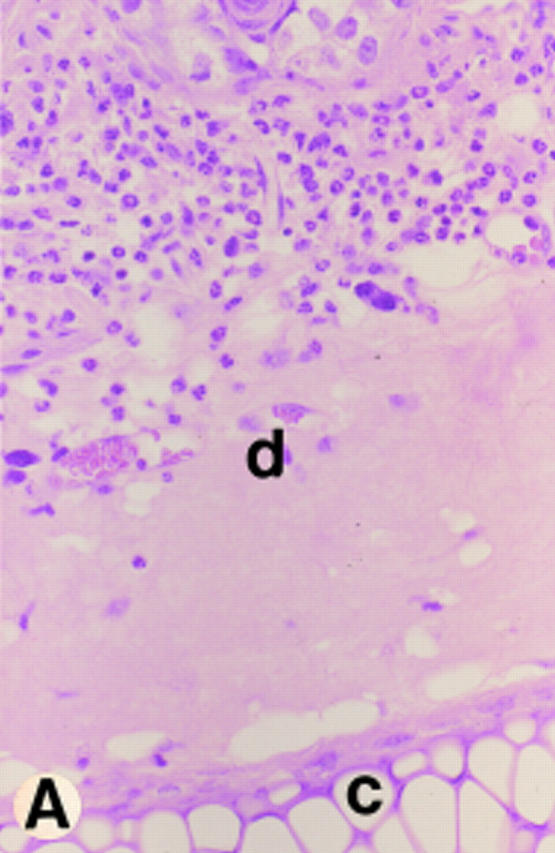

Neutrophil response to toxic dermatitis. Ears were challenged with topical application of 10 μl 2% DNFB 5 h before death. The dense neutrophil infiltrate in a wild-type mouse is shown in A and the lack of neutrophil response despite the presence of edematous interstitial tissue is shown in a CD18 null mouse in B. Hematoxylin and eosin staining at an original magnification of 400× is shown. Dermis (d) and cartilage (c) are labeled.
Intravital Microscopy Reveals Reduction of Firm Adhesion in Venules.
To elucidate the mechanism underlying the lack of neutrophil extravasation, intravital microscopy was performed. Firm cell attachment to the endothelial lining of the studied vessels did not occur in wild-type or CD18 null mice without stimulation by N-formyl-methionyl-leucyl-phenylalanine (fMLP; Fig. 6, A and B). Microinjection of fMLP resulted in a large increase in firm cell adherence in wild-type mice (Fig. 6, C and E), but no increase was observed in CD18 null mice upon fMLP stimulation (Fig. 6, D and E). Spontaneous rolling of cells occurred both in wild-type and in CD18 null mice, though in CD18 null to a significantly greater extent, most likely reflecting the dramatic increase in the peripheral leukocyte numbers (Fig. 6 F). Although rolling was significantly reduced in wild-type mice upon fMLP stimulation reflecting induction of firm adhesion, no significant decrease in the number of rolling cells was detected in CD18 null mice upon fMLP stimulation. The intravital microscopy and analysis of toxic dermatitis indicate that CD18 is required for leukocyte adhesion and emigration in these settings.
Figure 6.
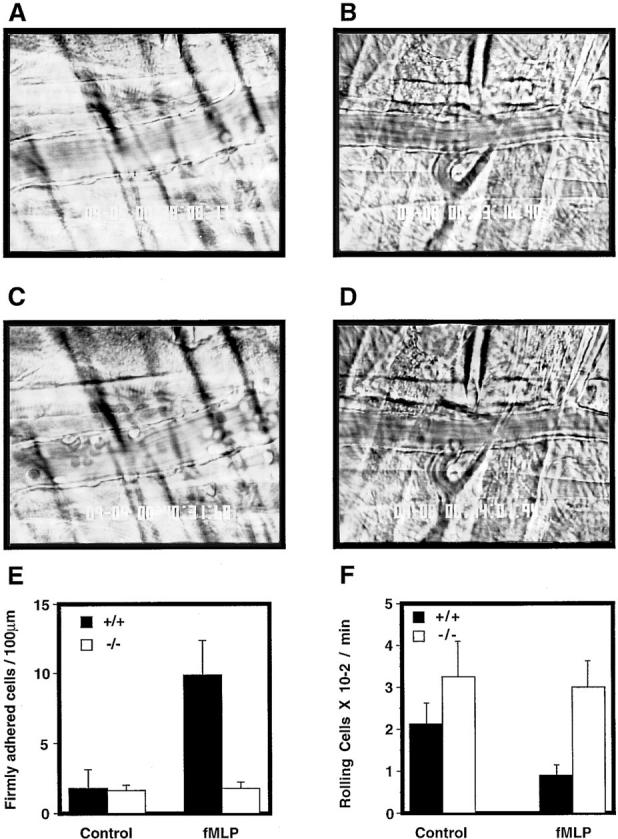
Firm attachment and rolling of leukocytes is altered in CD18 null mice. Intravital microscopy was performed as described in Materials and Methods. Venules with diameters between 20 and 40 μm were observed and recorded through a CCD camera. Almost no firm attachment of leukocyte occurred in a wild-type mouse before fMLP infusion (A). Infusion of fMLP resulted in a substantial increase in firmly attached leukocytes in a wild-type mouse (C ). No firm attachment of leukocytes to the endothelial lining was observed in CD18 null mice before or after fMLP stimulation (B and D, respectively). The number of firmly adherent leukocytes in the observed vessels was quantitated before fMLP infusion and 1 min after infusion (E). Results are presented as number of firmly adherent cells/100 μm of vessel length. Rolling leukocyte fluxes were determined before fMLP infusion and 1 min after infusion (F ). Results (mean ± SEM, n = 3) are expressed as the flux of rolling leukocytes per minute.
CD18 Plays a Major Role in TCR-dependent T Cell Proliferation.
We evaluated the importance of CD18 in T cell activation when the TCR was stimulated in the MLR or with staphylococcal enterotoxin A (SEA). Unfractionated spleen cells were irradiated and used as antigen-presenting stimulators, whereas T cells isolated from spleen were used as responder cells. Cells from CD18 null and wild-type mice were of mixed C57BL/6J and 129/Sv background (both H-2b), whereas cells from BALB/c mice express H-2d. T cells from wild-type littermates demonstrate a proliferative response to allogeneic stimulation with irradiated BALB/c cells; no proliferative response was observed with syngeneic stimulator cells (Fig. 7 A). There was no detectable proliferation of T cells derived from CD18 null mice with allogeneic or syngeneic stimulator cells (Fig. 7 B). The T cell response was evaluated further by quantitating the concentration-dependent proliferative response to SEA. When T cells from wild-type mice were exposed to SEA, the maximal [3H]thymidine incorporation ranged from 35,000 to 80,000 cpm at SEA concentrations between 1–10 ng/ml. However, even at an SEA concentration of 10 μg/ml, [3H]thymidine incorporation was below 10,000 cpm in T cells derived from CD18 null mice (Fig. 8 A). Stimulation of cells with phorbol myristate acetate and ionomycin demonstrated that cells from CD18 null mice were capable of proliferating normally, if the requirement for the T cell receptor was bypassed (Fig. 8 B). These data indicate that CD18 serves a critical role in generating a T cell proliferative response to MHC alloantigens or to SEA.
Figure 7.

Abrogation of mixed lymphocyte reaction in CD18 null mice. Splenocytes from three wild-type littermate controls (A) and from three CD18 null mice (B) were stimulated in a mixed lymphocyte reaction. Responder cells (4 × 105 cells per well) were mixed with increasing concentrations of allogeneic BALB/c (filled symbols) or with syngeneic wild-type or CD18 null spleen cells that had been irradiated (open symbols). Analysis of proliferation was performed after 96 h in culture as described in Materials and Methods. Data are presented as mean cpm ± SEM of triplicate cultures.
Figure 8.

Abrogation of T cell proliferation in response to staphylococcal enterotoxin A (SEA) in CD18 null mice. In A, splenocytes from three wild-type littermates (filled symbols) and from CD18 null mice (open symbols) were cultured without addition or with 0.1 ng to 20 μg/ml SEA. In B, splenocytes from wild-type (filled symbols) or CD18 null (open symbols) mice were stimulated with 100 ng/ml phorbol myristate acetate and 200 ng/ml ionomycin and analyzed for proliferation at the indicated times. Proliferation was quantitated as described in Materials and Methods and is expressed as mean cpm ± SEM of triplicate cultures for three mice of each genotype.
Discussion
The CD18 null mouse reported here displays a phenotype which is generally similar to that seen for human (4, 7) and bovine (5, 6) LAD I. The common features include leukocytosis, spontaneous skin infections, and impaired emigration of neutrophils at sites of infection. Impaired adherence of neutrophils and respiratory burst is similar in CD18 deficient mice and humans. Mutations in humans are heterogeneous, and hypomorphic mutations result in milder LAD I phenotypes. The hypomorphic mutation in the mouse is associated with 2–16% of normal levels of CD18 on leukocytes (8) and is analogous to mild or moderate forms of human LAD I. The hypomorphic mouse mutation may be more appropriate for chronic studies of the role of CD18 in inflammatory disease processes, while the null mutation reported here will be more valuable for determining whether CD18 is essential for a particular leukocyte function. Studies with mutant mice complement functional analyses with blocking antibodies most often confirming but occasionally questioning (28–31) the role of an adhesion molecule in specific leukocyte activities.
Mouse mutations in numerous leukocyte and endothelial cell adhesion molecules have been prepared including leukocyte integrins, immunoglobulin family members (ICAM-1, VCAM-1), selectins, and selectin ligands (32–35). Mutations in CD11a (10) and CD11b (11, 12) are of particular interest, since they would affect the subsets of CD18 functions attributable to LFA-1 and Mac-1, respectively. Mice with CD11a deficiency were found to have a moderate defect in peritoneal emigration at early times, reduced natural killer cytotoxicity, normal immunoglobulin levels, reduced responder cell function in MLR, normal CTL responses to virus, and impaired rejection of tumor cells (10). Mice deficient in CD11b show normal or increased peritoneal emigration, delayed apoptosis of extravasated leukocytes, impaired neutrophil degranulation and binding of neutrophils to fibrinogen, and reduced neutrophil phagocytosis and oxygen-free radical generation (11, 12).
Mice with deficiency of ICAM-1 or individual selectins are viable and do not develop spontaneous infections under laboratory conditions, but these mutants show varying abnormalities of leukocyte adhesion, migration, and rolling; see (32–35) for review and references. Mice with double deficiency of P-selectin and E-selectin do develop spontaneous cutaneous infections (36, 37), thus sharing some phenotypic features in common with LAD I. A rare human disorder affecting fucose metabolism causes leukocyte adhesion deficiency II (LAD II) apparently through an effect on carbohydrate structure of selectin ligand molecules (38).
The increased susceptibility to infection in CD18 null mice emphasizes the importance of granulocytes and macrophages as an antigen-independent first line of defense in response to pathogens or tissue injury. There is ample evidence that CD18 is important for the initial emigration of leukocytes, and it is not surprising that spontaneous cutaneous infections occur in the CD18 null mice, particularly given the previous information from humans and cattle. A systemic infectious challenge using intraperitoneal injection of S. pneumoniae demonstrated the increased infectious susceptibility of the CD18 null mice. These mice had essentially no ability to clear bacteremia and survive the infectious inoculation. Although mice with deficiency of ICAM-1 or individual selectins do not develop spontaneous infections as mentioned above, mice with individual deficiencies of E-selectin or P-selectin demonstrate increased morbidity and mortality using the S. pneumoniae inoculation protocol described here for the CD18 null mice (14). The increased mortality in CD18 null mice inoculated with S. pneumonia is not accompanied by a severe defect in neutrophil emigration, since approximately similar numbers of neutrophils were found in the peritoneal fluid compared with wild-type mice. This CD18-independent emigration is also seen in CD11b-deficient mice with a thioglycolate-induced peritonitis (11), and the data from CD18- and CD11b-deficient mice are consistent with the interpretation that neutrophils emigrating by CD18-independent pathways have different properties, do not function normally, and are not as effective in eliminating bacteria. Thus, although mice with individual deficiencies of leukocyte and endothelial cell adhesion molecules may not develop spontaneous infection under laboratory conditions, deficiency of these molecules probably causes more subtle susceptibility to infection in almost all cases. It is likely that susceptibility to infection provides a major pressure for evolutionary conservation of these molecules.
The complete deficiency of CD18 leads to severe abnormalities of leukocyte adhesion and activation. These in vitro abnormalities correlate with severe reductions in the ability of neutrophils to migrate in to sites of spontaneously occurring cutaneous infection or in response to toxic dermatitis. The deficiency of CD18 also leads to a substantial reduction of firm adherence as evaluated by intravital microscopy. However, the in vivo role of CD18 in leukocyte emigration is extremely complex with the clear existence of CD18-independent pathways. Using the CD18 null mice reported here, neutrophil emigration in response to irritant dermatitis due to croton oil was severely impaired, in agreement with our results with DNFB-induced dermatitis, but emigration was normal or enhanced during peritonitis induced by either thioglycolate or S. pneumoniae or with pneumonia induced by S. pneumoniae or E. coli (31). Although the normal or enhanced emigration of neutrophils in CD18 null mice is somewhat surprising, the data reported here and those of Mizgard et al. (31) are consistent with prominent dependence on CD18 for emigration into soft tissues, variable dependence on CD18 in peritonitis models, and lesser dependence on CD18 for pulmonary alveolar emigration. There is clear evidence for CD18- independent pathways of emigration, and perhaps these alternative mechanisms are more readily induced in animals with severe defects in the function of leukocyte and endothelial cell adhesion molecules as exemplified by the CD18 null mice and P-selectin/E-selectin double mutant mice (36, 37), both of which develop spontaneous skin disease.
The hypothesis has been forwarded that neutrophils initially infiltrating the inflammatory site, via the release of major T cell and macrophage chemoattractants, set the stage for the switch in the type of leukocyte infiltration typically occurring during the evolution of the inflammatory response from acute to chronic stage (39). The CD18 null mice demonstrate that the lack of initiating signals from egressed neutrophils does not prevent the development of the chronic mononuclear cell infiltrate in spontaneously occurring erosions and toxic dermatitis. However, cell populations of the chronic infiltrate are not able to prevent the ulcerative dermatitis.
A major finding resulting from the analysis of these mice was the recognition that CD18 deficiency causes a severe defect in T cell proliferation in response to stimulation of T cell receptors. Although CD11/CD18 integrins are accepted as playing a costimulatory role in the function of T cell receptors, their importance is often implied to be less than for CD28/B7. The defect in the CD18 null mice indicates an essential role for CD11/CD18 integrins in the response of T cell receptors under the conditions examined. CD18 null T cells fail to proliferate in response to the potent stimulus of SEA, which typically activates large subfamilies of T cells by interaction with common T cell receptor Vβ elements. Proliferative responses to allogeneic MHC determinants are similarly abolished. However, elevated serum Ig levels, particularly of certain IgG isotypes and to a lesser extent IgA, suggest that CD18 null T cell responses to at least some conventional peptide antigens occur and provide the cytokine and T cell membrane functions required to achieve B cell growth and isotype switching.
A significant role of the CD11a/CD18(LFA-1) receptor in SE response has been suggested previously. Antibodies blocking CD18, CD11a, or ICAM-1 each block SE-induced T cell growth and cytokine production (40, 41). Additionally, transfected or purified ICAM-1 restores SE-induced T cell growth under nonpermissive conditions. Other molecules that simply provide adhesion are ineffective, suggesting that distinct costimulatory functions are provided through LFA-1 binding (42, 43). Naive or resting, rather than memory T cells are particularly dependent upon LFA-1 costimulation (43). LFA-1 ligation causes increased expression of the well-recognized CD28/B7 costimulatory molecules on these cells (44) and may in part account for the role of CD28 in SE-induced proliferation (45–48). Thus the present work indicates that CD18+ receptors provide essential early signaling events in SE responses that are not bypassed by other costimulator systems of CD18 null T cells.
Similarly, antibody blocking and transfection studies have suggested a predominant role of the CD11a/CD18: ICAM-1 interaction in T cell responses to allogeneic MHC molecules (40, 44, 45, 49). Moreover, mice deficient in CD11a or ICAM-1 exhibit profound depression of MLR response when used as responder and stimulator populations respectively (10, 50). The present findings indicate that CD18 similarly plays a critical role in the capacity of T cells to respond to allogeneic MHC determinants. As in SE responses, naive alloreactive T cells appear to be critically dependent upon LFA-1 costimulation (44). In contrast, alloreactive memory T cells can exhibit apoptosis rather than growth upon coligation of LFA-1 and TCR (51). Thus, CD18 null T cells activated by bacterial or other antigens that bypass an initial role of CD18 may manifest dysregulated responses and thereby contribute to elevated cytokine and Ig production and chronic inflammation.
Acknowledgments
We thank Dr. Gregory Harriman for quantitating immunoglobulin levels, R. Geske for some histological studies, W. Schober for technical assistance, and G. Watson for preparation of the manuscript. We particularly thank J. Mizgerd and C. Doerschuk for sharing unpublished data and D. Bullard for helpful discussions.
This work was supported in part by grants from the National Institute of Health (AI 32177, AI 30036, and GM 15483) and K. Scharffetter-Kochanek was supported by a Heisenberg grant from the Deutsche Forschungsgemeinschaft.
Abbreviations used in this paper
- DNFB
dinitrofluorobenzene
- ES cell
embryonic stem cell
- ICAM-1
intercellular adhesion molecule-1
- LAD
leukocyte adhesion deficiency
- Mac-1
macrophage antigen-1
- OpZ
opsonized zymosan
- SE
staphylococcal enterotoxin
- ZAS
zymosan activated serum
Footnotes
Dr. Scharffetter-Kochanek's present address is Department of Dermatology, University of Cologne (Köln), Germany.
References
- 1.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 2.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Danilenko DM, Rossitto PV, Van der Vieren M, Trong HL, McDonough SP, Affolter VK, Moore PF. A novel canine leukointegrin, αdβ2, is expressed by specific macrophage subpopulations in tissue and a minor CD8+lymphocyte subpopulation in peripheral blood. J Immunol. 1995;155:35–44. [PubMed] [Google Scholar]
- 4.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 5.Arnaout MA. Leukocyte adhesion molecules deficiency: its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol Rev. 1990;114:145–181. doi: 10.1111/j.1600-065x.1990.tb00564.x. [DOI] [PubMed] [Google Scholar]
- 6.Shuster DE, Kehrli ME, Jr, Ackermann MR, Gilbert RO. Identification and prevalence of a genetic defect that causes leukocyte adhesion deficiency in Holstein cattle. Proc Natl Acad Sci USA. 1992;89:9225–9229. doi: 10.1073/pnas.89.19.9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson, D.C., T.K. Kishimoto, and C.W. Smith. 1995. Leukocyte adhesion deficiency and other disorders of leukocyte adherence and motility. In The Metabolic and Molecular Bases of Inherited Disease. C.R. Scriver, A.L. Beaudet, W.S. Sly, and D. Valle, editors. McGraw-Hill, Inc., New York. 3955–3994.
- 8.Wilson RW, Ballantyne CM, Smith CW, Montgomery C, Bradley A, O'Brien WE, Beaudet AL. Gene targeting yields a CD18-mutant mouse for study of inflammation. J Immunol. 1993;151:1571–1578. [PubMed] [Google Scholar]
- 9.Bullard DC, Scharffetter-Kochanek K, McArthur MJ, Chosay JG, McBride ME, Montgomery CA, Beaudet AL. A polygenic mouse model of psoriasiform skin disease in CD18 deficient mice. Proc Natl Acad Sci USA. 1996;93:2116–2121. doi: 10.1073/pnas.93.5.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJL, Duncan G, Wakeham A, Shahinian A, van der Heiden A, Bachmann MF, et al. LFA-1–deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. 1996;183:1415–1426. doi: 10.1084/jem.183.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, Von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Smith CW, Perrard J, Bullard DC, Tang L, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM. LFA-1 is sufficient in mediating neutrophil transmigration in Mac-1 deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 14.Munoz FM, Hawkins EP, Bullard DC, Beaudet AL, Kaplan SL. Host defense against systemic infection with S. pneumoniaeis impaired in E-, P-, and E-/P-selectin deficient mice. J Clin Invest. 1997;100:2099–2106. doi: 10.1172/JCI119744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunkel EJ, Ley K. Distinct phenotype of E-selectin-deficient mice. E-selectin is required for slow leukocyte rolling in vivo. Circ Res. 1996;79:1196–1204. doi: 10.1161/01.res.79.6.1196. [DOI] [PubMed] [Google Scholar]
- 16.Ley K, Gaehtgens P. Endothelial, not hemodynamic differences are responsible for preferential leukocyte rolling in venules. Circ Res. 1991;69:1034–1041. doi: 10.1161/01.res.69.4.1034. [DOI] [PubMed] [Google Scholar]
- 17.Shappell SB, Toman C, Anderson DC, Taylor AA, Entman ML, Smith CW. Mac-1 (CD11b/CD18) mediates adherence-dependent hydrogen peroxide production by human and canine neutrophils. J Immunol. 1990;144:2702–2711. [PubMed] [Google Scholar]
- 18.Anderson DC, Mace ML, Brinkley BR, Martin RR, Smith CW. Recurrent infection in glycogenesis type 1b: abnormal neutrophil motility related to impaired redistribution of adhesion sites. J Infect Dis. 1981;143:447–459. doi: 10.1093/infdis/143.3.447. [DOI] [PubMed] [Google Scholar]
- 19.Lee H-M, Rich S. Differential activation of CD8+ T cells by transforming growth factor-β11 . J Immunol. 1993;151:668–677. [PubMed] [Google Scholar]
- 20.Harriman GR, Kunimoto DY, Elliott JF, Paetkau V, Strober W. The role of IL-5 in IgA B cell differentiation. J Immunol. 1988;140:3033–3039. [PubMed] [Google Scholar]
- 21.Gallo RL, Grabbe S, Choi SS, Bleicher P, Granstein RD. Cyclosporin increases granulocyte/macrophage colony-stimulating factor (GM-CSF) activity and gene expression in murine keratinocytes. J Invest Dermatol. 1992;98:274–278. doi: 10.1111/1523-1747.ep12497858. [DOI] [PubMed] [Google Scholar]
- 22.Kirnbauer R, Köck A, Schwarz T, Urbanski A, Krutmann J, Borth W, Damm D, Shipley G, Anse JC, Luger TA. IFN-β, B cell differentiation factor 2, or hybridoma growth factor (IL-6) is expressed and released by human epidermal cells and epidermoid carcinoma cell lines. J Immunol. 1989;142:1922–1928. [PubMed] [Google Scholar]
- 23.Anderson DC, Miller LJ, Schmalstieg FC, Rothlein R, Springer TA. Contributions of the Mac-1 glycoprotein family to adherence-dependent granulocyte functions: structure-function assessments employing subunit-specific monoclonal antibodies. J Immunol. 1986;137:15–27. [PubMed] [Google Scholar]
- 24.Biffl WL, Moore EE, Moore FA, Barnett CCJ, Carl VS, Peterson VN. Interleukin-6 delays neutrophil apoptosis. Arch Surg. 1996;131:24–29. doi: 10.1001/archsurg.1996.01430130026005. [DOI] [PubMed] [Google Scholar]
- 25.Anderson DC, Schmalsteig FC, Finegold MJ, Hughes BJ, Rothlein R, Miller LJ, Kohl S, Tosi MF, Jacobs RL, Waldrop TC, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152:668–688. doi: 10.1093/infdis/152.4.668. [DOI] [PubMed] [Google Scholar]
- 26.Bowen TJ, Ochs HD, Altman LC, Price TH, Van Epps DE, Brautigan DL, Rosin RE, Perkins WD, Babior BM, Klebanoff SJ, Wedgwood RJ. Severe recurrent bacterial infections associated with defective adherence and chemotaxis in two patients with neutrophils deficient in a cell-associated glycoprotein. J Pediatr. 1982;101:932–940. doi: 10.1016/s0022-3476(82)80013-9. [DOI] [PubMed] [Google Scholar]
- 27.Weisman SJ, Berkow RL, Plautz G, Torres M, McGuire WA, Coates TD, Haak RA, Floyd A, Jersild R, Baehner RL. Glycoprotein-180 deficiency: genetics and abnormal neutrophil activation. Blood. 1985;65:696–704. [PubMed] [Google Scholar]
- 28.Kumasaka T, Quinlan WM, Doyle NA, Condon TP, Sligh J, Takei F, Beaudet AL, Bennett CF, Doerschuk CM. The role of ICAM-1 in endotoxin-induced pneumonia evaluated using ICAM-1 antisense oligonucleotides, anti–ICAM-1 monoclonal antibodies, and ICAM-1 mutant mice. J Clin Invest. 1996;97:2362–2369. doi: 10.1172/JCI118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doerschuk CM, Quinlan WM, Doyle NA, Bullard DC, Vestweber D, Jones ML, Takei F, Ward PA, Beaudet AL. The roles of P-selectin and ICAM-1 in acute lung injury as determined using anti-adhesion molecule antibodies and mutant mice. J Immunol. 1996;157:4609–4614. [PubMed] [Google Scholar]
- 30.Qin L, Quinlan WM, Doyle NA, Graham L, Sligh JE, Takei F, Beaudet AL, Doerschuk CM. The roles of CD11/CD18 and ICAM-1 in acute Pseudomonas aeruginosa-induced pneumonia in mice. J Immunol. 1996;157:5016–5021. [PubMed] [Google Scholar]
- 31.Mizgerd JP, Kubo H, Kutkoski GJ, Bhagwan SD, Scharffetter-Kochanek K, Beaudet AL, Doerschuk C. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J Exp Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bullard DC, Sandberg ET, Scharffetter-Kochanek K, Beaudet AL. Gene targeting for inflammatory cell adhesion molecules. Agents Actions. 1995;47:143–154. doi: 10.1007/978-3-0348-7343-7_13. [DOI] [PubMed] [Google Scholar]
- 33.Hynes RO, Wagner DD. Genetic manipulation of vascular adhesion molecules in mice. J Clin Invest. 1996;98:2193–2195. doi: 10.1172/JCI119027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frenette PS, Wagner DD. Insights into selectin function from knockout mice. Thromb Haemost. 1997;78:60–64. [PubMed] [Google Scholar]
- 35.Bullard, D.C., and A.L. Beaudet. 1996. Analysis of selectin deficient mice. In The Selectins: Initiators of Leukocyte Endothelial Adhesion. D. Vestweber, editor. Harwood Academic Publishers, Chur, Switzerland. 133–142.
- 36.Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 1996;84:563–574. doi: 10.1016/s0092-8674(00)81032-6. [DOI] [PubMed] [Google Scholar]
- 37.Bullard DC, Kunkel EJ, Kubo H, Hicks MJ, Lorenzo I, Doyle NA, Doerschuk CM, Ley K, Beaudet AL. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J Exp Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Etzioni A, Phillips LM, Paulson JC, Harlan JM. Leukocyte adhesion deficiency (LAD) II. CIBA Found Symp. 1995;189:51–58. doi: 10.1002/9780470514719.ch5. [DOI] [PubMed] [Google Scholar]
- 39.Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunol Today. 1995;16:21–26. doi: 10.1016/0167-5699(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 40.Damle NK, Klussman K, Leytze G, Linsley PS. Proliferation of human T lymphocytes induced with superantigens is not dependent on costimulation by the CD18 counter-receptor B7. J Immunol. 1993;150:726–735. [PubMed] [Google Scholar]
- 41.Nickoloff BJ, Mitra RS, Green J, Zheng X-G, Shimizu Y, Thompson C, Turka LA. Accessory cell function of keratinocytes for superantigens. Dependence on lymphocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. J Immunol. 1993;150:2148–2159. [PubMed] [Google Scholar]
- 42.Van Seventer GA, Newman W, Shimizu Y, Nutman TB, Tanaka Y, Horgan KJ, Gopal TV, Ennis E, O'Sullivan D, et al. Analysis of T cell stimulation by superantigen plus major histocompatibility complex class II molecules or by CD3 monoclonal antibody: costimulation by purified adhesion ligands VCAM-1, ICAM-1, but not ELAM-1. J Exp Med. 1991;174:901–913. doi: 10.1084/jem.174.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer H, Gjorloff A, Hedlund G, Hedman H, Lundgren E, Kalland T, Sjogren HO, Dohlsten M. Stimulation of human naive and memory T helper cells with bacterial superantigen. J Immunol. 1992;148:1993–1998. [PubMed] [Google Scholar]
- 44.Damle NK, Klussman K, Linsley PS, Aruffo A, Ledbetter JA. Differential regulatory effects of intercellular adhesion molecule-1 on costimulation by the CD28 counter-receptor B7. J Immunol. 1992;149:2541–2548. [PubMed] [Google Scholar]
- 45.Green JM, Zheng X-G, Shimizu Y, Thompson CB, Turka LA. T cell receptor stimulation, but not CD28 costimulation, is dependent on LFA-1-mediated events. Eur J Immunol. 1994;24:265–272. doi: 10.1002/eji.1830240141. [DOI] [PubMed] [Google Scholar]
- 46.Mittrücker HW, Shahinian A, Bouchard D, Kundig TM, Mak T-W. Induction of unresponsiveness and impaired T cell expansion by staphylococcal enterotoxin B in CD28-deficient mice. J Exp Med. 1996;183:2481–2488. doi: 10.1084/jem.183.6.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saha B, Harlan DM, Lee KP, June CH, Abe R. Protection against lethal toxic shock by targeted disruption of the CD28 gene. J Exp Med. 1996;183:2675–2680. doi: 10.1084/jem.183.6.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vella AT, Mitchell T, Groth B, Linsley PS, Green JM, Thompson CB, Kappler JW, Marrack P. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J Immunol. 1997;158:4714–4720. [PubMed] [Google Scholar]
- 49.Boussiotis VA, Freeman GJ, Gray G, Gribben J, Nadler LM. B7 but not intercellular adhesion molecule-1 costimulation prevents the induction of human alloantigen-specific tolerance. J Exp Med. 1993;178:1753–1763. doi: 10.1084/jem.178.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sligh JE, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in ICAM-1 deficient mice. Proc Natl Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Damle NK, Klussman K, Leytze G, Aruffo A, Linsley PS, Ledbetter JA. Costimulation with integrin ligands intercellular adhesion molecule-1 or vascular cell adhesion molecule-1 augments activation-induced death of antigen-specific CD4+T lymphocytes. J Immunol. 1993;151:2368–2379. [PubMed] [Google Scholar]