

Abstract
The complex pathophysiology of lung allergic inflammation and bronchial hyperresponsiveness (BHR) that characterize asthma is achieved by the regulated accumulation and activation of different leukocyte subsets in the lung. The development and maintenance of these processes correlate with the coordinated production of chemokines. Here, we have assessed the role that different chemokines play in lung allergic inflammation and BHR by blocking their activities in vivo. Our results show that blockage of each one of these chemokines reduces both lung leukocyte infiltration and BHR in a substantially different way. Thus, eotaxin neutralization reduces specifically BHR and lung eosinophilia transiently after each antigen exposure. Monocyte chemoattractant protein (MCP)-5 neutralization abolishes BHR not by affecting the accumulation of inflammatory leukocytes in the airways, but rather by altering the trafficking of the eosinophils and other leukocytes through the lung interstitium. Neutralization of RANTES (regulated upon activation, normal T cell expressed and secreted) receptor(s) with a receptor antagonist decreases significantly lymphocyte and eosinophil infiltration as well as mRNA expression of eotaxin and RANTES. In contrast, neutralization of one of the ligands for RANTES receptors, macrophage-inflammatory protein 1α, reduces only slightly lung eosinophilia and BHR. Finally, MCP-1 neutralization diminishes drastically BHR and inflammation, and this correlates with a pronounced decrease in monocyte- and lymphocyte-derived inflammatory mediators. These results suggest that different chemokines activate different cellular and molecular pathways that in a coordinated fashion contribute to the complex pathophysiology of asthma, and that their individual blockage results in intervention at different levels of these processes.
Keywords: chemokines, allergic inflammation, bronchial hyperresponsiveness, eosinophilia, leukocytes
Lung inflammation and bronchial hyperreactivity (BHR)1 are two distinct characteristics of asthma (1, 2). Eosinophils are considered to be the central proinflammatory leukocyte involved in the asthmatic reaction, due in part to secreted toxic granular proteins and membrane products that induce pulmonary damage and subsequently intensify BHR (1–3). Moreover, there is a correlation between levels of eosinophil-derived cation proteins in the airways of asthmatic patients and the severity of this disease (2). Despite the fact that the eosinophil is the predominant infiltrating cell type during asthma, other leukocytes may be critical in the initiation and amplification of the inflammatory response (4, 5). The prevention of both lung eosinophilia and BHR in T lymphocyte–deficient mice during antigen-induced inflammation supports a critical role for T cell cytokines such as IL-4 and IL-5 (6–9). The role that monocytes/macrophages play in lung allergic inflammation pertains to their ability to act as APCs and to generate a variety of mediators that promote both eosinophil activation and adhesion to endothelium (10). In this regard, macrophages secrete eosinophilic chemoattractants, including leukotriene (LT)B4, platelet-activating factor, and C5a, in addition to modulating the generation of some of these factors by eosinophils (10–12).
The immunological characterization of lung eosinophilia implicates chemokines in the initiation of lung allergic inflammation and the subsequent development of BHR (5, 13–17). Chemokines are a group of cytokines that promote leukocyte recruitment to inflammatory sites, stimulate leukocyte exocytosis, and induce hematopoiesis (18–20). Chemokine expression is high and readily modulated in the lung during the development of an inflammatory response (5, 16, 17). Among these, RANTES (regulated upon activation, normal T cell expressed and secreted), monocyte chemoattractant protein (MCP)-5, and eotaxin have been shown to induce eosinophil migration in vitro and/or to be involved in lung eosinophilia in humans, guinea pigs, and mice (5, 16, 13, 21–23). In addition to their activity on eosinophils, RANTES and MCP-5 are strong chemotactic factors both in vivo and in vitro for T lymphocytes and monocytes, respectively (16, 24). MCP-1, which is primarily involved in the recruitment of mononuclear phagocytes (25), is also strongly expressed in the lung during inflammation (5, 26). Signaling mediated by this noneosinophilic chemokine might also be involved in macrophage activation leading to a cascade of proinflammatory and tissue damage events (27).
In this report, we have used a mouse model of asthma in which the expression of the chemokines eotaxin, RANTES, macrophage-inflammatory protein (MIP)-1α, MCP-5, and MCP-1 is clearly modulated during disease progression (5). To understand the pathophysiology that results from the expression of multiple chemokines with partially overlapping functions, we have analyzed (a) the direct recruitment of specific cell types to the lung interstitium and airways exerted by these chemokines, (b) their role in the induction of BHR, (c) the effect they exert on the gene expression of other chemokines, and (d) the modulation by these chemokines of different components of the pathological response, such as cell activation or inflammatory mediator production.
Materials and Methods
Mice and In Vivo Procedures.
8–10-wk-old C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and kept in the specific pathogen–free mouse facility at Millennium Pharmaceuticals, Inc. The mouse model of lung inflammation used here consists of a sensitization phase (ovalbumin [OVA], 0.1 mg/mouse intraperitoneally on day 0; Sigma Chemical Co., St. Louis, MO) and an induction of the response phase (2% OVA for 5 min intranasally on day 8, and 1% OVA for 20 min intranasally on days 15–21). PBS (intraperitoneal and/or intranasal) was administered to mice as a negative control. For the blocking experiments, mice also received 20 μg/mouse of neutralizing polyclonal Abs against either eotaxin (anti-Eot), MCP-5 (anti–MCP-5), MIP-1α (anti–MIP-1α), or 5 μg/mouse of mAb against MCP-1/JE (anti–MCP-1/JE), or 25 μg/mouse of the RANTES antagonist, methionylated (Met)-RANTES. Anti– MIP-1α (R&D Systems, Inc., Minneapolis, MN) was selected for its ability to neutralize the in vitro bioactivity of recombinant murine MIP-1α and was used at the manufacturer's recommended dose. Anti-Eot Ab blocked the in vitro transmigration of eosinophils to eotaxin by 97% (5). Similarly, anti–MCP-5 Ab blocked MCP-5–induced eosinophil migration in vitro by 99% (16). The anti–MCP-1/JE Ab also showed a dose-dependent inhibition of MCP-1 bioactivity using in vitro chemotactic assays on monocytes (28). We have determined previously that the dose of antibodies used here blocked 100% of migration to each particular chemokine during in vivo peritoneal migration assays (data not shown) or during crescentic nephritis (29). Met-RANTES has been shown to inhibit both in vitro chemotaxis and calcium influx on the THP-1 cell line and T cells in response to RANTES and MIP-1α (30) and in vivo inflammation (29). These antibodies were administered intravenously 30 min before OVA provocation on days 8–21. In another series of blocking experiments, anti–MCP-1/JE mAb, anti-Eot, or anti–MCP-5 polyclonal Abs were also administered on days 8–16, days 15–21, days 19–21, or day 21. OVA-treated control mice were injected with the same amount of control Ab at the same time points indicated during treatment. Rabbit Ig fraction (DAKO Corp., Santa Barbara, CA) was used as control for poly-Eot and poly–MCP-5, normal hamster IgG (Jackson ImmunoResearch Labs, West Grove, PA) was used as control for anti–MCP-1/JE, and PBS as control for Met-RANTES. 1 or 3 h after OVA administration on day 15 or 21, mice were killed by CO2 asphyxiation and analyzed.
Bronchoalveolar lavage (BAL) was performed as described (23). In brief, the airways of the mice were lavaged via a trachea cannula with 1 ml of PBS. The resulting BAL fluid was immediately centrifuged (700 g, 5 min, 4°C), and BAL cells were then washed and resuspended in 1 ml of PBS.
The degree of bronchoconstriction (BHR) was measured 3 h after the last antigen challenge by recording respiratory pressure curves by whole body plethysmography (Buxco Electronics, Inc., Sharon, CT) in response to inhaled methacholine (MCh; Aldrich Chemical Co., Milwaukee, WI) at a concentration of 3 × 10−2 M for 1 min, as described previously (3, 31). BHR was expressed as enhanced pause (Penh), a calculated value, which correlates with measurement of airway resistance, impedance, and intrapleural pressure in the same mouse. Penh = (Te/Tr − 1) × (Pef/Pif), where Te is expiration time; Tr, relaxation time; Pef, peak expiratory flow; and Pif, peak inspiratory flow × 0.67 coefficient (32). The relaxation time is the time it takes for the box pressure to change from a maximum to a user-defined percentage of the maximum. Here, Tr measurement begins at the maximum box pressure and ends at 40%.
Immunohistochemical Phenotyping and Quantitation of Leukocytes.
Total BAL cell counts were performed, and aliquots (5 × 105 cells/slide) were pelleted onto glass slides by cytocentrifugation. To determine the number of eosinophils and neutrophils, slides were stained with Wright-Giemsa (Fisher Scientific Co., Pittsburgh, PA). T lymphocytes, B lymphocytes, and mononuclear phagocytes were identified by Thy 1.2 (53-2.1; PharMingen, San Diego, CA), IgM (II/41; PharMingen), and Moma-2 (Biosource International, Camarillo, CA) staining, respectively, as described (5). Percentage of eosinophils, lymphocytes, neutrophils, and macrophages was determined by counting their number in eight high power fields (×40, total area 0.5 mm2) per area randomly selected, and dividing this number by the total number of cells per high power field. To obtain the absolute number of each leukocyte subtype in the lavage, these percentages were multiplied by the total number of cells recovered from the BAL fluid.
Lung sections from the different experimental groups of mice were prepared as described (5). In brief, lungs were fixed in 10% neutral buffered formalin (NBF; J.T. Baker, Phillipsburg, NJ) and paraffin embedded. Sections (4 microns) were cut onto microscope slides and stained with hematoxylin/eosin according to standard protocols. To determine the sizes of pulmonary infiltrates in the experimental groups of mice, the area of lung tissue covered by infiltrate was calculated in sections at low power (×100) using NIH Image 1.56. At least six fields of at least 0.01 mm2 were scanned, and the mean percentage area was determined for each mouse of each experimental group, relative to the control mice for each experiment.
Determination of Chemokine Protein Expression within Lung Tissue.
The level of expression of eotaxin protein and MCP-5 protein was determined in sections from lungs of OVA-treated mice and controls. Sections were fixed and stained using a modified avidin/ biotin staining method. All incubations were carried out under humidified conditions, and slides were washed twice between steps for 5 min each in 0.1 M PBS supplemented with 0.2% gelatin. Sections were overlaid with 20% normal rabbit serum in PBS for 15 min and then incubated overnight at 4°C with either monoclonal anti-Eot diluted 1:2 in PBS with 0.1% BSA and 0.1% sodium azide (5) or anti-murine MCP-5 (ZY2A11) culture supernatant (undiluted). Endogenous peroxide was subsequently blocked by incubation for 20 min in methanol containing 0.3% hydrogen peroxide. Nonspecific staining due to cross-reaction with endogenous avidin or biotin was blocked by incubation with avidin solution followed by biotin solution, both for 20 min. Bound mAb was visualized by incubation with biotinylated rabbit anti–rat Ig diluted in 10% normal mouse serum PBS, and then incubated for 1 h each in streptavidin–peroxidase complex prepared according to the manufacturer's instructions (both from DAKO Corp.). Finally, slides were flooded with peroxidase substrate solution (20 mg diaminobenzidine in 10 ml PBS, containing 0.01% hydrogen peroxide) for 10 min before counterstaining with hematoxylin. Control slides were either stained with an isotype-matched negative control Ab instead of primary Ab or biotinylated anti–rat Ig or streptavidin complex were selectively omitted.
Measurement of Chemokine mRNA Expression.
Total RNA from the lungs of OVA-treated mice or control littermates at different time points was extracted by the single step method using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX).
Chemokine mRNA expression was determined by multiprobe RNase protection assay (RPA) using the RiboQuant RPA kit (PharMingen) as recommended by the supplier. The identity and quantity of each mRNA species in the original RNA sample were then determined based on the signal intensities given by the appropriately sized, protected probe fragment bands. The sample loading was normalized by the housekeeping gene, GAPDH, included in each template set.
Measurement of Cytokine, Inflammatory Mediator, and Ab Levels.
The release of these activating factors during OVA administration was determined by ELISA. BAL fluid was taken 1 h after antigen challenge on day 15 or 21. Serial dilutions of BAL fluid samples were assayed using commercial ELISA kits for IL-4, IL-5, TNF-α, and IFN-γ (Endogen, Inc., Boston, MA) and commercial enzyme immunoassay kits for LTB4, PGE2, thromboxane (TX)B2, and LTC4/D4/E4 (Amersham International, Buckinghamshire, UK). Total IgE titers were also measured by ELISA according to Ledermann et al. (33). Absorbance values were converted to concentrations of each factor in the BAL fluid (picograms or nanograms per milliliter) by interpolation in the respective standard curve.
Results
Neutralization of Specific Chemokines Expressed during OVA-induced Lung Allergic Inflammation.
Mice treated with OVA (see Materials and Methods; Fig. 1 A, bottom) show maximal lung monocyte/macrophage accumulation at early stages of the inflammatory response (3 h after OVA challenge on day 15), whereas eosinophil and T lymphocyte numbers increase in this organ at late stages of the response (3 h after OVA challenge on day 21; Fig. 1 A). The highest mRNA expression of MCP-1 (day 15, 3 h), as well as eotaxin and RANTES (day 21, 3 h) and MCP-5 and MIP-1α (day 15–21, 3 h), coincides with the kinetics followed by the infiltrating leukocytes described above (Fig. 1 A; reference 5). No MIP-1β mRNA expression was detected during OVA treatment (Fig. 1 A). Fig. 1 B shows that the production of both eotaxin and MCP-5 protein in response to OVA correlates with the mRNA expression pattern shown by these two chemokines, at least at the time points analyzed (days 15 and 21). In addition, 24 h after OVA administration on day 21, eotaxin and MCP-5 protein expression is clearly diminished compared with that detected 3 h after antigen challenge on the same day (data not shown). This suggests that the kinetics of chemokine mRNA expression parallels that observed, at least, for eotaxin and MCP-5 protein expression.
Figure 1.

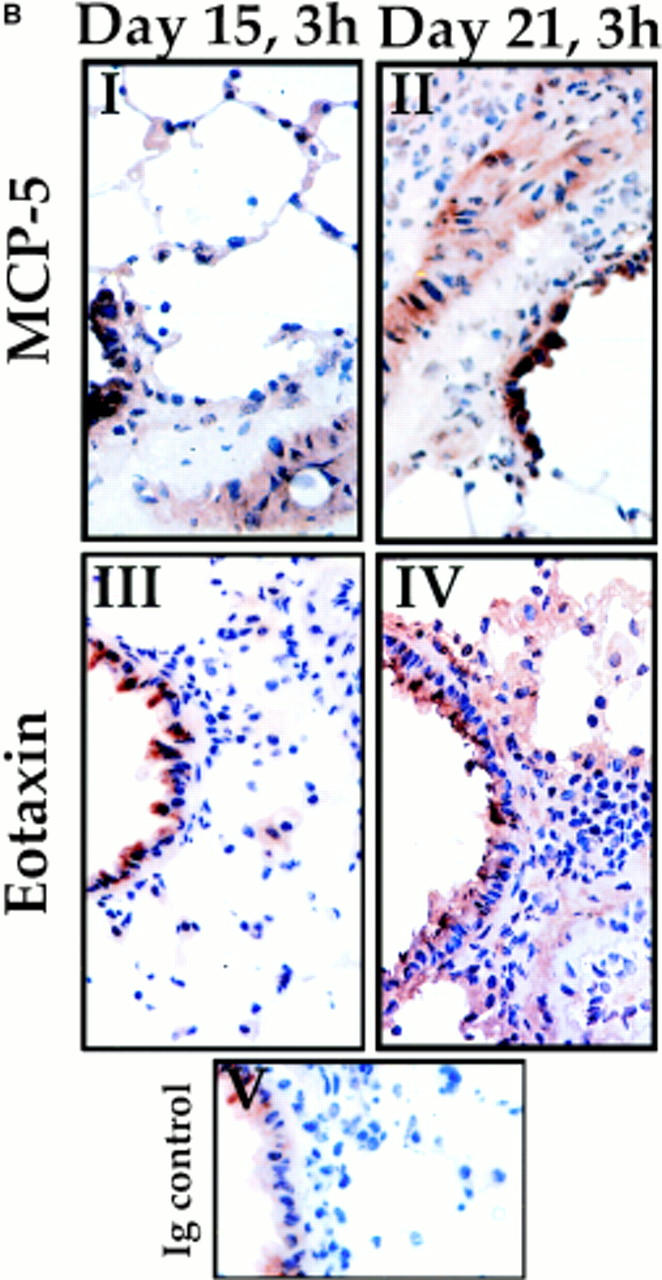
Chemokine expression and leukocyte infiltration in the lung of OVA-treated mice. (A) Eotaxin, RANTES, MCP-5, MCP-1, MIP-1α, and MIP-1β mRNA expression (gray bars) in the lung of each mouse were normalized to the 28S rRNA expression in the same organ of the same mice. The quantitation of total RNA was determined by hybridization of a 28S rRNAcDNA probe. Each bar represents data from five mice at the time points indicated (3 h after OVA challenge on days 15, 18, and 21) during treatment (bottom). Values are expressed as the mean ± SEM. The accumulation in the lung of BAL monocytes/macrophages (M/m), T lymphocytes (L), and eosinophils (E) during OVA treatment (3 h after antigen challenge on days 15, 18, and 21) is also shown. Although only BAL accumulation is presented here, there is correlation between these data and the accumulation in the interstitium (reference 23). Five data points at the different times indicated were collected per day as described previously (reference 23). Values are expressed as the mean ± SEM. i.n., intranasal. (B) Immunohistochemical staining of lung sections from OVA-treated mice on day 15 (3 h; I and III) or day 21 (3 h; II and IV) after OVA challenge. Sections were stained with anti–MCP-5 mAb (I and II; see Materials and Methods) or anti-Eot mAb (III and IV; see Materials and Methods). Note that positive staining (brown precipitate) is comparable between days 15 and 21 for MCP-5, but that there is clearly less eotaxin staining on day 15 compared with day 21. Ig-stained control is also shown (V).
To evaluate the specific contribution of eotaxin, an eosinophilic chemokine (13, 22, 23, 34), MCP-5, a monocyte and eosinophil chemokine (16), MIP-1α, a monocyte, lymphocyte, and eosinophil chemokine (14, 21, 35), and MCP-1, a monocyte chemokine (25, 36), to the development of lung inflammation in this OVA model, blocking experiments of these chemokines were performed using specific neutralizing Abs that have been characterized extensively in vivo and in vitro (references 5, 16, 28, and 29; and see Materials and Methods for details). Neutralization of chemokine receptors that are activated by RANTES, a T lymphocyte and eosinophil chemokine (21), and other ligands was achieved using the antagonist Met-RANTES (see Materials and Methods; reference 30). Neutralizing agents were delivered daily from days 8 to 21 in these first series of experiments, and analysis was always performed 3 h after OVA challenge on day 21. Since the location of infiltrating cells within the lung correlates strongly with the severity of the inflammatory response (3), leukocyte enumeration was performed in the airways (BAL fluid) and in the interstitium (lung sections) after OVA treatment.
Eotaxin neutralization during OVA treatment specifically affected eosinophils both in BAL fluid and pulmonary interstitium. This effect was more pronounced on day 21 (Fig. 2, and Tables 1 and 2). MCP-5 neutralization during OVA treatment strongly affected accumulation of BAL monocytes and eosinophils on day 15, but not on day 21 (Fig. 2, and Table 1). Interestingly, minimal OVA-induced leukocyte accumulation was detected in the lung interstitium at any time point after MCP-5 blockage (Table 2, and Fig. 3). Met-RANTES administration reduced OVA- induced monocyte accumulation in the BAL by half on day 21 (Table 1) and abrogated BAL lymphocyte and eosinophil accumulation during OVA treatment (Fig. 2, and Table 1). There was a corresponding decrease in inflammation in the pulmonary interstitium after Met-RANTES administration on day 21 (Table 2). MIP-1α neutralization reduced eosinophil recruitment into the airways and pulmonary interstitium by 20% only at late stages of the inflammatory response, but without affecting monocyte, macrophage, and lymphocyte numbers (day 21, Fig. 2 A, and Tables 1 and 2).
Figure 2.


BAL eosinophilia after blockage of chemokines during OVA-induced lung allergic inflammation. Chemokine blockage was performed daily before OVA provocation on days (d) 8–21. BAL eosinophil accumulation was evaluated 3 h after OVA administration on day 15 (A) or on day 21 (B). MCP-1, eotaxin, or MCP-5 blockage was also performed after each OVA intranasal challenge for the periods of time indicated on the x-axis, and exclusively analyzed on day 21 (C). Each circle represents a single PBS- or OVA-treated control mouse (open circle) or a single test mouse (filled circle). Bars represent the mean of each group. Significant difference between control and test groups of mice was determined using the Student's t test (P <0.001). Quantitative data is shown in Table 1 for this and other leukocyte subsets.
Table 1.
OVA-induced Leukocyte Infiltration in the Airways after Chemokine Blockage
| Chemokine blockage | Leukocyte subtype | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Macrophages | Monocytes | T lymphocytes | Eosinophils | |||||||||||||
| Day 15 | Day 21 | Day 15 | Day 21 | Day 15 | Day 21 | Day 15 | Day 21 | |||||||||
| Control (total | (180 ± 31) × 103 | (122 ± 12) × 103 | (38 ± 2) × 103 | (25 ± 3) × 103 | (27 ± 5) × 103 | (130 ± 22) × 103 | (80 ± 17) × 103 | (870 ± 92) × 103 | ||||||||
| numbers) | ||||||||||||||||
| αEotaxin | 0 | 17 ± 4 | 0 | 7 ± 1 | 9 ± 2 | 21 ± 4 | 27 ± 3 | 60 ± 7 | ||||||||
| αMCP-5 | 12 ± 3 | 11 ± 2 | 66 ± 8 | 11 ± 3 | 22 ± 5 | 0 | 76 ± 3 | 21 ± 8 | ||||||||
| Met-RANTES | 9 ± 1 | 17 ± 1 | 26 ± 4 | 44 ± 8 | 80 ± 2 | 95 ± 4 | 60 ± 10 | 91 ± 2 | ||||||||
| αMIP-1α | 0 | 2 ± 0.3 | 0 | 0 | 0 | 0 | 0 | 23 ± 2 | ||||||||
| αMCP-1 | 45 ± 10 | 58 ± 3 | 78 ± 11 | 53 ± 5 | 91 ± 3 | 89 ± 6 | 86 ± 8 | 78 ± 12 | ||||||||
Percentage of reduction in the number of leukocytes in the BAL fluid of OVA-treated mice after chemokine blockage. 30 min before each intranasal antigen administration, eotaxin, MCP-5, RANTES, or MCP-1 was neutralized as described in Materials and Methods. BAL fluid was obtained 3 h after OVA treatment on day 15 or 21, and the number of the different cell types was determined. Total numbers of macrophages, monocytes, T lymphocytes, and eosinophils present in the BAL fluid of OVA plus Ab control–treated mice are shown. Values are given as percentages referred to the OVA plus Ab control–treated mice, which are considered to be 100%.
Table 2.
OVA-induced Leukocyte Infiltration in the Lung Interstitium after Chemokine Blockage
| Chemokine blockage | Percent decrease in area covered by infiltrate | |
|---|---|---|
| Control | 0 | |
| αEotaxin | 90 ± 6 | |
| αMCP-5 | 75 ± 9 | |
| Met-RANTES | 73 ± 13 | |
| MIP-1α | 21 ± 5 | |
| αMCP-1 | 97 ± 23 |
Lung sections were obtained 3 h after OVA treatment on day 21 (Control), and the area covered by infiltrate was measured as described in Materials and Methods. Values after each chemokine blockage are given as percent decrease from control mice.
Figure 3.
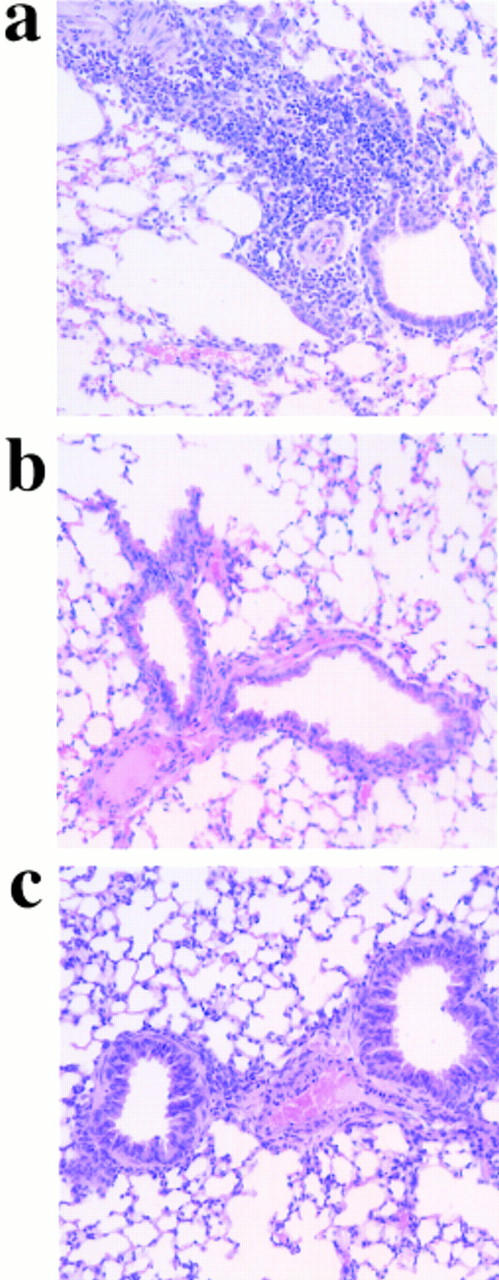
OVA-induced lung inflammation after chemokine blockage. On day 21, lung tissue was excised from OVA-treated mice injected with control (a), anti–MCP-1 (b), or anti–MCP-5 (c) antibodies. Chemokine blockage was performed daily before OVA provocation on days 8–21. One representative mouse out of ten is shown. Quantitative data corresponding to this set of experiments is shown in Table 2.
Neutralization of MCP-1 before each intranasal OVA challenge from days 8 to 21 reduced all four cell populations that migrate to the lung in response to OVA (Tables 1 and 2, and Fig. 3). Mononuclear phagocyte and eosinophil accumulation in the lung was reduced by 55 and 80%, respectively, after MCP-1 blockage (Fig. 2, and Table 1). OVA-induced pulmonary T lymphocyte accumulation during the inflammatory response was virtually abolished in the absence of MCP-1–mediated signals (Table 1).
To dissect the windows of action of the chemokines, blockage experiments were performed at different time points during OVA treatment, and mice were always analyzed 3 h after OVA challenge on day 21. Thus, when the anti–MCP-1 Abs were administered from days 15 to 21, only a small reduction in the numbers of infiltrating BAL leukocytes was detected (Fig. 2 C), suggesting a critical role of MCP-1 at early stages of the inflammatory response. However, blockage of MCP-1 neither at early stages (days 8–16) nor at late stages of the inflammatory response (days 19–21) is sufficient to decrease significantly BAL leukocyte numbers (Fig. 2 C). No differences in BAL cell numbers on day 21 were detected after eotaxin blockage for shorter periods (days 19–21 or day 21) compared with day 8–day 21 blockage (Fig. 2 C). However, when the anti-Eot Abs were administered on days 8–20 but not on day 21, no reduction in BAL eosinophilia was observed (Fig. 2 C). Signals delivered by MCP-1 during the whole process (days 8–21) are necessary to achieve OVA-induced lung inflammation, whereas eotaxin contributes to the development of eosinophilia by inducing a daily recruitment of eosinophils to the lung in this model. On day 21, similar OVA-induced inflammation was found after day 8–day 21, day 19–day 21, day 21, or day 8–day 20 MCP-5 blockage (Fig. 2 C).
BHR Induction after Chemokine Blockage.
To evaluate whether the specific reduction in lung inflammation induced by blockage of a particular chemokine correlates with a decrease in airway responsiveness, BHR was evaluated in OVA-treated mice after chemokine blockage. Fig. 4 A shows that MIP-1α neutralization reduced BHR by 30% compared with that observed in OVA-treated controls. This reduction correlates with the 20–25% decrease in eosinophil accumulation detected in both lung interstitium and airways of these mice after anti–MIP-1α Ab administration (Fig. 2, and Tables 1 and 2). In addition, eotaxin neutralization reduced BHR by half compared with that observed in OVA-treated controls (Fig. 4 A). This reduction correlates with the 60–70% decrease in eosinophil accumulation detected in the lung interstitium and airways of this experimental group of mice (Fig. 2, and Tables 1 and 2). However, when the anti-Eot Abs were administered on days 8–20 but not on day 21, no reduction in BHR was observed (Fig. 4 B). In contrast, blockage of eotaxin exclusively at the last day of the treatment (day 21) was sufficient to reduce BHR by 32% (Fig. 4 B). This indicates that eotaxin is involved in the induction of both BAL eosinophilia (Fig. 2 C) and BHR (Fig. 4 B) after each individual antigen provocation. The levels of BHR detected at day 21 in the OVA-treated mice after MCP-5 blockage—in which BAL eosinophilia was only reduced by 15% but interstitium eosinophilia was reduced by 87% (Figs. 2 and 3, and Tables 1 and 2)—were decreased by 60%, similar to the BHR baseline levels detected in PBS-treated littermates (Fig. 4 A). However, BHR was not affected when MCP-5 blockage was executed at late stages of the inflammatory response (day 21), although BAL and interstitium eosinophilia was comparable to that observed after day 8–day 21 blockage (Fig. 4 B). This indicates that signals delivered by MCP-5 at early stages of the response could be critical in the induction of BHR rather than the establishment of lung inflammation. BHR was decreased by 70% when MCP-1 was blocked from early stages of the OVA treatment (Fig. 4 A). OVA plus anti–MCP-1–treated mice and PBS-treated controls show similar degrees of bronchoconstriction before and after antigen provocation (Fig. 4 A). The effects of Met-RANTES administration on the development of BHR are pronounced and are described in detail elsewhere (31).
Figure 4.

Inhibition of OVA-induced airway hyperresponsiveness after chemokine blockage. Results are shown as the mean ± SEM for Penh before (gray bars) and after (black bars) methacholine provocation (n = 10, two independent experiments). Mice were exposed to an aerosol of methacholine for 1 min, and airway constriction was evaluated for the next 5 min. PBS- or OVA plus irrelevant Ab–treated mice were used as control for OVA-treated littermates in which MIP-1α, eotaxin, MCP-5, or MCP-1 was blocked from days (d) 8 to 21 (A) or at the time points indicated (B).
Lung Chemokine Expression after Chemokine Blockage.
The in vivo neutralization of eotaxin, MCP-1, or chemokines that bind RANTES receptor(s) either reduces or abrogates the OVA-induced accumulation of one or more leukocyte types both in BAL and lung interstitium (Figs. 2 and 3, and Tables 1 and 2). This could be related to changes in the pattern of expression of these chemokines. To evaluate whether eotaxin, MCP-1, or chemokines that bind RANTES receptor(s) control the expression of themselves and/or other chemokines, their mRNA expression in the lung was determined by RPA after the blockage of each chemokine during OVA treatment. Fig. 5 shows that after eotaxin neutralization, there was significant expression of OVA-induced eotaxin, RANTES, and MCP-1 on day 21. The mRNA expression level of these chemokines was comparable to those found in OVA-treated controls at the same time point (Fig. 5). In contrast, eotaxin and RANTES mRNA expression was reduced significantly in the lung of Met-RANTES–treated mice at each time point studied (Fig. 5, and data not shown). MCP-1 expression was not altered significantly by Met-RANTES in these mice (Fig. 5). Surprisingly, MCP-1 blockage, which prevents lung accumulation of eosinophils and lymphocytes in response to OVA, does not prevent the expression of the lymphocytic and/or eosinophilic chemokines RANTES and eotaxin (Fig. 5).
Figure 5.

Chemokine expression after chemokine blockage. On day 21, total RNA from lungs of OVA plus Ab control–, OVA plus anti-Eot–, OVA plus Met-RANTES–, or OVA plus anti–MCP-1–treated mice was extracted 3 h after antigen challenge. Chemokine blockage was performed daily before OVA provocation on days 8–21. In a control group, mice were treated with PBS (intranasally) and the irrelevant Ab (intravenously) instead of OVA (intranasally) and the chemokine-neutralizing Ab (intravenously), respectively. RANTES, eotaxin, and MCP-1 mRNA expression were determined by RPA. Both TCA and GAPDH mRNA expression were measured as negative and positive control, respectively. The intensity of the band for each chemokine was normalized to the intensity of the band of GAPDH in the control sample (not shown) and analyzed. Chemokine expression of two representative mice out of four is shown.
Production of Inflammatory Mediators after MCP-1 Blockage.
Since MCP-1 blockage does not affect the mRNA expression of the chemokines studied, the absence of leukocyte infiltration in the lung of OVA plus anti–MCP-1–treated mice could be due to the modulation of activating factors other than chemokines. These activating factors could be produced directly by monocytes or induced indirectly by this subset of leukocytes. Fig. 6 A shows that at 1 h after OVA administration, the production of LTB4 and PGE2 was increased in the BAL fluid of the OVA-treated mice on day 21 but not on day 15. However, blockage of MCP-1 completely inhibits OVA-induced production of these two inflammatory mediators (Fig. 6 A). Similarly, detectable levels of TXB2 were observed in the BAL fluid of OVA-treated mice, but not in OVA plus anti–MCP-1–treated littermates, on day 21 of treatment (Fig. 6 A). No differences between controls and test mice were detected in LTC4/D4/E4 levels at the time point analyzed (Fig. 6 A). The production of IgE in response to OVA was decreased fivefold in the BAL fluid after MCP-1 blockage at the time point indicated (Fig. 6 B). Similarly, the production of Th2-derived cytokines in the same fluid was decreased fourfold for IL-5 and threefold for IL-4 both on days 15 and 21 (Fig. 6 B). TNF-α production in the BAL fluid was similar in both groups of mice at both time points (Fig. 6 B). No IFN-γ production was detected after OVA or OVA plus anti–MCP-1 administration at the time points indicated (Fig. 6 B).
Figure 6.

Production of OVA-induced mediators of inflammation in the BAL fluid after MCP-1 blockage. 1 h after OVA administration on day 15 or 21, inflammatory mediator release was measured in the BAL fluid of OVA plus irrelevant Ab–treated control mice (open circles) and OVA plus anti–MCP-1(days 8–21)–treated mice (filled circles). Anti– MCP-1 or control Abs were administered daily before OVA provocation on days 8–21. Each circle represents a single mouse. Bars represent the mean of each group. Significant difference between control and test groups of mice was determined using the Student's t test (*P <0.01).
Discussion
We have characterized previously the expression of the chemokines eotaxin, MCP-5, RANTES, and MCP-1 (mRNA and/or protein), and correlated this with the leukocytes migrating to the lung during a murine model of lung inflammation (5, 16). From these experiments, we concluded that MCP-1 mRNA expression paralleled the accumulation of monocytes/macrophages in this organ, both events occurring predominantly at early stages of the response (day 15). Also, eotaxin mRNA expression paralleled lung eosinophilia predominantly at late stages (day 21). In contrast, other chemokines, such as RANTES or MCP-5, were expressed throughout the inflammatory reaction. This underlines the contribution of chemokines at different stages of the response.
From the work presented here, we first conclude that eosinophil recruitment and development of BHR in this model system involve the action of both eosinophilic (eotaxin, RANTES, MCP-5, and MIP-1α) and noneosinophilic chemokines (MCP-1). This indicates the absence of redundancy, since these chemokines seem to exert a critical role at different stages and on different pathways of the development of OVA-induced lung eosinophilia and BHR. Thus, MCP-5 recruits practically all the eosinophils present in the lung airways at early stages of the inflammatory response (day 15), while eotaxin attracts >60 and 80% of the BAL and interstitial eosinophils, respectively, at later stages of the response (day 21; Fig. 2, A and B, and Tables 1 and 2). In addition, MCP-1 mediates its critical influence throughout during the development of the inflammatory response (days 8–21), whereas eotaxin expression is important for eosinophil recruitment after each antigen provocation (Fig. 2, and Tables 1 and 2). MCP-5 also delivers signals at early stages of the response that are involved in the establishment of BHR. In fact, blockage of MCP-5 at late stages does not modify the degree of bronchoconstriction in response to OVA compared with that observed in the OVA plus Ab control–treated mice (Fig. 4 A). Furthermore, we also conclude that MCP-5 seems to affect leukocyte traffic through the interstitium, since the accumulation of these leukocytes in the interstitium—but not in the airways—was severely impaired after MCP-5 blockage (Figs. 2 and 3, and Tables 1 and 2). This could be due to a differential modulation of adhesion receptors by MCP-5 that results in retention of the leukocytes in the interstitium. In fact, it has been demonstrated that chemokines such as eotaxin affect the activation state of integrins and their ligands both in vivo and in vitro (37). MCP-5 blockage could also accelerate the transit of leukocytes from the lung interstitium to the airway spaces by modulating the expression of other inflammatory mediators. These hypotheses are currently being investigated in our laboratory.
The use of a RANTES receptor antagonist prevents completely lung eosinophilia at each time point during OVA treatment (Fig. 2, and Tables 1 and 2). Our data suggest that RANTES and/or other chemokines that bind RANTES receptor(s) are key lymphocytic chemokines in our model, because their blockage impedes pulmonary lymphocyte accumulation in response to OVA. Although both MIP-1α and MIP-1β are chemokines able to bind RANTES receptor(s), only mRNA expression of MIP-1α is modulated during this OVA model. In a series of elegant experiments, MIP-1α has been reported to reduce the intensity of the eosinophil recruitment to the lung and airways by 50% during Schistosoma mansoni egg antigen–induced allergic airway inflammation (38). MIP-1α neutralization does not affect leukocyte accumulation in the lung at early stages of the inflammatory response in the particular OVA model studied here (Fig. 2, and Tables 1 and 2). At late stages, only a 20% reduction in airway eosinophilia was detected after anti–MIP-1α Ab administration (Fig. 2, and Table 1). However, lung eosinophilia does not occur in the absence of CD4+ T lymphocytes (5), suggesting that RANTES receptor(s) may induce lung eosinophilia either by direct attraction of eosinophils or via the activation and pulmonary accumulation of CD4+ T lymphocytes, or both. In addition, OVA plus Met-RANTES–treated mice showed a marked reduction in RANTES and eotaxin mRNA expression in the lung during OVA treatment (Fig. 5). Thus, RANTES or other as yet unknown chemokines rather than MIP-1α or MIP-1β act via RANTES receptor(s) and control directly or indirectly the expression of other chemokines. Therefore, OVA-induced T cell and eosinophil infiltration in the lung is regulated by RANTES through itself and eotaxin. However, it is important to note that the reagent used here, Met-RANTES, may bind to more than one RANTES receptor and therefore inhibit the activity of other chemokines. Due to the complexity of this system, we have reported it in detail elsewhere (31).
Finally, the blockage of the mononuclear phagocyte chemokine MCP-1 throughout both the induction and later phases of the response affects these four leukocyte populations that migrate to the lung in response to OVA as well as blocking the induction of BHR (Figs. 2, 3, and 4, and Tables 1 and 2). Monocytes and macrophages are the first cell types to accumulate during OVA-induced lung inflammation in this model (5), and the strongest expression of MCP-1 occurs at early stages of the inflammatory response (Fig. 1; reference 5). Moreover, modification of the OVA-induced inflammatory response exclusively occurs when anti–MCP-1 mAb is administered from very early stages of the inflammatory process (Fig. 2, and Tables 1 and 2). In the absence of MCP-1–mediated signals, there is an ∼50% reduction in the number of lung monocytes/macrophages after OVA administration, but interestingly, there is an almost complete reduction in lymphocyte and eosinophil infiltration of the lung (Fig. 2, and Tables 1 and 2). Taken together, these data suggest that MCP-1 acts upstream of the inflammatory process and is a strong candidate to regulate the expression of the other chemokines during this physiological response. Nevertheless, comparable mRNA expression of eotaxin, RANTES, and MCP-1 was detected in the lung of OVA plus anti–MCP-1–treated mice and OVA-treated controls (Fig. 5). This indicates that MCP-1 is involved in other regulatory pathways rather than chemokine expression. Based on our present knowledge of MCP-1 function, it can be hypothesized that the regulatory pathways are critically affected by the reduction in monocyte/macrophage numbers after anti–MCP-1 treatment and/or by the depletion of a specific subset of the cells that is essential for this inflammatory process.
Both lung-resident macrophages distributed along the airways and circulating macrophages release mediators that can modulate the function of other cell types and amplify the lung inflammatory reactions (10, 39). Macrophages can release eosinophil chemoattractants, including LTB4 (10), or modulate the generation of LTC4 by eosinophils (12). They also produce PGE2 and TXB2, which are mediators of increased vascular permeability. In fact, levels of LTB4, PGE2, or TXB2 were detectable in the BAL fluid of OVA-treated mice but greatly diminished in the BAL fluid of OVA plus anti–MCP-1–treated littermates (Fig. 6 A). This indicates that the observed production of these factors is due either to infiltrating cells at this point, to increased activation of resident cells at late stages of the response, or to a combination of these two events. LTC4 was not detectable in the BAL fluid of either group of mice (Fig. 6 A). Macrophages can act as APCs and drive the proliferation of effector T cells that have been demonstrated to be required to achieve a functional response. In this scenario, macrophage-induced IL-4 production by T cells after antigen presentation drives the switch in Ig synthesis by B cells to IgE and potentiates the subsequent priming of mast cells by IgE plus antigen (40). MCP-1 blockage correlates with a clear decrease in the BAL levels of IgE and IL-4 after OVA treatment (Fig. 6 B). Similarly, IL-5 production is also affected by the MCP-1 blockage during OVA-induced lung allergic inflammation. These results indicate that MCP-1 function(s) (probably through monocyte/macrophage recruitment) are also involved in B and T cell activation. No IFN-γ production was detected in the BAL fluid of test or control mice, mainly because this model has a predominant Th2-type response. Macrophages and monocytes also represent the major source of inflammatory cytokines such as TNF (39). Despite the fact that only low levels of TNF-α were detected in the BAL fluid of OVA-treated mice, MCP-1 blockage reduced these levels by half at the time point of maximal macrophage and monocyte infiltration in the lung (day 15; Fig. 6 B).
In conclusion, this paper reports the functional relevance of chemokines at different stages of a complex inflammatory response in vivo. Chemokines not only ensure the physical presence of specific leukocytes at the site of inflammation by acting directly on them, but also regulate the expression of other chemokines and the activation of a variety of inflammatory mediators made by different cell types. Our results suggest that specific chemokines are involved in different cellular and molecular pathways that in a coordinated fashion contribute to the complex pathophysiology of asthma. Understanding all the components involved in lung allergic inflammation as well as the critical timing of their actions could represent a crucial step in the prevention and therapy of this disease. We believe that the in vivo results presented here may contribute to the support of clinical applications for the improvement of asthma therapy.
Acknowledgments
We thank Profs. Drazen and Butcher for critical reading of this manuscript, Drs. MacKay, Tepper, and Zlotnik for their comments and important suggestions, and Dr. L. Kremer, Centro Nacional Biotecnologia, for providing antibodies against MCP-5. We are also indebted to T. Delaney and N. Bikkal for skilled technical assistance, and M. Melzer for editorial assistance.
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- BHR
bronchial hyperresponsiveness
- Eot
eotaxin
- LT
leukotriene
- MCP
monocyte chemoattractant protein
- Met-RANTES
methionylated RANTES
- MIP
macrophage-inflammatory protein
- OVA
ovalbumin
- RANTES
regulated upon activation, normal T cell expressed and secreted
- RPA
RNase protection assay
- TX
thromboxane
Footnotes
This work was partially funded by Astra Draco AB.
References
- 1.Gleich GJ, Flavahan NA, Fujisawa T, Vanhoutte PM. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol. 1988;81:776–781. doi: 10.1016/0091-6749(88)90931-1. [DOI] [PubMed] [Google Scholar]
- 2.Gleich GJ. The eosinophil and bronchial asthma. Current understanding. J Allergy Clin Immunol. 1990;85:422–436. doi: 10.1016/0091-6749(90)90151-s. [DOI] [PubMed] [Google Scholar]
- 3.Eum S-Y, Hailé S, Lefort J, Huerre M, Vargaftig BB. Eosinophil recruitment into the respiratory epithelium following antigenic challenge in hyper-IgE mice is accompanied by interleukin 5-dependent bronchial hyperresponsiveness. Proc Natl Acad Sci USA. 1995;92:12290–12294. doi: 10.1073/pnas.92.26.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley BL, Azzawi M, Jacobson M, Assoufi B, Collins JV, Irani A-MA, Schwartz LB, Durham SR, Jeffrey PK, Kay AB. Eosinophils, T-lymphocytes, mast cells, neutrophils and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J Allergy Clin Immunol. 1991;88:661–667. doi: 10.1016/0091-6749(91)90160-p. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalo JA, Lloyd CM, Kremer L, Finger E, Martinez-A C, Siegelman MH, Cybulski M, Gutierrez-Ramos JC. Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of T cells, chemokines, and endothelial adhesion receptors. J Clin Invest. 1996;98:2332–2345. doi: 10.1172/JCI119045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gavett SH, Chen X, Kinkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 7.Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf M, Anderson GP. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- 8.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fels AOS, Pawlowski NA, Cramer EB. Human alveolar macrophages produce leukotriene B4. Proc Natl Acad Sci USA. 1982;79:7866–7870. doi: 10.1073/pnas.79.24.7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson JRW, Crea AEG, Clark TJH. Identification and characterization of a monocyte-derived neutrophil-activating peptide in corticosteroid-resistant bronchial asthma. J Clin Invest. 1989;84:1930–1941. doi: 10.1172/JCI114381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jose PJ, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, Truong O, Hsuan JJ, Williams TJ. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J Exp Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukacs NW, Strieter RM, Shaklee CL, Chensue SW, Kunkel SL. Macrophage inflammatory protein-1α influences eosinophil recruitment in antigen-specific airway inflammation. Eur J Immunol. 1995;25:245–251. doi: 10.1002/eji.1830250140. [DOI] [PubMed] [Google Scholar]
- 15.Lukacs NW, Strieter RM, Chensue SW, Kunkel SL. Activation and regulation of chemokines in allergic airway inflammation. J Leukocyte Biol. 1996;59:13–17. doi: 10.1002/jlb.59.1.13. [DOI] [PubMed] [Google Scholar]
- 16.Jia GQ, Gonzalo JA, Lloyd C, Kremer L, Lu L, Martinez C, Wershil BK, Gutierrez-Ramos J-C. Distinct expression and function of the novel mouse chemokine monocyte chemotactic protein-5 in lung allergic inflammation. J Exp Med. 1996;184:1939–1951. doi: 10.1084/jem.184.5.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacLean JA, Ownbey R, Luster AD. T cell-dependent regulation of eotaxin in antigen-induced pulmonary eosinophilia. J Exp Med. 1996;184:1461–1469. doi: 10.1084/jem.184.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller MD, Krangel MS. Biology and biochemistry of the chemokines: a family of chemotactic and inflammatory cytokines. Crit Rev Immunol. 1992;12:17–46. [PubMed] [Google Scholar]
- 19.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 20.Peled A, Gonzalo J-A, Lloyd C, Gutierrez-Ramos J-C. The chemotactic cytokine eotaxin acts as a granulocyte-macrophage colony-stimulating factor during lung inflammation. Blood. 1998;91:1909–1916. [PubMed] [Google Scholar]
- 21.Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J Exp Med. 1992;176:1489–1495. doi: 10.1084/jem.176.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothenberg ME, Luster AD, Leder P. Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc Natl Acad Sci USA. 1995;92:8960–8964. doi: 10.1073/pnas.92.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalo J-A, Jia G-Q, Aguirre V, Friend D, Coyle AJ, Jenkins NA, Lin GS, Katz H, Litchman A, Copeland N, et al. Mouse eotaxin expression parallels eosinophil accumulation during lung allergic inflammation but it is not restricted to a Th2-type response. Immunity. 1996;4:1–14. doi: 10.1016/s1074-7613(00)80293-9. [DOI] [PubMed] [Google Scholar]
- 24.Ebisawa M, Yamada T, Bickel C, Klunk D, Schleimer RP. Eosinophil transendothelial migration induced by cytokines. III. Effect of the chemokine RANTES1. J Immunol. 1994;153:2153–2160. [PubMed] [Google Scholar]
- 25.Rollins BJ, Yoshimura T, Leonard EJ, Pober J. Cytokine-activated human endothelial cells synthesize and secrete monocyte chemoattractant protein, MCP-1. Am J Pathol. 1990;136:1229–1233. [PMC free article] [PubMed] [Google Scholar]
- 26.Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel S. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formation: relationship to local inflammation, Th cell expression, and IL-12 production. J Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- 27.Gunn MD, Nelken NA, Liao X, Williams LT. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J Immunol. 1997;158:376–383. [PubMed] [Google Scholar]
- 28.Luo Y, Laning J, Hayashi M, Hancock PR, Rollins B, Dorf ME. Serologic analysis of the mouse beta chemokine JE/monocyte chemoattractant protein-1. J Immunol. 1994;153:3708–3716. [PubMed] [Google Scholar]
- 29.Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TNC, Salant DJ, Gutierrez-Ramos J-C. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proudfoot AEI, Power CA, Hoogewerf AJ, Montjovent MO, Borlat F, Offord RE, Wells TNC. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 31.Coyle, A.J., J. Kips, A.E.I. Proudfoot, and T.N.C. Wells. 1998. Inhibition of lung inflammation and altered airway responsiveness by Met-RANTES, a novel chemokine receptor antagonist. J. Exp. Med. In press.
- 32.Gelfand EW, Irvin CG. T lymphocyte: setting the tone of the airways. Nat Med. 1997;3:382–383. doi: 10.1038/nm0497-382. [DOI] [PubMed] [Google Scholar]
- 33.Ledermann F, Schlienger C, Wagner K, Heusser C. A sensitive and efficient induction system for murine IgE. Single cell analysis at the clonal level. J Immunol Methods. 1991;141:263–275. doi: 10.1016/0022-1759(91)90153-7. [DOI] [PubMed] [Google Scholar]
- 34.Ponath PD, Qin S, Ringler DJ, Clark-Lewis I, Wang J, Kassam N, Smith H, Shi X, Gonzalo J-A, Newman W, et al. Cloning of the human eosinophil chemoattractant, eotaxin. Expression, receptor binding, and functional properties suggest a mechanism for the selective recruitment of eosinophils. J Clin Invest. 1996;97:604–612. doi: 10.1172/JCI118456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uguccioni M, D'Apuzzo M, Loetscher M, Dewald B, Baggiolini M. Actions of the chemotactic cytokines MCP-1, MCP-2, MCP-3, RANTES, MIP-1α and MIP-1β on human monocytes. Eur J Immunol. 1995;25:64–68. doi: 10.1002/eji.1830250113. [DOI] [PubMed] [Google Scholar]
- 36.Rollins BJ, Morrison ED, Stiles CD. Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc Natl Acad Sci USA. 1988;85:3738–3742. doi: 10.1073/pnas.85.11.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia, G.-Q., J.-A. Gonzalo, D. Wagner, A.J. Coyle, M. Cybulski, and J.-C. Gutierrez-Ramos. 1997. Selective eosinophil transendothelial migration triggered by eotaxin via modulation of Mac-1/ICAM-1 and VLA-4/VCAM-1 interactions. Eur. J. Immunol. In press. [DOI] [PubMed]
- 38.Lukacs NW, Strieter RM, Warmington K, Lincoln P, Chensue SW, Kunkel SL. Differential recruitment of leukocyte populations and alteration of airway hyperreactivity by C-C family chemokines in allergic airway inflammation. J Immunol. 1997;158:4398–4404. [PubMed] [Google Scholar]
- 39.Elias JA, Schreiber AD, Gostilo K. Differential interleukin-1 elaboration by unfractionated and density fractionated human alveolar macrophages and blood monocytes. J Immunol. 1985;135:3198–3204. [PubMed] [Google Scholar]
- 40.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–5. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]