

Abstract
We have examined the role of the immunomodulatory cytokine transforming growth factor (TGF)-β in the resolution and pathology of malaria in BALB/c mice. Circulating levels of TGF-β, and production of bioactive TGF-β by splenocytes, were found to be low in lethal infections with Plasmodium berghei. In contrast, resolving infections with P. chabaudi chabaudi or P. yoelii were accompanied by significant TGF-β production. A causal association between the failure to produce TGF-β and the severity of malaria infection was demonstrated by treatment of infected mice with neutralizing antibody to TGF-β, which exacerbated the virulence of P. berghei and transformed a resolving P. chabaudi chabaudi infection into a lethal infection, but had little effect on the course of P. yoelii infection. Parasitemia increased more rapidly in anti–TGF-β–treated mice but this did not seem to be the explanation for the increased pathology of infection as peak parasitemias were unchanged. Treatment of P. berghei–infected mice with recombinant TGF-β (rTGF-β) slowed the rate of parasite proliferation and prolonged their survival from 15 to up to 35 d. rTGF-β treatment was accompanied by a significant decrease in serum tumor necrosis factor α and an increase in interleukin 10. Finally, we present evidence that differences in TGF-β responses in different malaria infections are due to intrinsic differences between species of malaria parasites in their ability to induce production of TGF-β. Thus, TGF-β seems to induce protective immune responses, leading to slower parasite growth, early in infection, and, subsequently, appears to downregulate pathogenic responses late in infection. This duality of effect makes TGF-β a prime candidate for a major immunomodulatory cytokine associated with successful control of malaria infection.
Keywords: transforming growth factor β, malaria, tumor necrosis factor α, interleukin 10, immunomodulation
Experimental infection of mice with various species and strains of rodent malaria parasites has facilitated dissection of the immunological events associated with both parasite clearance and the pathology of malaria infection. Resolution of a primary infection with the nonlethal parasites Plasmodium yoelii 17X, P. chabaudi chabaudi, and P. chabaudi adami is dependent on IFN-γ (1–3); TNF-α and IFN-γ act synergistically to optimize nitric oxide production (4, 5), which is involved in parasite killing (6). The difference between lethal and nonlethal murine malarias can be explained, at least in part, by the ability of the mice to mount an early IFN-γ response (7, 8) and/or an early TNF-α response (2); this may, in turn, be linked to early IL-12 production (4).
However, proinflammatory cytokines can also contribute to the pathology of rodent malaria. In mice infected with lethal strains of P. berghei, neutralization of IFN-γ (9), or blocking IFN-γ signaling by disruption of the IFN-γ receptor gene (10), delays or completely abrogates mortality, whereas overproduction, or sustained production, of IFN-γ or TNF-α predisposes to severe pathology in both P. chabaudi chabaudi– and P. vinckei–infected mice (2, 9, 11). Thus, it seems that an early proinflammatory cytokine response mediates protective immunity, whereas a late response contributes to pathology.
In nonlethal infections, inflammatory responses may be actively downregulated by antiinflammatory cytokines. One candidate cytokine is IL-10. IL-10–deficient mice infected with P. chabaudi chabaudi AS showed increased mortality compared to normal littermates, even though peak parasitemias were not significantly different (12). However, similar IL-10–deficient mice infected with nonlethal P. yoelii or with the relatively avirulent P. chabaudi adami 556KA showed the same response to infection as wild-type mice (1). In P. berghei ANKA infections, susceptible strains of mice show increased expression of IFN-γ mRNA and reduced expression of mRNA for TGF-β compared to resistant strains of mice (13), suggesting that TGF-β may play a role in downregulation of pathogenic proinflammatory cytokines.
TGF-β, which is produced by a wide range of cells including macrophages and T cells (14), has both pro- and antiinflammatory properties, depending on its environment and concentration (15). Importantly, TGF-β suppresses production of TNF-α and nitric oxide from macrophages (16, 17) and suppresses production of IFN-γ and TNF-α from NK cells (18). It has recently been proposed that these effects may be mediated via enhanced IL-10 production by macrophages (19), eventually leading to a shift in the immune response away from a Th1-like response and towards a Th2-like response (20).
Although murine malaria models do not replicate all the features of human malaria, there are strong correlations between the patterns of cytokine production seen in infected mice and humans. In certain circumstances, IFN-γ responses are associated with protective immunity to P. falciparum (21, 22), but IFN-γ levels are higher in clinical cases of malaria than in asymptomatic cases (23, 24), and there is evidence of a causal association between IFN-γ secretion and fever (25). Similarly, TNF-α mediates parasite killing by macrophages (26, 27), but severe P. falciparum malaria is accompanied by high levels of circulating TNF-α (28, 29), and polymorphisms within the promoter region of the TNF-α gene have been linked to an increased risk of cerebral malaria (30). Together, these observations indicate that in humans, as in mice, there is a critical balance to be found in terms of the inflammatory response to malaria infection. Understanding how this balance is maintained may provide new approaches to control of malarial parasitemia and prevention of severe disease.
To investigate the role of TGF-β in the pathogenesis of malaria, we have measured TGF-β production from splenic mononuclear cells of mice infected with both nonlethal (P. chabaudi chabaudi A/J and P. yoelii 17X) and lethal (P. berghei NK65) rodent malarias and have examined the effect of neutralizing antibodies to TGF-β, or recombinant TGF-β, on the course of malaria infections in vivo. We conclude that levels of TGF-β are inversely correlated with the severity of malaria infections in mice and that TGF-β plays an essential role in downregulating the production of potentially pathogenic proinflammatory cytokines. Furthermore, differences in TGF-β production in mice infected with different Plasmodium species appear to be due to intrinsic differences in the ability of parasite antigens to induce TGF-β production from macrophages.
Materials and Methods
Parasites
P. berghei (NK65), P. chabaudi chabaudi (A/J), and P. yoelii (17X) were obtained from Professor David Walliker, WHO Malaria Repository, University of Edinburgh, Edinburgh, UK. P. berghei is highly virulent in mice (31), susceptibility to P. chabaudi chabaudi varies between strains of inbred mice but resolves spontaneously in BALB/c mice (32), and P. yoelii is generally avirulent, although a lethal strain (17XL/YM) has been derived from the avirulent 17X strain (33). Cryopreserved parasites were thawed, injected intraperitoneally into BALB/c mice, and maintained by regular passage into naive mice.
Parasitized mouse erythrocytes (20–40% parasitemia) were purified by layering onto 72% Percoll (Pharmacia Biotech AB, Uppsala, Sweden) and centrifuged at 2,300 rpm for 15 min. Schizonts were recovered from the gradient interface and washed in RPMI (GIBCO BRL, Paisley, UK).
Mice
4–6-wk-old male BALB/c mice were obtained from Harlan (Oxford, UK). The drinking water of experimental mice was supplemented with 2.5 g/liter p-aminobenzoic acid to ensure that parasite growth was not inhibited by a lack of essential nutrients (34).
Experimental Malaria Infections
Mice were infected with either 104 (P. berghei) or 105 (P. berghei, P. chabaudi chabaudi, or P. yoelii) parasite-infected erythrocytes, in 100 μl of PBS by intraperitoneal injection. Control mice received an equal number of uninfected erythrocytes. Parasitemia was monitored every second day by Giemsa-stained thin blood smears obtained from tail bleeds.
For estimation of cytokine production from spleen cells, and to obtain serum for cytokine assays, test and control mice were killed at regular intervals; blood was obtained by venepuncture and spleens were retained for cellular assays. Blood was allowed to clot and serum was stored at −20°C until required.
To examine the effect of neutralization of TGF-β on the course of malaria infection, mice were each given 50 μg of an IgG1 monoclonal antibody to TGF-β, which neutralizes all three TGF-β isoforms (1835-01; Genzyme Diagnostics, Cambridge, MA), by intraperitoneal injection 1 d before malaria infection and on days 2, 5, and 7 after infection. Control mice received 50 μg of polyclonal mouse IgG1 (Serotec, Oxford, UK).
To examine the effect of increased TGF-β levels in malaria- infected mice, mice were infected with P. berghei and given either 5 ng or 20 ng of recombinant TGF-β1 (R&D Systems Europe Ltd., Abingdon, UK) in 100 μl of PBS by intraperitoneal injection on the day of infection and then daily for another 4 d. Control mice received PBS only.
Mononuclear Cell Cultures
Mononuclear cells were obtained from macerated spleens by centrifugation over a 5-ml gradient of mouse lymphocyte separation medium (Harlan-Seralab, Loughborough, UK). After washing in RPMI, cells were resuspended in complete medium (RPMI containing 2 g/liter NaCO2, 2mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, all from ICN Biomedicals, Costa Mesa, CA) and allowed to adhere to plastic petri dishes coated with mouse serum for 90 min at 37°C. Nonadherent cells were washed away, adherent cells (∼90% macrophages and ∼10% lymphocytes) were recovered by incubation for 30 min at 4°C in calcium- and magnesium-free saline (HBSS; Sigma Chemical Co., St. Louis, MO) and counted, and 2.5 × 106 adherent cells were added to each well of a 48-well microtiter plate. Cells were cultured in 350 μl of complete medium at 37°C in 5% CO2 in air for up to 48 h either without further stimulation or in the presence of 5 × 105 live malaria parasites, uninfected red blood cells, or the mitogen Con A (5 μg/ml; Sigma Chemical Co.). Cell culture supernatants were collected and stored at −20°C until required.
TGF-β Bioassay
This assay, based on the ability of TGF-β to inhibit the in vitro proliferation of mink lung epithelial cells (Mv-I-Lu; 35), measures the combined effects of all TGF-β isoforms. Mv-I-Lu cells (ATCC clone CCL64; European Collection of Cell Cultures, Wiltshire, UK) were maintained by weekly passage in Eagle's minimum essential medium (Sigma Chemical Co.) supplemented with 0.1 M nonessential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS; all from Sigma Chemical Co.). Cells were detached from the culture flask by incubation at 37°C with 0.5% trypsin/5.3 mM EDTA, washed, and resuspended in complete medium, containing 6% serum substitute (Ultroser; GIBCO BRL) instead of FBS, at 2 × 104 cells/ml. 104 cells, in a volume of 50 μl, were aliquoted into each well of a 96-well, flat-bottomed microculture plate and left to adhere for 2 h.
Culture supernatants were tested for TGF-β activity either with or without prior acid-activation. Acid-activation converts the inactive precursor form of TGF-β into biologically active TGF-β, allowing total TGF-β to be measured; in contrast, only biologically active TGF-β is measured in non–acid-activated samples. For acid activation, 10 μl 1 M HCl was added to 100 μl supernatant, incubated at 4°C for 30 min, and neutralized by dropwise addition of 20 μl 1 M NaOH. 50 μl of test sample or TGF-β1 standard (recombinant murine TGF-β1, IC50 0.03–0.05 ng/ml; Sigma Chemical Co.) and 50 μl fresh medium (with 6% Ultroser) were added to triplicate wells of Mv-I-Lu cells and incubated for 48 h at 37°C in 5% CO2 in air; [3H-]thymidine (0.5 μCi/well; Amersham Life Sciences, Little Chalfont, UK) was added for the last 18 h of culture. For cell harvesting, cells were incubated with 50 μl trypsin/EDTA for 15–30 min at 37°C and then harvested onto glass fiber filters; incorporation of 3H was assessed by liquid scintillation counting. Concentration of TGF-β in the sample was calculated by reference to the percentage inhibition of 3H incorporation caused by different concentrations of the TGF-β1 standard. The specificity of the assay was confirmed by blocking the inhibition of cell growth with a neutralizing monoclonal antibody to all isoforms of murine TGF-β (20 μg/ml; 1D11.16; Genzyme).
ELISAs
TGF-β1.
A sandwich ELISA was used to measure total TGF-β1 (latent and mature) in mouse serum (36). Plates were coated overnight with 4 μg/ml monoclonal anti-TGF-β1 antibody (clone MCA797, Serotec, Oxford, UK), washed, blocked with 4% bovine serum albumin (BSA; Sigma Chemical Co.) and samples (100 μl) added to duplicate wells and incubated at 37°C for 1 h. After washing, bound TGF-β1 was detected with biotinylated, polyclonal chicken antibody to mouse TGF-β (2 μg/ml) (R&D Systems) and streptavidin-peroxidase (Sigma) and visualized with 2,2′-azino-bis(-3ethylbenzthiazoline-6-sulfonic acid) (ABTS, Sigma) and hydrogen peroxide (Sigma). A standard curve was generated using rTGF-β1 (R&D Systems). The assay was sensitive over a range of TGF-β1 concentrations from 0.01 to 100 ng/ml.
IFN-γ, TNF-α, IL-10, and IL-4.
Sandwich ELISAs were conducted with commercial reagents (PharMingen, San Diego, CA) according to the manufacturer's instructions. Briefly, plates were coated with 2 μg/ml of capture antibody and blocked with 4% BSA in PBS containing 0.05% Tween 20 (Sigma). Samples or standards, 100 μl, were added to duplicate wells and incubated at 37°C for 1 h. Plates were washed and bound cytokine was detected with 2 μg/ml biotinylated antibody and streptavidin-peroxidase. Plates were developed as described above. Cytokine standards (PharMingen) were tested over the following ranges: IFN-γ, 1.5 to 3,200 U/ml; TNF-α, 0.01 to 1,500 ng/ml; IL-10, 0.04 to 1,500 ng/ml; IL-4, 20 to 5,000 pg/ml.
Statistical Analysis
Differences between the means, of groups of five mice, were determined by Student's t test.
Results
TGF-β Production during Fatal and Resolving Murine Malaria Infections
After infection with either 104 or 105 infected erythrocytes, P. berghei infection was universally fatal by day 20 (104) or days 13–15 (105) after infection. In contrast, P. yoelii and P. chabaudi chabaudi infections resolved spontaneously; parasitemia peaked around days 14–16 and was cleared by day 23 after infection.
Mice were killed at various time points after infection and spleens were removed. Spontaneous production of both latent and bioactive TGF-β by splenic mononuclear cells was assessed by bioassay. In P. berghei–infected mice there was a steady and significant decline in total and bioactive TGF-β production from day 10 after infection onwards (day 0 versus day 20, total TGF-β student's t test (t) = 7.47, degrees of freedom (df ) = 8, P <0.001; bioactive TGF-β t = 81.7, df = 8, P <0.0001), with levels of TGF-β being inversely related to parasitemia (Fig. 1 a). In both P. chabaudi chabaudi– and P. yoelii–infected mice there was a transient fall in bioactive TGF-β production as parasitemia increased; this was statistically significant for P. chabaudi chabaudi at day 7 (t = 3.77, df = 8, P = 0.005) and for P. yoelii at day 12 (t = 4.99, df = 8, P = 0.005), but total TGF-β production increased steadily and significantly throughout the course of infection (day 0 versus day 23, t >3.9, df = 8, P = 0.004 in both cases; Fig. 1, b and c).
Figure 1.
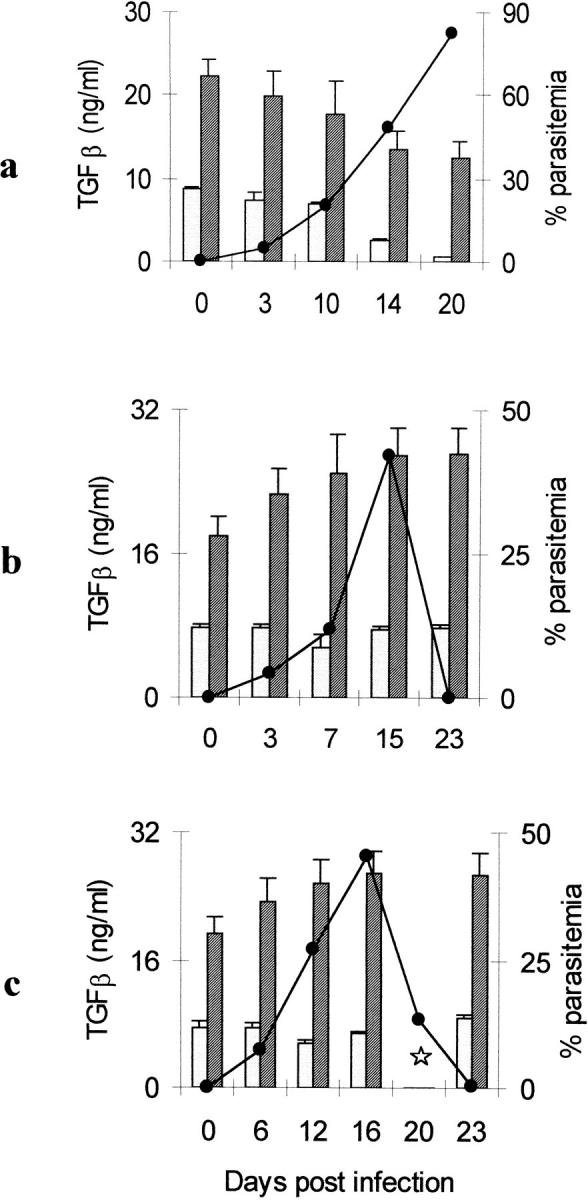
TGF-β production by splenic mononuclear cells during murine malaria infections, measured by bioassay after 48 h in vitro culture. Mice were infected with (a) 104 P. berghei–, (b) 105 P. chabaudi chabaudi–, or (c) 105 P. yoelii–infected erythrocytes on day 0. n = 5 mice/group; star, no data; hatched bar, total TGF-β; dotted bar, bioactive TGF-β; black circle, percentage of parasitemia.
In a parallel experiment, levels of circulating serum TGF-β1 in infected mice were measured by ELISA (Fig. 2). This assay measures both latent and bioactive TGF-β1. In P. berghei–infected mice, plasma TGF-β1 levels declined significantly between days 8 and 20 (t = 6.3, df = 7, P <0.001). In contrast, plasma TGF-β1 levels in mice infected with P. chabaudi chabaudi or P. yoelii decreased transiently during the first week of infection (day 0 versus day 3, t = 3.7, df = 8, P = 0.006 for P. chabaudi chabaudi; t = 2.34, df = 8, P <0.049 for P. yoelii) but then rose steadily for the remainder of the infection (day 0 versus day 20, t >5.9, df = 8, P <0.001 in both cases).
Figure 2.

Serum TGF-β1 levels in malaria-infected mice, measured by ELISA. 5 mice/group. Mice were infected with (•) 104 P. berghei, (♦) 105 P. yoelii, (▪) 105 P. chabaudi chabaudi, or were left uninfected (□).
Taken together, these data suggest that TGF-β may play a crucial role in preventing the severe pathology of malaria. Where TGF-β levels are maintained at normal or above normal levels, the mice survive, but where TGF-β levels are suppressed, the mice die.
The Role of TGF-β in Murine Malaria Infections
Neutralization of TGF-β.
To test the hypothesis that TGF-β is required to prevent the severe pathology associated with some murine malarias, mice were treated with a neutralizing antibody to all mouse TGF-β isoforms immediately before and during infection with P. berghei, P. chabaudi chabaudi, or P. yoelii. Anti–TGF-β antibody, or isotype-matched control IgG, was administered 1 d before infection and on days 2, 5, and 7 after infection (Fig. 3).
Figure 3.

Effect of treatment with anti–TGF-β monoclonal antibody (given on days −1, 2, 5, and 7 after infection) on the course of malaria parasitemia (mean % parasitemia ± SD; a–c) and mouse survival (d–f) with (a and d) 105 P. berghei, (b and e) 105 P. chabaudi chabaudi, and (c and f) 105 P. yoelii. •, anti–TGF-β–treated; ○, control, treated with irrelevant IgG1.
In this experiment, P. berghei–infected mice received 105 infected erythrocytes, leading to death between days 10 and 13. Anti–TGF-β treatment significantly enhanced the rate of increase P. berghei parasitemia (Fig. 3 a, treated versus untreated mice, day 7, t = 10.2, df = 8, P <0.0001), and all the infected mice died on either day 6 or 8 after infection, 4 or 5 d earlier than in mice who received the control antibody (Fig. 3 d). The anti–TGF-β antibody did not, by itself, cause any observable side-effects, as treated, uninfected mice remained perfectly healthy (data not shown).
Neutralization of TGF-β converted the normally nonlethal P. chabaudi chabaudi infection into a rapidly lethal infection. Infected mice began dying at day 6 after infection and all died by day 12 (Fig. 3 e). Although parasitemia increased more rapidly in anti–TGF-β–treated mice than in untreated mice (day 10, t = 4.74, df = 8, P <0.001) and the peak parasitemia occurred ∼2 d earlier in treated mice, the peak was not significantly higher than in untreated mice (Fig. 3 b; t = 0.92, df = 8, P = 0.39).
In contrast, neutralization of TGF-β had little effect on the overall course of P. yoelii infection. Parasitemia rose slightly earlier in treated mice and was significantly higher at day 12 (treated versus untreated, t = 3.58, df = 8, P = 0.007), but the peak parasitemia was not significantly different (day 16, t = 1.31, df = 8, P = 0.23). Parasitemia resolved spontaneously (Fig. 3 c) and all the mice survived (Fig. 3 f ).
These data show very clearly that TGF-β plays a crucial role in protecting mice with P. chabaudi chabaudi infection from the severe pathology of malaria and suggest that the lack of a TGF-β response in mice infected with P. berghei may explain the severe pathology associated with this parasite. Interestingly, neutralization of TGF-β has little effect on the outcome of P. yoelii infection, suggesting that this parasite interacts very differently with the host's immune system compared with P. berghei or P. chabaudi chabaudi. The data also suggest that TGF-β may contribute to the control of parasite growth as, in all cases, neutralization of TGF-β led to increased rates of parasite proliferation. However, the effect of anti-TFG-β antibody on mouse survival is probably distinct from its effect on parasitemia, as parasitemia increased more rapidly in all infections, but (a) death was accelerated only in P. berghei and P. chabaudi chabaudi infections and (b) P. chabaudi chabaudi–infected mice died at parasitemias that are compatible with survival in control mice.
Treatment of P. berghei–infected Mice with TGF-β1.
To test the hypothesis that the severity of P. berghei malaria in BALB/c mice is associated with the failure to upregulate TGF-β production during infection, mice were infected with 105 P. berghei–infected erythrocytes and treated with either 5 or 20 ng/mouse of rTGF-β1 daily for 5 d.
In mice receiving rTGF-β1, the parasitemia rose less quickly than in control mice (Fig. 4 a). Importantly, death was significantly delayed in treated mice; mice receiving 20 ng TGF-β1 died 12 d later than untreated mice, whereas mice receiving 5 ng/day died ∼20 d later (Fig. 4 b). However, it was not possible to separate the effect of TGF-β on parasite growth from the effect on mouse survival; in mice treated with 20 ng rTGF-β, parasitemia rose more quickly, and death occurred earlier, than in mice receiving 5 ng TGF-β.
Figure 4.
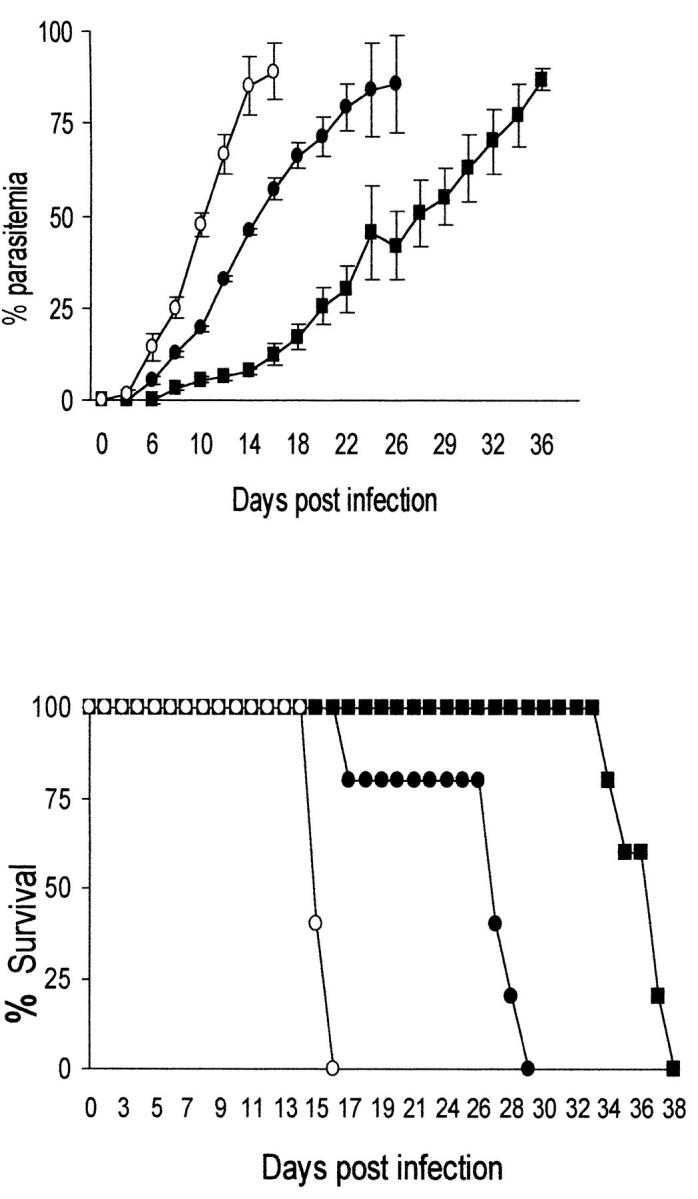
Effect of treatment with rTGF-β1 on the course of P. berghei parasitemia (a) and mouse survival (b). Mice were infected with 105 P. berghei–infected erythrocytes and treated with 5 or 20 ng rTGF-β1 per mouse daily for 5 d. •, 20 ng/d of rTGF-β1; ▪, 5 ng/d of rTGF-β1; ○, control, PBS-treated.
Interaction between TGF-β and Other Cytokines
TNF-α and IFN-γ.
It is known that much of the pathology of malaria is mediated by proinflammatory cytokines such as TNF-α and IFN-γ (25, 28, 29). TGF-β is a known antiinflammatory agent that acts to downregulate the production of proinflammatory cytokines (37). We hypothesized that the severe pathology of P. berghei infection was due to overproduction of proinflammatory cytokines in the absence of TGF-β. Circulating levels of TNF-α and IFN-γ in the plasma of infected mice were measured by ELISA.
In mice infected with P. berghei, serum TNF-α levels rise significantly over the first 10 d of infection (Fig. 5 a; day 3 versus day 10, t = 3.26, df = 8, P = 0.012). As predicted, when P. berghei–infected mice are treated with rTGF-β, there is a highly significant decrease in circulating levels of TNF-α (Fig. 5 a; t >4.8, df = 8, P <0.001 at days 3, 7, and 10), and when mice are given anti–TGF-β antibody, serum TNF-α levels are significantly higher (day 7 after infection, control mice, mean TNF-α = 5.4 ± 0.5 ng/ml; anti–TGF-β mice, mean = 9.15 ± 0.92 ng/ml; t = 7.92, df = 7, P <0.0001).
Figure 5.

Serum cytokine levels in P. berghei–infected (105 infected erythrocytes) mice, measured by ELISA. White bar, uninfected, control mice; dotted bar, infected, untreated mice; black bar, infected mice treated with 20 ng of rTGF-β1 for 5 d.
In contrast, rTGF-β1 or anti–TGF-β antibody had an unexpected effect on levels of circulating IFN-γ in P. berghei–infected mice. rTGF-β1 treatment lead to a small, although statistically significant, increase in serum IFN-γ levels (Fig. 5 b), whereas anti–TGF-β antibody had no significant effect on IFN-γ levels (day 7 after infection, control mice, mean IFN-γ = 40.5 ± 3.1 U/ml; anti–TGF-β–treated mice, mean = 44.3 ± 2.25 U/ml; t = 1.97, df = 7, P = 0.1).
IL-4 and IL-10.
An alternative means by which TGF-β may downregulate inflammatory cytokines is by augmentation of the production of other antiinflammatory cytokines. We therefore measured plasma levels of IL-4 and IL-10 in P. berghei–infected mice and assessed the effect of rTGF-β or anti-TGF-β antibody. Neither IL-4 nor IL-10 levels changed significantly over the course of infection with P. berghei (Fig. 5, c and d). Treatment with rTGF-β over 5 d lead to an apparent steady decline in IL-4 levels (Fig. 5 c), but the difference between the two groups was statistically significant only on day 10 (t = 3.7, df = 8, P <0.01). In contrast, rTGF-β leads to a rapid and highly significant increase in IL-10 production in infected mice (Fig. 5 d) that persisted until at least day 10 after infection (days 3, 7, and 10, t >2.33, df = 8, P <0.05 in all cases). Anti–TGF-β antibody treatment had no significant effect on IL-4 or IL-10 levels (data not shown).
In Vitro Induction of TGF-β by Live Malaria Parasites
The failure of mice infected with P. berghei to sufficiently upregulate TGF-β production was investigated in vitro. Plastic adherent splenic mononuclear cells from uninfected mice were incubated in vitro for up to 36 h with parasitized erythrocytes, uninfected erythrocytes, or mitogen, and the concentration of total and bioactive TGF-β in the culture supernatants was measured by bioassay (Table 1).
Table 1.
TGF-β Production by Splenic Adherent Mononuclear Cells (2 × 106/well) from Uninfected BALB/c Mice Incubated with Live Malaria Parasites (5 × 105/well)
| TGF-β concentration (ng/ml) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 h | 2 h | 6 h | 10 h | 18 h | 36 h | |||||||
| Medium | 0* | 0 | 0 | 0.32 ± 0.07 | 0.59 ± 0.28 | 2.1 ± 0.20 | ||||||
| (1.23 ± 0.08)‡ | (2.91 ± 1.05) | (4.22 ± 1.07) | ||||||||||
| mRBC | 0 | 0 | 0 | 0.64 ± 0.30 | 0.72 ± 0.44 | 1.85 ± 0.09 | ||||||
| (1.40 +0.09) | (1.78 ± 0.75) | (3.82 ± 1.95) | (5.13 ± 2.10) | |||||||||
| Pb live | 0 | 0 | 0.26 ± 0.08 | 1.66 ± 0.30 | 1.18 ± 0.08 | 1.15 ± 0.28 | ||||||
| (2.01 ± 0.90) | (2.90 ± 1.12) | (3.11 ± 1.41) | (2.78 ± 1.50) | |||||||||
| Pc Live | 0 | 0 | 0.66 ± 0.07 | 1.4 ± 0.07 | 4.6 ± 0.53 | 5.4 ± 1.12 | ||||||
| (3.63 ± 1.40) | (6.23 ± 1.40) | (8.12 ± 1.31) | (7.93 ± 2.03) | |||||||||
| Con A | 0 | 0.37 ± 0.08 | 1.31 ± 0.27 | 1.56 ± 0.30 | 4.70 ± 0.81 | 4.3 ± 1.32 | ||||||
| (1.40 ± 0.34) | (3.82 ± 1.14) | (7.36 +1.82) | (13.43 ± 1.98) | (11.07 ± 2.34) | ||||||||
Results represent averages of data from three separate experiments. mRB, mouse erythrocytes, Pc, P. chabaudi, Pb, P. berghei.
0 values represent below-detection limits of the bioassay.
Figures in parentheses represent TGF-β values determined for acid-activated sample supernatants.
The mitogen Con A induced production of TGF-β within 2–6 h, and TGF-β levels peaked at 18 h. Live P. chabaudi chabaudi parasites were almost as effective as Con A, inducing TGF-β production within 6 h, with levels also peaking after 18 h in culture. In contrast, P. berghei parasites induced little if any TGF-β production.
Discussion
We have shown that murine infection with P. chabaudi chabaudi or P. yoelii, but not P. berghei, is accompanied by increased TGF-β production, as assessed by raised serum levels of TGF-β1 in vivo and spontaneous secretion of TGF-β in vitro by splenic mononuclear cells from infected mice. We have also shown that TGF-β levels during murine malaria infection are inversely correlated with severity of disease, in that lethal infections are accompanied by low levels of TGF-β and self-resolving infections are accompanied by high levels of TGF-β. These findings have been confirmed by treatment of mice with either neutralizing antibodies to TGF-β or rTGF-β1. These findings are consistent with the only other data regarding TGF-β in murine malaria, which simply showed that P. berghei (ANKA)– induced TGF-β mRNA levels were lower in susceptible strains of mice than in resistant strains (13).
Given the wealth of evidence that the pathology of murine malaria is mediated by the inflammatory cytokines IFN-γ and TNF-α (2, 9, 11), the most likely explanation for our observations was that TGF-β acted to downregulate either the production or the activity of these cytokines. We were able to show quite clearly that rTGF-β1 treatment led to a decrease in circulating levels of TNF-α but had much less effect on levels of IFN-γ. These observations are consistent with the known targets of the antiinflammatory activities of TGF-β, namely its ability to suppress translation of TNF-α mRNA in macrophages (37) and to inhibit transcription of genes for cytokines such as IL-8 (38) and macrophage chemoattractant protein (MCP1; 39). In contrast, there is little evidence that TGF-β has a direct effect on macrophage-derived IFN-γ; rather, TGF-β antagonizes the effects of IFN-γ downstream of IFN-γ itself, for example by inhibiting the induction of MHC class II expression (40).
Although our data are consistent with a direct effect of TGF-β on TNF-α production, we cannot rule out an indirect effect via IL-10 (19, 20). IL-10 is induced by TGF-β (19) and has been shown to suppress production of TNF-α in activated macrophages (37). Consistent with this hypothesis, IL-10 levels were not raised in the serum of P. berghei–infected mice, but IL-10 was clearly upregulated when infected mice were treated with rTGF-β1. A role for IL-10 in ameliorating the pathology of P. chabaudi chabaudi infection has been shown in IL-10–deficient mice (12) but, interestingly, IL-10 does not seem to play a similar role in avirulent P. yoelii or P. chabaudi adami infections where IL-10–deficient mice were able to resolve their infections normally (1). This mirrors our observation that the pathology of P. yoelii was not affected by neutralization of TGF-β activity and suggests that the pathology of P. yoelii infection has a very different aetiology. However, IL-10 is unlikely to be the only pathway for TGF-β activity as P. chabaudi chabaudi infection is much more severe in mice treated with anti–TGF-β antibody, where all the mice died, than in IL-10–deficient mice, where mortality is restricted to female mice and even then only 50% of the female mice die (12). However, in both IL-10–deficient and anti–TGF-β– treated mice, death occurs without a significant increase in peak parasitemia, indicating that failure to control P. chabaudi chabaudi replication is not the cause of death.
Our demonstration that TGF-β upregulates IL-10 production without downregulating IFN-γ offers an explanation for the observation that both IFN-γ and IL-10 levels are raised in acute P. falciparum infection in humans (41). Little is known of TGF-β responses in human malarias; supernatants of human mononuclear cells cocultured with P. falciparum–infected erythrocytes contained high levels of TGF-β (42), but TGF-β serum levels are lower in patients with acute P. falciparum malaria than in healthy controls (42a).
In addition to downregulation of TNF-α production, TGF-β may inhibit the development of malarial pathology by direct effects on parasite sequestration. Adherence of infected erythrocytes to cerebral capillary endothelium, via cellular adhesion molecules such as intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, has been proposed to contribute to the development of cerebral malaria (43, 44). Mice in which the TGF-β1 genes have been disrupted show enhanced expression of both ICAM-1 and VCAM-1 (45), suggesting that TGF-β may play a role in downregulating expression of these adhesion molecules thus reducing the risk of cerebral malaria.
Although our study provides strong evidence that TGF-β can protect against the severe pathology of P. berghei and P. chabaudi chabaudi malaria, our data also indicate that TGF-β may play a role in controlling parasite growth, at least in the early stages of infection, as anti–TGF-β antibody led to more rapid parasite growth and a 5-d course of rTGF-β1 resulted in slower parasite growth. In the case of P. chabaudi chabaudi infection, the effect of TGF-β on survival is clearly distinct from its effect on parasite growth; this distinction is less clear in P. berghei infection. The apparent ability of TGF-β to help control parasite growth may relate to the fact that, early in an immune response, low concentrations of TGF-β actually promote inflammation, recruit monocytes and macrophages to the site of injury, and activate them to become phagocytic (15). In support of this hypothesis, we have shown that TGF-β1 increases phagocytosis of P. falciparum–infected erythrocytes by human peripheral blood mononuclear cells in vitro (Omer, F.M., and E. Riley, manuscript in preparation). Thus, inhibition of TGF-β early in the infection may inhibit macrophage activation and nonspecific parasite clearance, as has been shown for other pathogens such as Candida albicans (46), pneumococcus (47), and HIV (48). In contrast, high concentrations of TGF-β are antiinflammatory but also inhibit the activity of inducible nitric oxide synthase (49), thus reducing the ability of macrophages to control the growth of intracellular pathogens such as Trypanosoma cruzi (50) and Leishmania amazonensis (51). Thus, the balance between controlling parasite replication and avoiding immunopathology appears to depend on controlling local and systemic concentrations of TGF-β.
In summary, the bimodal activities of TGF-β–promoting inflammation and parasite clearance early in infection while downregulating inflammation and pathology later in infection—make it a very strong contender for being a major immunoregulatory molecule, maintaining the balance between the protective and pathogenic effects of other inflammatory cytokines during malaria infections. If this supposition is correct, one would predict that severe malaria in humans is associated with reduced capacity to produce TGF-β. Studies are currently underway in our laboratory to test this hypothesis. However, this theory begs the question as to why some infections are associated with low TGF-β production. The preliminary data presented here, indicating that P. berghei parasites fail to induce TGF-β production from normal mouse macrophages, suggests that genetic differences between parasites may be responsible. Possible explanations for the failure of P. berghei to induce TGF-β include the failure to induce TGF-β per se, induction of high levels of IL-12 or IFN-γ, both of which have been shown to act as negative regulators of TGF-β production (52), or induction of TGF-β inhibitors such as α2-macroglobulins (49). Identification of the mechanisms of TGF-β antagonism by P. berghei may provide clues as to the pathogenesis of severe malaria in humans.
Acknowledgments
We thank David McGuinness for statistical advice, Richard Carter and Niklas Ahlborg for constructive comments on the manuscript, and David Walliker and Pedro Cravo for providing malaria parasites.
This study was funded by the Wellcome Trust. F.M. Omer is supported by a Wellcome Trust International Fellowship.
References
- 1.van der Heyde HC, Pepper B, Batchelder J, Cigel F, Weidanz WP. The time course of selected malarial infections in cytokine-deficient mice. Exp Parasitol. 1997;85:206–213. doi: 10.1006/expr.1996.4132. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs P, Radzioch D, Stevenson MM. A Th1-associated increase in tumor necrosis factor alpha expression in the spleen correlates with resistance to blood-stage malaria in mice. Infect Immun. 1996;64:535–541. doi: 10.1128/iai.64.2.535-541.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Favre N, Ryffel B, Bordmann G, Rudin W. The course of Plasmodium chabaudi chabaudiinfections in interferon-gamma receptor–deficient mice. Parasite Immunol. 1997;19:375–383. doi: 10.1046/j.1365-3024.1997.d01-227.x. [DOI] [PubMed] [Google Scholar]
- 4.Stevenson MM, Tam MF, Wolf SF, Sher A. IL-12–induced protection against blood-stage Plasmodium chabaudi ASrequires IFN-γ and TNF-α and occurs via a nitric oxide–dependent mechanism. J Immunol. 1995;155:2545–2556. [PubMed] [Google Scholar]
- 5.Jacobs P, Radzioch D, Stevenson MM. In vivo regulation of nitric oxide production by tumor necrosis factor alpha and gamma interferon, but not by interleukin-4, during blood stage malaria in mice. Infect Immun. 1996;64:44–49. doi: 10.1128/iai.64.1.44-49.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nussler AK, Rénia L, Pasquetto V, Miltgen F, Matile H, Mazier D. In vivo induction of the nitric oxide pathway in hepatocytes after injection with irradiated malaria sporozoites, malaria parasites or adjuvants. Eur J Immunol. 1993;23:882–887. doi: 10.1002/eji.1830230417. [DOI] [PubMed] [Google Scholar]
- 7.Shear HL, Srinavasan R, Nolan T, Ng C. Role of IFN-γ in lethal and nonlethal malaria in susceptible and resistant hosts. J Immunol. 1989;143:2038–2044. [PubMed] [Google Scholar]
- 8.de Souza JB, Williamson KH, Otani T, Playfair JHL. Early gamma interferon responses in lethal and nonlethal murine blood stage malaria. Infect Immun. 1997;65:1593–1598. doi: 10.1128/iai.65.5.1593-1598.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waki S, Uehara S, Kanbe K, Ono K, Suzuki M, Nariuchi H. The role of T cells in pathogenesis and protective immunity to murine malaria. Immunology. 1992;75:646–651. [PMC free article] [PubMed] [Google Scholar]
- 10.Rudin W, Favre N, Bordmann G, Ryffel B. Interferon-γ is essential for the development of cerebral malaria. Eur J Immunol. 1997;27:810–815. doi: 10.1002/eji.1830270403. [DOI] [PubMed] [Google Scholar]
- 11.Kremsner PG, Neifer S, Chaves MF, Rudolph R, Bienzle U. Interferon-γ induced lethality in the late phase of Plasmodium vinckeimalaria despite effective parasite clearance by chloroquine. Eur J Immunol. 1992;22:2873–2878. doi: 10.1002/eji.1830221118. [DOI] [PubMed] [Google Scholar]
- 12.Linke A, Kuhn R, Muller W, Honarvar N, Li C, Langhorne J. Plasmodium chabaudi chabaudi: differential susceptibility of gene-targeted mice deficient in IL-10 to an erythrocytic-stage infection. Exp Parsitol. 1996;84:253–263. doi: 10.1006/expr.1996.0111. [DOI] [PubMed] [Google Scholar]
- 13.de Kossodo S, Grau GE. Profiles of cytokine production in relation with susceptibility to cerebral malaria. J Immunol. 1993;151:4811–4820. [PubMed] [Google Scholar]
- 14.Assosian RK, Fleurdelys BE, Stevenson HC, Miller PJ, Madtes DK, Raines EW, Ross R, Sporn MB. Expression and secretion of type β transforming growth factor by activated human macrophages. Proc Natl Acad Sci USA. 1987;84:6020–6024. doi: 10.1073/pnas.84.17.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahl SM, McCartney-Francis N, Mergenhagen SE. Inflammatory and immunomodulatory roles of TGF-β. Immunol Today. 1989;10:258–261. doi: 10.1016/0167-5699(89)90136-9. [DOI] [PubMed] [Google Scholar]
- 16.Espevik T, Figari IS, Shalaby MR, Lackides GA, Lewis GD, Shepard HM, Palladino MA., Jr Inhibition of cytokine production by cyclosporin A and transforming growth factor β. J Exp Med. 1987;166:571–576. doi: 10.1084/jem.166.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding A, Nathan CF, Grayear J, Derynck R, Stuehr DJ, Srimai S. Macropahge deactivating factor and transforming growth factor-β1, -β2 and -β3 inhibit induction of macrophage nitrogen oxide synthesis by interferon-γ. J Immunol. 1990;145:940–944. [PubMed] [Google Scholar]
- 18.Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-β1. J Immunol. 1995;155:1066–1073. [PubMed] [Google Scholar]
- 19.Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A. TGF-β enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol. 1995;155:4926–4932. [PubMed] [Google Scholar]
- 20.Maeda H, Shiraishi A. TGF-β contributes to the shift toward Th2-type responses through direct and IL-10– mediated pathways in tumor-bearing mice. J Immunol. 1996;156:73–78. [PubMed] [Google Scholar]
- 21.Ferreira A, Schofield L, Enea V, Schellekens H, Van der Meide P, Collins WE, Nussenzweig RS, Nussenzweig V. Inhibition of development of exoerythrocytic forms of malaria parasites by IFN-gamma. Science. 1986;232:881–884. doi: 10.1126/science.3085218. [DOI] [PubMed] [Google Scholar]
- 22.Herrera M, Rosero F, Herrera S, Caspers P, Rotmann D, Sinigaglia F, Certa U. Protection against malaria in Aotusmonkeys immunised with a recombinant blood stage antigen fused to a universal T cell epitope: correlation of serum gamma-interferon levels with protection. Infect Immun. 1992;60:154–158. doi: 10.1128/iai.60.1.154-158.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riley EM, Jakobsen PH, Allen SJ, Wheeler JG, Bennett S, Greenwood BM. Immune responses to soluble exoantigens of Plasmodium falciparummay contribute to both pathogenesis and protection in clinical malaria: evidence from a longitudinal, prospective study of semi-immune African children. Eur J Immunol. 1991;21:1019–1025. doi: 10.1002/eji.1830210424. [DOI] [PubMed] [Google Scholar]
- 24.Mshana RN, Boulandi J, Mshana NM, Mayombo J, Mendome G. Cytokines in the pathogenesis of malaria: levels of IL-1β, IL-4, IL-6, TNF-α and IFN-γ in plasma of healthy individuals and malaria patients in a holoendemic area. J Clin Lab Immunol. 1991;34:131–139. [PubMed] [Google Scholar]
- 25.Harpaz R, Edelman R, Wasserman SS, Levine MM, Davis JR, Sztein MB. Serum cytokine profiles in experimental human malaria. Relationship to protection and disease course after challenge. J Clin Invest. 1992;90:515–523. doi: 10.1172/JCI115889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouharoun-Tayoun H, Oeuvray C, Lunel F, Druilhe P. Mechanisms underlying the monocyte-mediated antibody-dependent killing of Plasmodium falciparumasexual blood stages. J Exp Med. 1995;182:409–418. doi: 10.1084/jem.182.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rockett KA MM, Awburn, Aggarwal BB, Cowden WB, Clark IA. In vivo induction of nitrite and nitrate by tumour necrosis factor, lymphotoxin and interleukin 1: possible roles in malaria. Infect Immun. 1992;60:3725–3730. doi: 10.1128/iai.60.9.3725-3730.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert PH. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med. 1989;320:1586–1591. doi: 10.1056/NEJM198906153202404. [DOI] [PubMed] [Google Scholar]
- 29.Kwiatkowski D, Hill AVS, Sambou I, Twumasi P, Castracane J, Manogue KR, Cerami A, Brewster D, Greenwood BM. TNF concentration in fatal, non- fatal cerebral and uncomplicated Plasmodium falciparummalaria. Lancet. 1990;336:1201–1204. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 30.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwiatkowski D. Variation in the TNF-α promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–511. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 31.Cox, F.E.G. 1988. Major animal models in malaria research: rodent. In Malaria: Principles and Practice of Malariology. W.H. Wernsdorfer and I. McGregor, editors. Churchill Livingstone, London. 1503–1543.
- 32.Stevenson MM, Lyanga JJ, Skamene E. Murine malaria: genetic control of resistance to Plasmodium chabaudi. . Infect Immun. 1982;38:80–88. doi: 10.1128/iai.38.1.80-88.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoelii M, Hargreaves B, Carter R, Walliker D. Sudden increase in virulence in a strain of Plasmodium yoelii. . Ann Trop Med Parasitol. 1975;69:173–178. doi: 10.1080/00034983.1975.11686998. [DOI] [PubMed] [Google Scholar]
- 34.Peters W. Chemotherapy of Plasmodium chabaudiinfection in albino mice. Ann Trop Med Parasitol. 1967;61:52–56. doi: 10.1080/00034983.1967.11686457. [DOI] [PubMed] [Google Scholar]
- 35.Danielpour D, Dart LL, Flanders KC, Roberts AB, Sporn MB. Immunodetection and quantitation of the two forms of transforming growth factor-β (TGF-β1 and TGF-β2) secreted by cells in culture. J Cell Physiol. 1989;138:79–86. doi: 10.1002/jcp.1041380112. [DOI] [PubMed] [Google Scholar]
- 36.Lucas C, Fendly BM, Mukku FR, Wong WL, Palladino MA. Generation of antibodies and assays for transforming growth factor β. Methods Enzymol. 1991;198:303–336. doi: 10.1016/0076-6879(91)98031-z. [DOI] [PubMed] [Google Scholar]
- 37.Bogdan C, Paik J, Vodovotz Y, Nathan C. Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-β and interleukin-10. J Biol Chem. 1992;267:23301–23308. [PubMed] [Google Scholar]
- 38.Smith WB, Noack L, Khew-Goodall Y, Isenmann S, Vadas MA, Gamble JR. Transforming growth factor-β1inhibits the production of IL-8 and the transmigration of neutrophils through activated endothelium. J Immunol. 1996;157:360–368. [PubMed] [Google Scholar]
- 39.Kitamura M. Identification of an inhibitor targeting macrophage production of monocyte chemoattractant protein-1 as TGF-β1 . J Immunol. 1997;159:1404–1411. [PubMed] [Google Scholar]
- 40.Nandan D, Reiner NE. TGF-β attenuates the class II transactivator and reveals an accessory pathway of IFN-γ action. J Immunol. 1997;158:1095–1101. [PubMed] [Google Scholar]
- 41.Wenisch C, Parschalk B, Narzt E, Looareesuwan S, Graninger W. Elevated serum levels of IL-10 and IFN-γ in patients with acute Plasmodium falciparummalaria. Clin Immunol Immunopathol. 1995;74:115–117. doi: 10.1006/clin.1995.1017. [DOI] [PubMed] [Google Scholar]
- 42.Wahlgren M, Abrams JS, Fernandez V, Bejarano M, Azuma M, Torii M, Aikawa M, Howard RJ. Adhesion of Plasmodium falciparum–infected erythrocytes to human cells and secretion of cytokines (IL-1–β, IL-1Rα, IL-6, IL-8, IL-10, TGF β, TNF α, G-CSF, GM-CSF) Scand J Immunol. 1995;42:626–636. doi: 10.1111/j.1365-3083.1995.tb03705.x. [DOI] [PubMed] [Google Scholar]
- 42a.Wenisch C, Parschalk B, Burgmann H, Looareesuwan S, Graninger W. Decreased serum levels of TGF-β in patients with acute Plasmodium falciparummalaria. J Clin Immunol. 1995;15:69–73. doi: 10.1007/BF01541734. [DOI] [PubMed] [Google Scholar]
- 43.Ockenhouse CF, Tegoshi T, Maeno Y, Benjamin C, Ho M, Ei K, Khan, Thway Y, Win K, Aikawa M, Lobb RR. Human vascular endothelial cell adhesion receptors for Plasmodium falciparum–infected erythrocytes: roles for endothelial leukocyte adhesion molecule 1 and vascular cell adhesion molecule 1. J Exp Med. 1992;176:1183–1189. doi: 10.1084/jem.176.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. . Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 45.Nakabayashi T, Letterio JJ, Geiser AG, Kong L, Ogawa N, Zhao W, Koike T, Fernandes G, Dang H, Talal N. Up-regulation of cytokine mRNA, adhesion molecule proteins, and MHC class II proteins in salivary glands of TGF-β1knockout mice. J Immunol. 1997;158:5527–5535. [PubMed] [Google Scholar]
- 46.Spaccapelo R, Romani L, Tonnetti L, Cenci E, Mencacci A, Del Sero G, Tognellini R, Reed SG, Puccetti P, Bistoni F. TGF-β is important in determining the in vivo patterns of susceptibility or resistance in mice infected with Candida albicans. . J Immunol. 1995;155:1349–1360. [PubMed] [Google Scholar]
- 47.Pfister HW, Frei K, Ottand B, Koedel U, Tomasz A, Fontana A. Transforming growth factor β2 inhibits cerebrovascular changes and brain edema formation in the tumor necrosis factor α–independent early phase of experimental pneumococcal meningitis. J Exp Med. 1992;176:265–268. doi: 10.1084/jem.176.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poli G, Kinter AL, Justement JS, Bressler P, Kehrl JH, Fauci AS. Transforming growth factor β suppresses human immunodeficiency virus expression and replication in infected cells of the monocyte/macrophage lineage. J Exp Med. 1991;173:589–597. doi: 10.1084/jem.173.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webb DJ, Wen J, Lysiak JL, Umans L, Van Leuven F, Gonias SL. Murine α-macroglobulins demonstrate divergent activities as neutralizers of transforming growth factor-β and as inducers of nitric oxide synthesis. J Biol Chem. 1996;271:24982–24988. doi: 10.1074/jbc.271.40.24982. [DOI] [PubMed] [Google Scholar]
- 50.Silva JS, Twardzik DR, Reed SG. Regulation of Trypansoma cruziinfections in vitro and in vivo by transforming growth factor β (TGF-β) J Exp Med. 1991;174:539–545. doi: 10.1084/jem.174.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barral-Neto M, Barral A, Brownwell CE, Skeiky YAW, Ellingsworth LR, Twardzik DR, Reed SG. Transforming growth factor β in leishmanial infections: a parasite escape mechanism. Science. 1992;257:545–548. doi: 10.1126/science.1636092. [DOI] [PubMed] [Google Scholar]
- 52.Marth T, Strober W, Seder RA, Kelsall BL. Regulation of transforming growth factor-β production by interleukin-12. Eur J Immunol. 1997;27:1213–1220. doi: 10.1002/eji.1830270524. [DOI] [PubMed] [Google Scholar]