

Abstract
By differential screening of tumor necrosis factor α (TNF-α) and lipopolysaccharide (LPS)- activated endothelial cells (ECs), we have identified a cDNA clone that turned out to be a member of the inhibitor of apoptosis (iap) gene family. iap genes function to protect cells from undergoing apoptotic death in response to a variety of stimuli. These iap genes, hiap1, hiap2, and xiap were found to be strongly upregulated upon treatment of ECs with the inflammatory cytokines TNF-α, interleukin 1β, and LPS, reagents that lead to activation of the nuclear transcription factor κB (NF-κB). Indeed, overexpression of IκBα, an inhibitor of NF-κB, suppresses the induced expression of iap genes and sensitizes ECs to TNF-α–induced apoptosis. Ectopic expression of one member of the human iap genes, human X-chromosome–linked iap (xiap), using recombinant adenovirus overrules the IκBα effect and protects ECs from TNF-α– induced apoptosis. We conclude that xiap represents one of the NF-κB–regulated genes that counteracts the apoptotic signals caused by TNF-α and thereby prevents ECs from undergoing apoptosis during inflammation.
Keywords: activation, inhibitor of apoptosis gene family, endothelial cells, adenovirus, nuclear factor κB
Endothelial cells (ECs) are located at the strategic interface between blood stream and tissue and regulate local exchange of cells and nutrients. They are critically involved in local and systemic inflammatory responses at the sites of transmigration of immune cells such as neutrophils, monocytes, and lymphocytes. The concentration of inflammatory cytokines at the site of transmigration is expected to be high, and in fact inflammatory cytokine–mediated activation of ECs is responsible for the attraction, adhesion, and extravasation of white blood cells to the inflamed tissue.
Stimulation of cells with TNF-α, a potent inflammatory cytokine, generates two types of signals: one that initiates programmed cell death (1), and one that leads to activation of the transcription factor NF-κB (2), and subsequently to the inflammatory response. The overall result in a specific cell type is dependent on the balance of the two signals. Direct inhibition of NF-κB or of the upstream parts of its signaling pathway during TNF-α activation results in apoptosis in a variety of cell types originally resistant to TNF-α– induced apoptosis (3, 4). Furthermore, fibroblasts and macrophages from NF-κB subunit p65–deficient mice are more sensitive to TNF-α–induced apoptosis (5). Therefore, it has been proposed that activation of NF-κB induces the expression of genes that counteract apoptotic signals and prevent cell death.
Members of the inhibitor of apoptosis (iap) gene family have been demonstrated to suppress apoptosis induced by a variety of stimuli in different cell types (6–13, and for review see reference 14). The iap genes have also been shown to play a role in TNF-α–induced programmed cell death. Different iap gene family members appear to interfere with the cell death–triggering cascade at different levels. hiap1 and hiap2 can bind to the TNFR-associated factor 2 (TRAF2), a molecule that is associated with the cytoplasmic part of the TNFR complex and is essential for the activation of NF-κB (9, 15). Both have also been shown to be direct inhibitors of cell death proteases caspase 3 and caspase 7 (16). Another iap gene family member, the X-chromosome–linked iap (xiap), protects embryonic kidney 293T cells from bax-triggered apoptosis by inhibiting the same proteases, but in contrast it has not been found to be associated with members of the TRAF family (16, 17).
The studies presented here demonstrate that three human iap gene family members (xiap, hiap1, and hiap2) are strongly upregulated in TNF-α–stimulated primary ECs, which are resistant to TNF-α–induced apoptosis. However, adenovirus-mediated overexpression of IκBα (18, 19), an inhibitor of NF-κB, renders primary ECs sensitive to TNF-α–induced apoptosis and at the same time inhibits iap gene upregulation. Thus, iap gene expression appears to be dependent on NF-κB activation. Importantly, we show that ectopic expression of xiap is sufficient to overcome the IκBα effect in IκBα-overexpressing ECs and protects these cells from TNF-α–induced apoptosis.
Materials and Methods
Cell Culture
Cell culture flasks were coated with 1% gelatine for 30 min at 37°C. Human umbilical vein endothelial cells (HUVECs) and human skin microvascular endothelial cells (HSMECs) were grown in medium M199 supplemented with 20% bovine calf serum (HyClone, Logan, UT), endothelial cell growth factor supplement (Technoclone, Vienna, Austria), penicillin, streptomycin, fungizone, and heparin (3 U/ml). Confluent cells were split in a 1:3 ratio and used up to the sixth passage.
U937 cells were cultivated in RPMI-1640 medium supplemented with 10% FCS, l-glutamine, penicillin, and streptomycin. Cells were split 1:10 when grown to a density of 106 cells/ml.
Northern Analysis
Total RNA was isolated using Trizol reagent (GIBCO BRL, Gaithersburg, MD). 10 μg total RNA was separated on a 1.3% formaldehyde agarose gel. Samples were run in 0.02 M MOPS (3-[N-morpholino]propanesulfonic acid), pH 7.0, 5 mM sodium acetate, 1 mM EDTA. The gel was blotted overnight using 10× SSC onto a GeneScreen Plus nylon membrane (Dupont-NEN, Boston, MA), dried, and fixed by UV-light (UV-cross-linker 120.000 μJ; Stratagene Inc., La Jolla, CA). Membranes were hybridized with α-[32P]dATP-labeled (Terminal Transferase, Boehringer Mannheim, Mannheim, Germany) oligonucleotides specific to hiap1 (5′-agaaatgtttcagtggcattcaatcaacccaaagatgtaatgtgtgactcatgaagcttct-3′), hiap2 (5′-aagatttccaccacaaaaagaaatcaatgatagactcttatgtagaatttactacactttc -3′ ), xiap (5′-gaagggtggtgggtgggaaacaacacagctccctaggaagagcacaggatagtcacggggg-3′), and naip (5′-actgcatctaggcccagaagagcagacagctctggcagcaaattgtgatcaaactgggaga-3′) using Quickhyb-solution (Stratagene, Inc.) at 65°C. Membranes were washed twice for 15 min at room temperature in 1% SDS/3× SSC/20 mM sodium phosphate buffer, pH 7.2, and twice for 30 min at 65°C in 1% SDS/1× SSC. Signals were analyzed on a PhosphorImager SF (Molecular Dynamics, Sunnyvale, CA).
Adenovirus Construction and Infection
Adenovirus IκBα has been described previously (26), and construction of xiap adenovirus was done by firstly introducing a fragment encoding the myc peptide sequence MEQKLISEEDL into the adenovirus transfer vector pACCMVpLpASR+ (20). Subsequently, a 1,600-bp BamHI/XbaI cDNA fragment containing the entire coding region of human xiap was ligated and the construct was cotransfected together with pJM17, a plasmid containing the adenoviral genome with a deletion in the E1 region into 293 cells (21). Plaques appearing after 10 d of culture were subcloned on 293 cells and were tested for xiap expression on immunoblots using anti-myc mAb 9E10 (22). Purification of a large batch of the recombinant adenovirus was done by two consecutive cesium chloride centrifugations as previously described (23).
Postconfluent HSMECs and HUVECs were washed once with complete PBS and incubated at a multiplicity of infection of 100 with the respective adenovirus constructs in PBS. After 30 min at 37°C, the adenovirus was washed off and fresh medium was added. Cells were maintained for an additional 2 d before being assayed.
Analysis of DNA Fragmentation
Electrophoresis of Genomic DNA.
Cells were incubated for 3 h at 55°C (100 mM NaCl, 10 mM Tris Hcl, pH 8.0, 25 mM EDTA, 0.5% SDS, 0.47 mg/ml Proteinase K), and then incubated with 100 μg/ml RNAseA for 1 h at 37°C. After phenol– chloroform extraction and isopropanol precipitation, the DNA was dissolved in 50 μl Tris/EDTA and resolved on 1.3% agarose gel.
Quantification of Fragmented DNA.
For quantification of apoptosis fragmented DNA was determined by sandwich ELISA with antihistone coated microtiter plates and peroxidase-conjugated anti-DNA antibodies using the Cell Death Detection ELISA system from Boehringer Mannheim, according to the manufacturer's protocol.
Flow Cytometry
48 h after infection cells were treated with TNF-α (500 U/ml) for 6 h or left untreated. Cells were harvested, fixed in 70% ethanol, and the proportion of cells undergoing apoptosis was determined by flow cytometric analysis (FACSort®, Becton Dickinson, San Jose, CA) after staining with propidium iodide. Cells with a DNA content <2 N appear in the sub-G1 region (M1).
Results and Discussion
Using a modified differential screening technique to identify and clone genes regulated by inflammatory mediators in porcine aortic ECs (PAECs) (23a) we have obtained a porcine homologue (piap) of the human iap gene family. Initially identified as a TNF-α–inducible gene, piap was found also to respond to the inflammatory stimuli LPS and to a lesser degree to IL-1β. Subsequently, we have tested whether members of the human iap gene family (xiap [hILP, MIHA], hiap1 [ciap2, MIHC], and hiap2 [ciap1, MIHB]; references 6–12) show similar responses to inflammatory cytokines. Using oligonucleotides specific for the different iap genes, we performed Northern blot analysis of HSMECs (Fig. 1) and HUVECs (data not shown). We demonstrate that, apart from the neuronal inhibitor of apoptosis (naip) that is not expressed in ECs, the xiap, hiap1, and hiap2 genes were strongly upregulated in response to TNF-α in HSMECs and HUVECs.
Figure 1.

Northern blot analysis of iap gene expression in HSMECs. 10 μg of total RNA from either nontreated or TNF-α–treated (500 U/ml) HSMECs was loaded in each lane and hybridized to oligonucleotides specific to hiap1, hiap2, xiap, and naip. The predicted transcript size corresponds to the published one (7) for hiap1 (6.5 kb), hiap2 (4.5 kb), and xiap (9 kb). To confirm the equal loading of RNA, membranes were stripped and reprobed with GAPDH.
Treatment of HSMECs or HUVECs with TNF-α for up to 24 h did not lead to apoptosis, whereas the well- established TNF-α–sensitive monocytic cell line U937 became apoptotic under these experimental conditions. iap gene expression has been shown to inhibit apoptosis induced by a variety of apoptotic stimuli (12). Thus, we speculated that induced iap gene expression may prevent ECs from undergoing programmed cell death in response to TNF-α.
TNF-α is a proinflammatory cytokine whose pleiotropic biological effects are signaled through two distinct cell surface receptors, TNFR 1 and TNFR 2 (2). It is known to be a potent activator of NF-κB that has been shown to be the central mediator of gene regulation in the inflammatory response of activated ECs leading to leukocyte adhesion and thrombosis (24, 25). Therefore, we tested whether NF-κB was involved in upregulation of iap genes in response to inflammatory stimuli. Having shown previously that expression of IκBα from a recombinant adenovirus vector abolishes NF-κB-dependent upregulation of inflammatory genes such as IL-1β, IL-6, IL-8, and vascular cell adhesion molecule 1 in LPS-stimulated ECs (26), we used this adenovirus-IκBα construct to investigate whether NF-κB inhibition also impairs iap gene expression. HUVECs and HSMECs were infected with either a control adenovirus or the recombinant adenovirus IκBα (27). After 2 d, cells were stimulated with TNF-α for 4 h and probed for xiap, hiap1, and hiap2 expression. As shown in Fig. 2, the expression of all three iap genes tested in adenovirus IκBα– infected ECs was suppressed, indicating that the upregulation of iap genes is controlled by activation of NF-κB.
Figure 2.

Northern blot analysis of iap gene expression in adenovirus IκBα–infected ECs. HUVECs and HSMECs were not infected, were infected with a control adenovirus, or were infected with the recombinant adenovirus IκBα construct. Cells were either left untreated or treated with TNF-α (500 U/ ml) for 4 h. The membranes were probed with oligonucleotides specific to hiap1, hiap2, and xiap. Expression of IκBα was controlled by reprobing the membranes with an IκBα-cDNA. Equal loading was confirmed by hybridization with a GAPDH cDNA probe. Adv, adenovirus.
We then raised the question whether blocking the activation of NF-κB would actually sensitize ECs to TNF-α–induced apoptosis. Indeed, ECs infected with the recombinant adenovirus IκBα construct started to die ∼6 h after TNF-α stimulation. To demonstrate that the apoptotic program is involved in cell death, genomic DNA was isolated from dying cells. As shown in Fig. 3, genomic DNA from IκBα-expressing and TNF-α–treated cells, but not from control virus–infected or nontreated cells, showed the DNA fragmentation pattern characteristic for apoptosis. Thus, inhibition of NF-κB activation renders ECs TNF-α sensitive, indicating that induction of apoptosis in ECs can occur independent of NF-κB.
Figure 3.

DNA fragmentation in adenovirus IκBα–infected and TNF-α–stimulated HUVECs. HUVECs were not infected, were infected, with a control adenovirus, or were infected with the recombinant adenovirus-IκBα construct. Noninfected cells and infected cells were left untreated or treated with TNF-α (500 U/ml) for 6 h. Appearance of fragmented genomic DNA was analysed by 1.3% agarose gel electrophoresis. Left lane: 1-kb ladder molecular weight standard; right lane: 123-bp ladder molecular weight standard. Non-inf.: noninfected cells; AdV: adenovirus.
These data suggested that TNF-α–induced expression of iap genes could be required to protect ECs from undergoing apoptosis. To directly demonstrate the ability of iap genes to prevent ECs from TNF-α–induced apoptosis, we coinfected HUVECs with recombinant adenovirus constructs expressing myc-tagged xiap and IκBα, respectively. Infection with recombinant adenovirus IκBα alone and stimulation with TNF-α–induced apoptosis in HUVECs (Fig. 4 B, c and d). Coexpression of xiap and IκBα (Fig. 4 B, f ) reduced the percentage of apoptotic cells to background levels obtained in TNF-α–treated or nontreated HUVECs (Fig. 4 B, a and b). A recombinant adenovirus expressing green fluorescent protein (27) was used as a control to show that adenovirus infection itself had no influence on apoptosis induced by TNF-α in IκBα-overexpressing cells (Fig. 4 B, g and h). Expression of myc-tagged xiap in infected HUVECs was demonstrated by Western blots stained with anti-myc mAb (Fig. 4 A).
Figure 4.
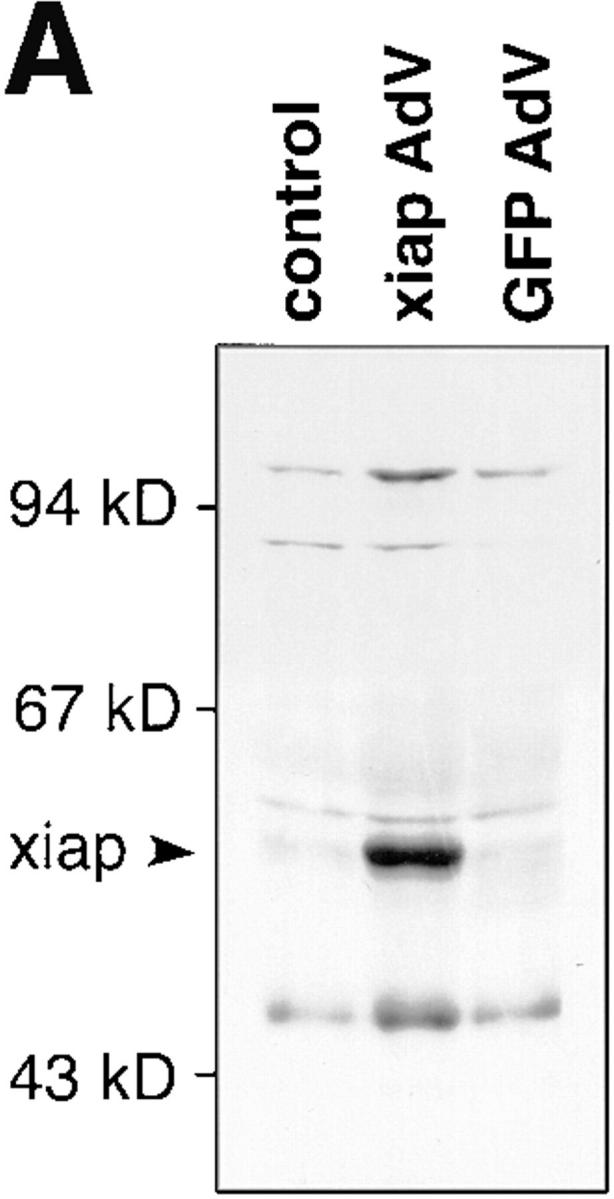


Inhibition of apoptosis by ectopic xiap expression. (A) Lysates of noninfected or infected HUVECs were separated by SDS-PAGE, blotted onto nylon membranes, and stained for myc-tagged XIAP protein. AdV, adenovirus; GFP, green fluorescent protein. (B) HUVECs were infected with IκBα alone (c and d), together with xiap (e and f ), or together with GFP (g and h) recombinant adenovirus. 48 h after infection cells were treated with TNF-α (500 U/ml) for 6 h or left untreated and analyzed by FACS® after propidium iodide staining. Cells with a DNA content <2 N appear in the sub-G1 region (M1). The percentage of cells found in the M1 region is indicated. The data show one out of three representative experiments.
Since the monocytic cell line U937 is sensitive to TNF-α–induced apoptosis when compared to primary ECs, we analyzed whether this cell line also differs with respect to TNF-α–inducible upregulation of iap genes. U937 and HUVECs were treated with TNF-α for 4, 6, and 9 h. At the same time points we monitored and quantified apoptosis by analysis of fragmented genomic DNA using an ELISA assay for histone-associated DNA fragments. Fig. 5 C shows that xiap gene was barely expressed in nontreated U937 cells and expression could not be induced by TNF-α. Consistently, U937 cells became significantly apoptotic after 4 h (Fig. 5 D). In contrast, as shown in Fig. 5, A and B, xiap was upregulated in HUVECs and no increase in fragmented DNA could be assayed in response to TNF-α. Identical results were obtained for hiap1 and hiap2 in U937 cells and HSMECs (data not shown).
Figure 5.


Lack of TNF-α–inducible xiap gene expression correlates with apoptosis in U937 cells. Northern blot analysis of xiap gene expression in HUVECs (A) and U937 cells (C). HUVECs and U937 cells were treated for 4, 6, and 9 h with TNF-α. To confirm equal loading of RNA, membranes were stripped and reprobed with GAPDH. TNF-α–induced genomic DNA fragments from HUVECs (B) and U937 cells (D) were determined by colorimetric enzyme immunoassay. Columns represent the mean of three independent experiments. SD is indicated by error bars. TNF-α (500 U/ml); c, nontreated cells.
Our findings provide several lines of evidence that the iap gene products are regulated by NF-κB and that xiap appears to be sufficient to protect primary ECs from undergoing apoptosis in response to TNF-α: (a) iap genes are expressed in response to TNF-α, IL-1β, and LPS, respectively; (b) inhibition of NF-κB activation suppresses inducible iap gene expression; (c) inhibition of NF-κB activation by overexpressing its inhibitor IκBα renders ECs sensitive to TNF-α–induced apoptosis; and (d) ectopic expression of xiap in IκBα-overexpressing ECs overrules the IκBα/ TNF-α effect.
These data show that ECs and presumably other cells have developed cellular mechanisms that protect them from apoptosis and keep them able to function properly in an inflammatory situation. Fast activation of NF-κB in response to proinflammatory signals, like TNF-α, would be an appropriate mechanism to ensure the prompt expression of antiapoptotic gene(s). This hypothesis is supported by the demonstration that NF-κB p65 is necessary to protect fibroblasts from TNF-α–induced apoptosis (5).
Whether under physiological circumstances the expression of xiap is sufficient or whether simultanous expression of all three iap genes (or other genes such as A20 [28], manganese superoxide dismutase [29], plasminogen activator– inhibitor type 2 [30], A1 [31], or other as yet undefined genes) is required to protect ECs from TNF-α–induced apoptosis remains open. Chu et al. (32) have shown recently that hiap1 expression is dependent on activation of NF-κB in Jurkat cells and hiap1 protein is able to protect these cells from apoptosis. However, in contrast to primary ECs, hiap2 showed a steady state level of expression in Jurkat cells and was not controlled by NF-κB. The data indicate that expression of the iap gene family members and their involvement in protection from apoptosis varies in certain cell types and follows a rather complex scheme. iap gene expression appears to be specific for the cell type and the given stimulus. This view is supported by our finding that iap gene expression seems to be not involved in the TNF-α response of the monocytic cell line U937. These cells become partially apoptotic upon TNF-α treatment but do not express iap genes, suggesting that other protective mechanisms are operative. Recent reports demonstrated that hiap1/2 can interfere at different levels with the apoptotic program. hiap1 and hiap2 associate via TRAF 2 with the TNFR 2, leading to NF-κB activation (9), and hiap2 is also part of the TNFR 1 signaling complex (15). On the other hand, hiap1 and hiap2 as well as xiap directly inhibit caspase 3 and caspase 7 activity, two members of the caspase family of cell death proteases, in embryonic kidney 293T cells (16, 17). However, inhibition by xiap is two to three orders of magnitude more potent, suggesting xiap as the physiological inhibitor of caspase 3 and 7 (16). These data and our finding that xiap expression is sufficient to prevent TNF-α–induced apoptosis in ECs support the concept that xiap plays a central role in inhibition of programmed cell death. It remains to be established whether xiap operates via an identical mechanism in ECs as in 293T cells and which other cell-type specific and stimulus-dependent mechanisms exist.
Unexpectedly, iap gene expression is also induced by LPS and IL-1β. Pretreatment of a human fibrosarcoma line (HT1080V) with the nonapoptotic, NF-κB–inducing IL-1 protects these cells from apoptosis induced by the later addition of TNF-α even in the presence of a protein synthesis inhibitor (3). In cells expressing a super-repressor form of the NF-κB inhibitor IκBα, IL-1β does not have this protective effect, suggesting that IL-1β also induces the expression of NF-κB–regulated antiapoptotic genes. A mechanism to overrule apoptotic signals during inflammation would enable ECs to respond properly by upregulation of inflammatory mediators such as tissue factor and adhesion molecules and at the same time to survive inflammation in order to maintain homeostasis of the inflamed tissue and initiate the healing process.
Acknowledgments
We thank Drs. R. Kroismayr and T. Ebel for critical reading of the manuscript, and C. Kaun for providing HSMECs and HUVECs.
This work was supported by grants to J. Lipp from the Austrian Science Foundation, project SFB 005-05, and from the Jubilaeumsfonds of the Austrian National Bank, project 5548.
Footnotes
Ichiro Kumabashiri's current address is Department of Internal Medicine, Keiju General Hospital, Tomiokamachi 94, Nanao, Japan.
References
- 1.Hale AJ, Smith CA, Sutherland LC, Stoneman VEA, Longthorne VL, Culhane AC, Williams GT. Apoptosis: molecular regulation of cell death. Eur J Biochem. 1996;236:1–26. doi: 10.1111/j.1432-1033.1996.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Barinaga M. Forging a path to cell death. Science. 1996;273:735–737. doi: 10.1126/science.273.5276.735. [DOI] [PubMed] [Google Scholar]
- 3.Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy–induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 4.Van Antwerpen DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α–induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 6.Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67:2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birnbaum MJ, Clem RJ, Miller LK. An apoptosis inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol. 1994;68:2521–2528. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 9.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 10.Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner-Johnston A, Lefebvre C, Kang X, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80:167–178. doi: 10.1016/0092-8674(95)90461-1. [DOI] [PubMed] [Google Scholar]
- 11.Duckett CS, Nava VE, Gedrich RW, Clem RJ, Van Dongen JL, Gilfillan MC, Shiels H, Hardwick JM, Thompson CB. A conserved family of cellular genes related to the baculovirus iapgene and encoding apoptosis inhibitors. EMBO (Eur Mol Biol Organ) J. 1996;15:2685–2694. [PMC free article] [PubMed] [Google Scholar]
- 12.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE, MacKenzie A, Korneluk RG. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 13.Uren AG, Pakusch M, Hawkins CJ, Puls KL, Vaux DL. Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor–associated factors. Proc Natl Acad Sci USA. 1996;93:4974–4978. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clem RJ, Duckett CS. The iap genes: unique arbitrators of cell death. Trends Cell Biol. 1997;7:337–339. doi: 10.1016/S0962-8924(97)01088-X. [DOI] [PubMed] [Google Scholar]
- 15.Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci USA. 1996;93:13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP and c-IAP proteins are direct inhibitors of specific caspases. EMBO (Eur Mol Biol Organ) J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 18.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 20.Gomex-Foix AM, Coats WS, Baques S, Alam T, Gerard RD, Newgard CB. Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J Biol Chem. 1992;267:25129–25134. [PubMed] [Google Scholar]
- 21.McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 22.Evan GI, Lewis KG, Ramsay G, Bishop M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham FL, Van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 23a.Stehlik C, de Martin M, Binder BR, Lipp J. Cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NF-κB. Biochem Biophys Res Commun. 1998;243:827–832. doi: 10.1006/bbrc.1998.8185. [DOI] [PubMed] [Google Scholar]
- 24.Collins T. Biology of disease: endothelial nuclear factor-κB and the initiation of the atherosclerotic lesion. Lab Invest. 1993;68:499–508. [PubMed] [Google Scholar]
- 25.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T .Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB (Fed Am Soc Exp Biol) J. 1995;9:899–909. [PubMed] [Google Scholar]
- 26.Wrighton CJ, Hofer-Warbinek R, Moll T, Eytner R, Bach FH, de Martin R. Inhibition of endothelial cell activation by adenovirus-mediated expression of IκBα, an inhibitor of the transcription factor NF-κB. J Exp Med. 1996;183:1013–1022. doi: 10.1084/jem.183.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Martin R, Raidl M, Hofer E, Binder BR. Adenovirus-mediated expression of green fluorescent protein. Gene Ther. 1997;4:493–495. doi: 10.1038/sj.gt.3300408. [DOI] [PubMed] [Google Scholar]
- 28.Opipari AW, Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424–12427. [PubMed] [Google Scholar]
- 29.Wong GHW, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923–931. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Baglioni C. Protection from tumor necrosis factor–mediated cytolysis by overexpression of plasminogen activator inhibitor type-2. J Biol Chem. 1991;266:20960–20964. [PubMed] [Google Scholar]
- 31.Karsan A, Yee E, Kaushansky K, Harlan JM. Cloning of a human bcl-2 homologue: inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996;87:3089–3096. [PubMed] [Google Scholar]
- 32.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor– induced cell death by inhibitor of apoptosis c-iap2 is under NF-κB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]