

Abstract
The B cell–specific transmembrane protein RP-105 belongs to the family of Drosophila toll-like proteins which are likely to trigger innate immune responses in mice and man. Here we demonstrate that the Src-family protein tyrosine kinase Lyn, protein kinase C β I/II (PKCβI/II), and Erk2-specific mitogen-activated protein (MAP) kinase kinase (MEK) are essential and probably functionally connected elements of the RP-105–mediated signaling cascade in B cells. We also find that negative regulation of RP-105–mediated activation of MAP kinases by membrane immunoglobulin may account for the phenomenon of antigen receptor–mediated arrest of RP-105–mediated B cell proliferation.
Keywords: RP-105, B lymphocytes, signal transduction, mice
Activation of B cells during adaptive immune responses requires coordinated signaling through the surface expressed antigen receptor and coreceptors such as CD19, CD21, or CD22 (1). The combined antigen receptor- and coreceptor-derived signals define the degree of B cell activation and the strength of humoral immune responses (2). In contrast to adaptive immune responses, innate immune responses are antigen receptor-independent and induced by invariant molecular structures in pathogens (pathogen-associated molecular pattern, PAMPs)1 via pattern-recognition receptors (PRRs; reference 3). The common feature of B cell-activating PAMPs such as bacteria cell wall lipopolysaccharide (4), viral hemagglutinins (5, 6), or CpG-rich bacterial DNA (7) lies in their ability to induce polyclonal B cell activation as defined by strong proliferative responses associated with upregulation of the surface expressed MHC class II and costimulatory receptor molecules CD80 (B7.1) and CD86 (B7.2) (8).
Responses of this type were found recently to be mediated by a human homologue of the Drosophila toll protein (9). The expression of a constitutively active form of human toll in a monocytic cell line leads to induction of expression of inflammatory cytokines such as IL-1, IL-8, IL-6, IFN-γ, as well as to the expression of the costimulatory molecule CD80 (9). Human toll belongs to the family of leucine-rich PRRs which also comprises the LPS receptor CD14 (10) and the toll-like protein RP-105 (11). RP-105 is a 105-kD transmembrane protein expressed on the surface of mature B cells in mice (12) and B lymphocytes and dendritic cells in humans (13, 14). As in toll protein, the extracellular domain of RP-105 is characterized by the presence of multiple tandemly repeated leucine-rich motifs separated from the single transmembrane domain by a carboxy-flanking region (11). The similarity between toll and RP-105 is further strengthened by the presence of conserved cysteine residues in the carboxy-flanking region of toll and RP-105 (11). These cysteine residues are essential for the regulation of signal transduction through toll (9) and, possibly, RP-105.
Antibody-mediated cross-linking of RP-105 in vitro induces a strong proliferative response in B cells that can be inhibited by surface IgM (sIgM) cross-linking (15). Thus, the simultaneous treatment of B cells with anti-RP-105 and anti-IgM or incubation of anti-RP-105–induced B cell blasts with anti-IgM leads to cell growth arrest and apoptotic death (15). The described signaling properties of RP-105 suggest a possible role of this protein in regulation of B cell activation during immune responses and invite questions about the mechanisms of RP-105–mediated signal transduction. Using a combination of biochemical and genetic approaches we analyzed the mechanism of RP-105– mediated signaling. Our data demonstrate that the Src-family protein tyrosine kinase Lyn, protein kinase C β I/II (PKCβI/II) and Erk2-specific mitogen-activated protein (MAP) kinase kinase MEK are essential and probably functionally connected elements of the RP-105–mediated signaling cascade. We also find that negative regulation of anti-RP-105–induced activation of MAP kinases by membrane immunoglobulin may account for the arrest of RP-105– induced proliferation mediated by the antigen receptor.
Materials and Methods
Mice.
The lyn-/-, fyn-/-, and PKCβI/II-/- mice were described previously (16–18). Lyn-/- mice that had developed splenomegaly were not used. The Blk-/- mice were generated by using ES cells in which exon 8, encoding the tyrosine kinase domain of Blk was replaced by neor gene (Texido, G., manuscript in preparation). The B cells expressing the dominant negative mutant of MEK (dnMEK) were derived in vivo from chimeric RAG-2–deficient mice, in which the lymphoid system was reconstituted with the ES cells carrying multiple copies of the dnMEK transgene under the control of B cell–specific regulatory elements (Carsetti, R., A. Tarakhovsky, manuscript in preparation).
Cells and Antibodies.
Unless otherwise indicated tissue culture media used was RPMI 1640 supplemented with 5% FCS, 2 mM pyruvate, 2 mM glutamine, and 50 μM β-mercaptoethanol. Splenic B lymphocytes were purified as described (16, 18). Goat or rabbit anti–mouse IgM (Jackson ImmunoResearch Laboratories, West Grove, PA) was used for the induction of sIgM-mediated protein tyrosine phosphorylation and Ca2+ mobilization in B cells. AffiPure goat anti–mouse IgM (2.5 μg/ml; Dianova, Hamburg, Germany), IL-4 (25 U/ml; Genzyme Corp., Boston, MA) and monoclonal anti-RP-105 antibody (12) were used for the activation of B cells in vitro. Anti-Lyn and anti-Syk polyclonal antisera were generated and used as described (19, 20). Rabbit anti-Shc was a gift from Dr. Mary Crowley (Scripps Institute, La Jolla, CA). Anti-phosphotyrosine mAb-4G10 was from Upstate Biotechnology Inc. (Lake Placid, NY). Polyclonal anti-Vav, anti-Erk2, anti-JNK1/anti-JNK-2, and anti-p38 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Culture supernatant of anti-FcγRII-III (mAb2.4G2) was obtained from cells from American Type Culture Collection (ATCC, Rockville, MD).
Analysis of B Cell Proliferation and Upregulation of Activation Markers.
Purified splenic B cells (5 × 106/ml) were cultured for 24 h in 24-well flat-bottom plates in media supplemented with 10% FCS in the absence or presence of anti-RP-105. After incubation, cells were stained with phycoerythrin-conjugated antibodies to B220/CD45R (RA3-6B2), fluorescein-conjugated antibodies to B7.2 (CD86; PharMingen, San Diego, CA), or MHC class II (M5/114) and analyzed by two-color flow cytometry on a FACScan® (Becton Dickinson & Co., Sparks, MD). For dose- dependent proliferative response, purified splenic B cells were cultured at 2 × 105/well or at 4 × 105/well in 96-well flat-bottom plates for 36 h followed by the addition of [3H]thymidine (1 μCi/well) for the next 8 h. The cells were harvested on filters and the incorporation of [3H]thymidine in cell DNA was measured as described (18).
In Vitro Kinase Assays.
After stimulation with anti-IgM or anti-RP-105, the B cells were lysed and Erk2, JNK1/2 or p38 MAP kinase isoforms were immunoprecipitated from B cell lysates by corresponding polyclonal antibodies (16). Assessment of MAP kinase isoform activity was carried out as described (21). The phosphorylation of substrates was quantified by PhosphorImager analysis. After analysis the membranes were reprobed with antibodies to each respective kinase to confirm equivalent immunoprecipitation in each sample. Lyn immunoprecipitation, immunoblot analysis and determination of Lyn protein kinase activity were carried out as described (22). Immunoprecipitates were washed with kinase buffer (20 mM Tris, pH7.2, 10 mM MgCl2, 10 mM MnCl2, 0.1% NP-40) and resuspended in 50 μl of the same buffer containing 0.5 μg of acid-denatured rabbit muscle enolase (Sigma Chemical Co., St. Louis, MO) and 10 μCi of γ-[32P]ATP. The phosphorylation reaction was performed at room temperature and stopped by addition of Laemmli buffer. Aliquots of the reaction mix were separated on 10% PAGE, transferred to nitrocellulose membrane and Lyn-mediated enolase phosphorylation was quantitated by PhosphorImager analysis.
Flow Cytometry and Calcium Mobilization.
FACS® analysis was performed on a FACScan® (Becton Dickinson & Co.) and the data were analyzed using CellQuest v3.1 software (Beckton Dickinson & Co.). The analysis of Ca2+ mobilization was carried out as described (21). In some experiments, Fcγ receptors (FcγR) on B cells were blocked by preincubation of splenocytes with the anti–FcγRII-III mAb 2.4G2. The stained cells were washed and resuspended in media/Hepes. After establishing base line fluorescence in the FITC (530 nm) channel, cells were stimulated by addition of anti-IgM or anti-RP-105 and data were collected continuously over a 14-min interval. Results were plotted as the mean Fluo-3 fluorescence at 20-s intervals.
Results
Early Signaling Events Induced by Anti–RP-105 Antibodies.
Incubation of purified splenic B cells with anti-RP-105 antibody leads to the activation of B cells as determined by the upregulation of surface MHC class II (Fig. 1, top), the costimulatory molecule B7.2 (Fig. 1, middle) and a strong dose-dependent proliferative response (Fig. 1, bottom). The possible mechanisms of RP-105–mediated B cell activation were addressed by analyzing RP-105–mediated protein tyrosine phosphorylation and Ca2+ mobilization. Both of these events are known to precede ligand-induced upregulation of costimulatory molecules and proliferation of B cells (23, 24).
Figure 1.
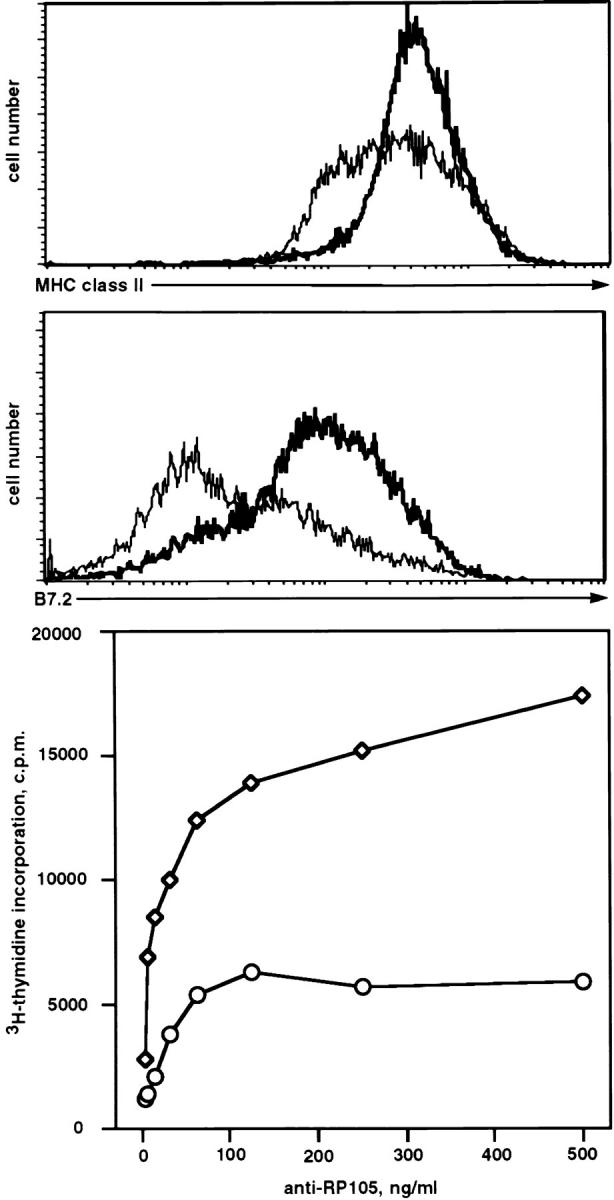
RP-105–mediated activation of splenic B cells in vitro. Splenic B cells were purified and stimulated as described in Materials and Methods. Histograms show the surface expression levels of MHC class II (top) or costimulatory molecule CD86 (middle) on B cells incubated for 24 h in the absence (thin line) or presence of anti-RP-105 antibody (5 μg/ml) (bold line). For the analysis of anti-RP-105–mediated proliferation, purified splenic B cells were cultured either at 2 × 105/well (circles) or at 4 × 105/well (squares) with the indicated amounts of anti-RP-105 for 36 h. [3H]thymidine (1 μCi/well) was added 8 h before cell harvesting. The proliferation of B cells was determined by the analysis of the amount of [3H]thymidine incorporated into the DNA of stimulated cells.
In contrast to the strong induction of protein tyrosine phosphorylation in anti-IgM-stimulated cells, the treatment of B cells with anti-RP-105 at the concentration optimal for B cell proliferation (5 μg/ml) results in a very modest increase in tyrosine phosphorylation of proteins with molecular masses ranging from 60 to 95 kD (Fig. 2 a). Moreover, proteins such as Syk, Vav, or Shc, which serve as common substrates for various receptor-linked protein tyrosine kinases (PTKs; reference 23), do not undergo any major change in degree of phosphorylation upon RP-105 cross-linking in comparison to the changes seen after treatment with anti-IgM (Fig. 2 b and data not shown).
Figure 2.

Induction of protein tyrosine phosphorylation by anti-RP-105. Purified wild-type splenic B cells were stimulated with goat anti-IgM (10 μg/ml) or anti-RP-105 (5 μg/ml) for the indicated periods of time and cell lysates were prepared. (a) Whole cell lysates (15 μg of protein) were resolved on 10% SDS-PAGE, transferred to nitrocellulose membranes and phosphorylation of the transferred proteins was determined by incubation of membranes with anti-phosphotyrosine antibody 4G10. The two predominant bands seen in all samples at molecular masses ∼55 kD represent p53/p56lyn. Equal protein loading was verified by reprobing the blots with the anti-Syk antibody. (b and c) Whole cell lysates were immunoprecipitated with anti-Syk antibody, the immunoprecipitates were resolved on 10% PAGE and the amount (b) and phosphorylation (c) of the immunoprecipitated Syk was analyzed by immunoblotting with 4G10 mAb. Two protein bands on c reflect a difference in the phosphorylation of Syk.
Activation of B cells by various agonists such as anti-IgM, CD40 ligand, or anti-CD38 is accompanied by Ca2+ mobilization from intracellular stores (25–27). In sharp contrast to the rapid rise in cytosolic Ca2+ concentration induced by anti-IgM, the incubation of B cells with anti–RP-105 causes a very slow and gradual increase in cytosolic Ca2+ concentration (Fig. 3 a). Although blockade of FcγR dramatically increases the duration of anti-IgM–induced Ca2+ mobilization, this treatment has essentially no effect on anti-RP-105– induced Ca2+ mobilization (Fig. 3 a). These data indicate that the FcγR does not regulate anti-RP-105–induced Ca2+ mobilization. The role of Ca2+ in RP-105–mediated B cell activation was further addressed by analyzing the effect of cyclosporin A (CsA) on anti-RP-105–induced proliferation. This drug inhibits the Ca2+-dependent phosphatase calcineurin which controls the phosphorylation and, therefore, nuclear translocation of transcription factor NF-AT in lymphocytes (28, 29). Despite the striking differences in kinetics and amplitude of Ca2+ mobilization induced by anti-RP-105 and anti-IgM, the anti-RP-105–induced proliferation of B cells is inhibited by CsA with the same efficiency as the proliferation of B cells induced by anti-IgM in combination with IL-4 (Fig. 3 c). These data suggest that Ca2+-dependent calcineurin activation plays an essential role in the induction of B cell proliferation by anti-RP-105.
Figure 3.



Ca2+ mobilization is essential for anti-RP-105–induced B cell proliferation. (a) Purified splenic B cells were stimulated with rabbit anti-IgM (10 μg/ml) or anti-RP-105 (5 μg/ml) (either with or without prior blocking of FcγR with anti-FcγRII-III-mAb). Mobilization of intracellular Ca2+ was analyzed by flow cytometry as described in Materials and Methods. Antibodies were added at the time points indicated by the arrow. Closed symbols represent cells stimulated with rabbit anti-IgM (diamonds and triangles, with or without prior blocking of FcγR, respectively); open symbols represent stimulation with anti-RP-105 (circles and squares, with or without prior blocking of FcγR, respectively). (b) Splenic B cells were stimulated with anti-IgM (10 μg/ml), anti-RP-105 (5 μg/ml) or anti-IgM in combination with anti-RP-105 (at the concentrations mentioned above) and calcium mobilization was analyzed. Closed triangles represent stimulation with anti-IgM alone, open circles represent stimulation with anti-RP-105 alone, and open squares with crosses represent stimulation with both anti-IgM and anti-RP-105. (c) Inhibition of the RP-105–induced B cell proliferation by cyclosporin A (CsA). B cells (5 × 105/well) were stimulated with anti-IgM (2.5 μg/ml) and IL-4 (25 U/ml; squares) or with anti-RP-105 (5 μg/ml) (circles) in the presence or absence of the indicated concentrations of CsA for 36 h. Cyclosporin A (Sigma Chemical Co.) was dissolved at 2.5 mg/ml in ethanol and then further diluted in culture medium. Each symbol shows the degree of inhibition (%) of [3H]thymidine incorporation into the DNA of stimulated purified splenic B cells in the presence of CsA. Representative result from one of three independent experiments is shown. The triplicate values for proliferation were as follows: unstimulated B cells 2,978 ± 126; B cells stimulated with: anti-IgM plus IL-4 5,625 ± 583; anti-RP-105 26,537 ± 2,203 cpm.
Engagement of various surface-expressed B cell receptors such as the antigen receptor, the CD19/CD21 complex, CD22, or CD40 leads to the activation of MAP kinases ERK2, SAPK/JNK, and p38 (1, 30). Treatment of B cells with anti-RP-105 results in a dose-dependent activation of these MAP kinase isoforms (Fig. 4, a–c, lanes 1, 3–5). Activation of Erk2 and p38 reaches a maximum within 15 min of RP-105 treatment and diminishes at 45 min, whereas JNK1/2 shows sustained activation (Fig. 5, a–c, left). Averaged over multiple experiments, activation of MAPK isoforms was much stronger after anti-RP-105 treatment than after anti-IgM stimulation (Table 1). However, the kinetics of activation of MAPK isoforms by anti-RP-105 is slower as compared with the previously reported anti-IgM– induced MAPK activation (16). Although anti-IgM treatment of B cells leads to maximal induction of Erk2 activity within 3–5 min, the maximal Erk2 activation by anti-RP-105 requires ∼15 min (Fig. 5, a–c, left).
Figure 4.

The dose-dependent activation of MAP kinase isoform by RP-105 and anti-IgM–mediated negative regulation of RP-105–mediated MAPK activation. Purified splenic B cells from wild-type C57BL/6 and lyn−/− mutant mice (4 × 106 cells/ml of media) were stimulated for 15 min with anti-IgM, anti-RP-105 at the indicated concentrations or with the combination of both. The cell lysates (50 μg of protein) were immunoprecipitated with anti-Erk2 (a), anti-JNK1/2 (b), or anti-p38 (c), and the immunoprecipitates were used for in vitro kinase assays (see Materials and Methods). Phosphorylated substrates were resolved on a 10% PAGE and quantitated by PhosphorImager analysis. The n-fold activation was determined relative to the levels of MAP kinase activity in unstimulated cells.
Figure 5.

The kinetics of RP-105–mediated activation of MAPK isoforms in wild-type and lyn−/− B cells. Purified splenic B cells from wild type C57BL/6 and lyn−/− mice (4 × 106 cells/ml of media) were stimulated with anti-IgM (10 μg/ml) or anti-RP-105 (5 μg/ml) for the indicated periods of time and cell lysates were prepared. Immunoprecipitations and determination of Erk2 (a), JNK1/2 (b), and p38 (c) kinase activities were carried out as described for Fig. 4.
Table 1.
Activation of MAPK Isoforms in Wild-type and lyn− /− B Cells
| Genotype | Stimulus | n-Fold activation compared to unstimulated cells | ||||||
|---|---|---|---|---|---|---|---|---|
| MAPK isoform | ||||||||
| Erk2 | JNK1/2 | p38 | ||||||
| C57BL/6 | anti-RP-105 | 21 ± 6 | 10 ± 2 | 6 ± 1 | ||||
| anti-IgM | 6 ± 2 | 3 ± 0.8 | 3 ± 1 | |||||
| Lyn −/− | anti-RP-105 | 6 ± 1 | 3 ± 0.8 | 2 ± 0.2 | ||||
| anti-IgM | 23 ± 9 | 14 ± 3 | 4 ± 1 | |||||
Enzymatic activation of MAPK isoforms was determined after 15 min of B cell stimulation as described in the legend to Fig. 5. Enzymatic activity in resting wild-type cells was given an arbitrary value of 1. In each experiment splenic B cells isolated from at least four mice were used. The n-fold induction of kinase activity (mean ± standard error) from three to five separate experiments are presented.
The Essential Roles of Lyn, PKCβI/II, and MAP Kinase Kinase in Anti-RP-105–induced B Cell Proliferation.
The low level of anti-RP-105–induced protein tyrosine phosphorylation does not exclude the involvement of PTKs in RP-105–mediated signaling. Indeed, the dramatic reduction of anti-RP-105–induced proliferation of xid B cells demonstrates the essential role of protein tyrosine kinase Btk in RP-105–mediated signaling (12). The activation of Btk is partially regulated by Src-family PTKs (31, 32). In B cells the Src-family kinases are represented mostly by Blk, Fyn and Lyn (33–35). The role of Src-family PTKs in RP-105– mediated signaling was addressed by analyzing anti-RP-105–induced activation of B cells deficient for Blk, Fyn, or Lyn. RP-105–induced proliferation is unaltered in Blk- and Fyn-deficient B cells (Fig. 6). In sharp contrast to Blk- and Fyn-deficient B cells, the proliferative responses of Lyn-deficient B cells are dramatically reduced as compared with responses of the wild-type (Fig. 6). The surface expression levels of RP-105 in Lyn-deficient B cells are similar to those in wild-type B cells (data not shown), excluding that reduction of RP-105 signaling is due to the downregulation of RP-105 expression in the absence of Lyn.
Figure 6.

Proliferative responses of blk−/−, fyn−/−, lyn−/−, PKCβI/ II−/−, and dnMEK B cells to anti-RP-105. Splenic B cells isolated from wild-type control mice (open symbols) or mutant mice (filled symbols) were cultured in the absence (triangles) or presence (circles) of anti-RP-105 (5 μg/ml). Each symbol represents the mean value of triplicate measurements in individual experiments. For the analysis of blk−/−, PKCβI/II−/−, and dnMEK, 129/Sv mice-derived B cells were used as a control. The C57BL/6 mice-derived B cells were used as a control for the analysis of responses of lyn−/− B lymphocytes.
In view of the essential role of Lyn in RP-105–mediated B cell proliferation, the activation of Lyn by anti-RP-105 was analyzed by a sensitive immunocomplex kinase assay (22). Incubation of wild-type B cells with anti-RP-105 or anti-IgM results in increase of protein tyrosine kinase activity of Lyn as determined by phosphorylation of the exogenous substrate enolase and increase of Lyn autophosphorylation (Fig. 7 a). The kinase activity of Lyn in anti-IgM–treated B cells reaches the maximum after 1 min and declines to the basal level of activity after 15 min of incubation (Fig. 7 a). In contrast, the activation of Lyn by RP-105, although not as high as in anti-IgM–treated cells, remains constant for 30 min at least (Fig. 7). Notably, the amounts of immunoreactive Lyn did not vary between the samples (Fig. 7 b).
Figure 7.

Anti-RP-105–induced activation of Lyn kinase. Purified splenic B cells from wild-type C57BL/6 mice were treated with either goat anti-IgM (10 μg/ml) or anti-RP-105 (5 μg/ml) for the indicated periods of time. 50 μg of lysate protein were used for anti-Lyn immunoprecipitation/immune complex kinase assay with enolase added as an exogenous substrate (see Materials and Methods). (a) The products of the in vitro kinase reactions were resolved on a 10% PAGE and relative phosphorylation of enolase (normalized to unstimulated cells) was quantitated by PhosphorImager analysis. (b) The amount of Lyn in whole cell lysates (15 μg) was determined by immunoblot analysis with anti-Lyn antibody.
The defect of anti-RP-105–induced activation of Lyn-deficient B cells and the previously described impairment of anti-RP-105–induced proliferation of xid B cells (12) suggest a functional link between Lyn and Btk within the RP-105–dependent signaling cascade. In B lymphocytes and mast cells Btk appears to be associated with protein kinase C β I/II (PKCβI/II) (36). The physiological importance of Btk-PKCβI/II interaction in B cell function is underscored by the demonstration of essentially identical profiles of B cell signaling defects in PKCβI/II-deficient and xid B cells (18). To determine whether PKCβI/II plays a role in RP-105–mediated B cell activation, the anti-RP-105–induced proliferation of PKCβI/II-deficient B cells was analyzed. The PKCβI/II-deficient B cells express RP-105 at levels similar to those in control B lymphocytes (data not shown). Similar to xid B cells, which do not proliferate in response to anti-RP-105 (12), treatment of PKCβI/II-deficient B cells with anti-RP-105 does not induce detectable proliferative responses (Fig. 6).
Activation of MAP kinase isoforms by anti-RP-105 and impaired RP-105–mediated activation of Lyn-deficient, xid, or PKCβI/II-deficient B cells suggests a link between the Lyn-Btk/PKCβI/II signaling chain and MAP kinases in the RP-105–dependent signaling cascade. Indeed, the treatment of Lyn-deficient B cells with anti-RP-105 is accompanied by a significantly weaker activation of MAP kinase isoforms as compared with wild-type B cells (Fig. 5 and Table 1). A similar result was obtained by the analysis of anti-RP-105–induced MAP kinase activation in xid B cells (data not shown). The role of MAP kinases in anti-RP-105–induced B cell activation was further addressed by the analysis of anti-RP-105–induced proliferation of splenic B cells expressing the dominant-negative form of MAP kinase kinase (MEK; Carsetti, R., and A. Tarakhovsky, manuscript in preparation). B cells expressing the dominant negative mutant of MEK (dnMEK) were derived in vivo from chimeric RAG-2–deficient mice in which the lymphoid system was reconstituted by ES cells carrying multiple copies of the dnMEK transgene under the control of B cell–specific regulatory elements (Carsetti, R., and A. Tarakhovsky, manuscript in preparation). Expression of dnMEK in B cells suppresses the activation of MAP kinase by stimuli such as anti-IgM or phorbol ester in combination with ionomycin (Carsetti, R., and A. Tarakhovsky, manuscript in preparation). Incubation of dnMEK-expressing B cells with anti-RP-105 antibody at a concentration inducing strong proliferation of wild-type control B cells does not induce the proliferation of transgenic B cells (Fig. 6).
Counterregulation by sIgM of RP-105–mediated MAP Kinase Activation and Calcium Mobilization.
In spite of the strong proliferation-inducing potential of anti-RP-105, the simultaneous antibody-mediated ligation of sIgM and RP-105 in vitro leads to B cell growth arrest and death (15). The degree of MAP kinase activation and/or changes in the pattern of activated MAPK isoforms induced by engagement of various receptor molecules define the fate of responding cells (37). To determine whether the negative regulation of RP-105 signaling by sIgM could be detected at the level of MAP kinase isoform activation, purified splenic B cells were incubated with variable amounts of anti-RP-105 in the presence or absence of anti-IgM. Antibody-mediated cross-linking of RP-105 results in a dose-dependent increase of the activities of Erk2, JNK1/2, and p38 (Fig. 4). The level of MAP kinase isoform activation by anti-RP-105 at a concentration optimal for B cell proliferation (5 μg/ml) is significantly higher than that induced by anti-IgM. However, simultaneous incubation of B cells with anti-RP-105 and anti-IgM reduces the amplitude of MAP kinase isoform activation to levels characteristic for anti-IgM–induced MAP kinase activation alone (Fig. 4). The dominance of antigen receptor-mediated signaling is similarly observed in lyn−/− B cells, where low levels of anti-RP-105–induced MAPK activation become significantly higher upon costimulation with anti-IgM (Fig. 4).
The negative regulation of RP-105 signals by signals through the antigen receptor were also seen at the level of Ca2+ mobilization. Coincubation of B cells with anti-IgM and RP-105 induced a Ca2+ mobilization response that is indistinguishable from Ca2+ mobilization in cells treated with anti-IgM alone (Fig. 3 b).
Discussion
This study advances the understanding of two questions regarding the activation of B cells by RP-105. First, can a biochemical pathway of RP-105–mediated intracellular signaling be identified? Second, what is the mechanism of negative regulation of RP-105–mediated B cell activation by the antigen receptor? Using genetic and biochemical approaches we revealed a critical role of Lyn in RP-105– mediated B cell activation. Lyn is one of the most abundant Src-family PTKs in B cells and is known to be associated with the antigen receptor and the CD19 coreceptor (38, 39). Although an association of Lyn with RP-105 has not been detected (Miyake, K., unpublished observation), the rapid though modest increase of kinase activity of Lyn upon anti-RP-105 treatment suggests a proximal location of Lyn with regard to the putative RP-105 signaling complex. The activation of Lyn kinase by RP-105 does not result in a significant increase of the tyrosine phosphorylation of various other cellular proteins in the activated cells. This result is not completely unexpected in view of the minimal changes in tyrosine phosphorylation of various proteins in anti-IgM–stimulated Lyn-deficient B cells as compared with wild-type B cells (16). Hence, both in B cell antigen receptor- and RP-105–dependent signaling pathways Lyn might be responsible for the phosphorylation of substrates the expression and/or degree of phosphorylation of which do not allow their detection by anti-phosphotyrosine immunoblot analysis of crude cell lysates.
The defective RP-105–mediated activation of Lyn-deficient and xid B cells suggests a possible link between Lyn and Btk in the RP-105 signaling cascade. Among the Src-family PTKs expressed in B cells, Lyn seems to play a leading role in antigen receptor-mediated phosphorylation and activation of Btk (31). The lack of effect of Blk and Fyn on RP-105–induced B cell activation suggests that among the three major Src-family PTKs in B cells Lyn is likely to be responsible for Btk activation by RP-105.
As with Lyn-deficient and xid B cells, PKCβI/II-deficient cells cannot be successfully activated by anti-RP-105. The ability of Btk to bind PKCβI/II and the virtual identity of immunodeficiency syndromes in xid and PKCβI/II-deficient mice (18) strongly support the existence of a Btk/ PKCβI/II signaling module and its importance for B cell activation. In B cells, PKCβI/II together with PKCα represent the subfamily of PKCs the activation of which is dependent on Ca2+ and diacylglycerol (40). Importantly, PKCβI/II appears to be activated by significantly lower concentrations of Ca2+ than PKCα (41, 42). Therefore, it seems likely that the very slow and gradual Ca2+ mobilization induced by anti-RP-105 would be sufficient to induce the activation of PKCβI/II, but not to induce the activation of PKCα. This hypothesis is currently under investigation.
Several lines of evidence support the importance of MAP kinase in RP-105–mediated activation of B cells. First, the activation of B cells by RP-105 leads to activation of MAP kinase isoforms Erk2, p38, and Jun kinase (JNK1/2). Second, anti-RP-105 fails to induce proliferation of B cells expressing the dominant negative form of Erk-specific (43, 44) MAPK kinase (MEK). The impaired RP-105–mediated activation of all three MAP kinase isoforms in the absence of Lyn and our preliminary data on the lack of MAP kinase activation in xid B cells support a possible connection between the RP-105 and MAP kinase signaling cascades via the Lyn-Btk/PKCβI/II module.
The similarity between toll and RP-105 as well as the ability of anti-RP-105 antibody to induce polyclonal activation of B cells in vitro supports a possible involvement of RP-105 in the regulation of innate immune responses. The activation of lymphocytes during innate immune response plays an important role in the efficient recruitment of activated B cells into antigen-driven adaptive responses (3). However, it seems conceivable that the switch from innate to adaptive immune responses may require the existence of mechanisms promoting antigen-specific responses at the expense of polyclonal B cell activation. In this study we have tried to address the mechanism of negative regulation of RP-105–mediated B cell activation by antigen receptor-derived signal(s). Involvement of Lyn, Btk, PKCβI/II, and MAP kinases in both sIgM- and RP-105–mediated activation implies the existence of a signaling pathway(s) common for both of the receptors. In contrast to Btk and PKCβI/II, which both play positive roles in anti-RP-105– and IgM-mediated B cell activation, Lyn appears to have different functions in sIgM- and RP-105–mediated signaling. Although the sIgM-mediated MAP kinase activation is negatively controlled by Lyn (16), the presence of Lyn is essential for RP-105–induced MAP kinase activation. The relatively stronger activation of Lyn kinase by anti-IgM than by anti-RP-105 may reflect more efficient recruitment of Lyn to the B cell antigen receptor (BCR) complex as compared with the putative RP-105 signaling complex. In such a case the simultaneous ligation of BCR and RP-105 will reduce the amount of Lyn that could be used by RP-105 signaling complex and block the RP-105–induced MAP kinase activation. This, in turn, may lead to the onset of the antigen receptor-specific pattern of MAP kinase activation and reprogramming of B cell responses. Assuming that RP-105 activation in vivo is regulated by specific ligand(s), the antagonistic relation between antigen receptor- and RP-105–mediated signaling predicts a temporal and/or spatial separation of putative RP-105 ligand- and antigen– induced B cell activation during immune responses.
Acknowledgments
We thank Roger Perlmutter for the fyn−/− mice. We thank Tim Bender, Jonathan Howard, Kaoru Saijo, Christian Schmedt for discussion and critical reading of the manuscript, and Sigrid Irlenbusch for technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft through SFB 243 and by the National Institutes of Health (DK50267 and HL54476).
Abbreviations used in this paper
- BCR
B cell antigen receptor
- CsA
cyclosporin A
- dnMEK
double negative mutant of MEK
- FcγR
Fcγ receptors
- MAP
mitogen-activated protein
- MEK
MAP kinase kinase
- PAMPS
pathogen-associated molecular pattern
- PKCβI/II
protein kinase C β I/II
- PRR
pattern recognition receptor
- PTK
protein tyrosine kinase
- sIgM
surface IgM
References
- 1.Tooze RM, Doody GM, Fearon DT. Counterregulation by the coreceptors CD19 and CD22 of MAP kinase activation by membrane immunoglobulin. Immunity. 1997;7:59–67. doi: 10.1016/s1074-7613(00)80510-5. [DOI] [PubMed] [Google Scholar]
- 2.O'Rourke L, Tooze R, Fearon DT. Co-receptors of B lymphocytes. Curr Opin Immunol. 1997;9:324–329. doi: 10.1016/s0952-7915(97)80077-5. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 4.Wright SD. CD14 and innate recognition of bacteria. J Immunol. 1995;155:6–8. [PubMed] [Google Scholar]
- 5.Rott O, Cash E. Influenza virus hemagglutinin induces differentiation of mature resting B cells and growth arrest of immature WEHI-231 lymphoma cells. J Immunol. 1994;152:5381–5391. [PubMed] [Google Scholar]
- 6.Cash E, Charreire J, Rott O. B-cell activation by superstimulatory influenza virus hemagglutinin: a pathogenesis for autoimmunity? . Immunol Rev. 1996;152:67–88. doi: 10.1111/j.1600-065x.1996.tb00911.x. [DOI] [PubMed] [Google Scholar]
- 7.Krieg AM. An innate immune defense mechanism based on the recognition of CpG motifs in microbial DNA. J Lab Clin Med. 1996;128:128–133. doi: 10.1016/s0022-2143(96)90004-9. [DOI] [PubMed] [Google Scholar]
- 8.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the DrosophilaToll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 10.Ferrero E, Hsieh CL, Francke U, Goyert SM. CD14 is a member of the family of leucine-rich proteins and is encoded by a gene syntenic with multiple receptor genes. J Immunol. 1990;145:331–336. [PubMed] [Google Scholar]
- 11.Miyake K, Yamashita Y, Ogata M, Sudo T, Kimoto M. RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J Immunol. 1995;154:3333–3340. [PubMed] [Google Scholar]
- 12.Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J Exp Med. 1994;180:1217–1224. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miura Y, Miyake K, Yamashita Y, Shimazu R, Copeland NG, Gilbert DJ, Jenkins NA, Inazawa J, Abe T, Kimoto M. Molecular cloning of a human RP105 homologue and chromosomal localization of the mouse and human RP105 genes (Ly64 and LY64) Genomics. 1996;38:299–304. doi: 10.1006/geno.1996.0632. [DOI] [PubMed] [Google Scholar]
- 14.Fugier-Vivier I, de Bouteiller O, Guret C, Fossiez F, Banchereau J, Mattei MG, Ait-Yahia S, Garcia E, Lebecque S, Liu YJ. Molecular cloning of human RP-105. Eur J Immunol. 1997;27:1824–1827. doi: 10.1002/eji.1830270734. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita Y, Miyake K, Miura Y, Kaneko Y, Yagita H, Suda T, Nagata S, Nomura J, Sakaguchi N, Kimoto M. Activation mediated by RP105 but not CD40 makes normal B cells susceptible to anti-IgM–induced apoptosis: a role for Fc receptor coligation. J Exp Med. 1996;184:113–120. doi: 10.1084/jem.184.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan VWF, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity. 1997;7:69–81. doi: 10.1016/s1074-7613(00)80511-7. [DOI] [PubMed] [Google Scholar]
- 17.Appleby MW, Kerner JD, Chien S, Maliszewski CR, Bondada S, Perlmutter RM, Bondada S. Involvement of p59fynT in interleukin-5 receptor signaling. J Exp Med. 1995;182:811–820. doi: 10.1084/jem.182.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase C β-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 19.Law DA, Chan VWF, Datta SK, DeFranco AL. B-cell antigen receptor motifs have abundant signaling capabilities and bind the tyrosine kinases PTK72, Lyn and Fyn. Curr Biol. 1993;3:645–657. doi: 10.1016/0960-9822(93)90062-s. [DOI] [PubMed] [Google Scholar]
- 20.Richards JD, Gold MR, Hourihane SL, DeFranco AL, Matsuuchi L. Reconstitution of B cell antigen- receptor induced signaling events in a nonlymphoid cell line by expressing the syk protein-tyrosine kinase. J Biol Chem. 1996;271:6458–6466. doi: 10.1074/jbc.271.11.6458. [DOI] [PubMed] [Google Scholar]
- 21.Chan VWF, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol. 1998;8:545–553. doi: 10.1016/s0960-9822(98)70223-4. [DOI] [PubMed] [Google Scholar]
- 22.Lowell CA, Soriano P, Varmus HE. Functional overlap in the Src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 1994;8:387–398. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- 23.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 24.Jensen, W.A., C.M. Pleiman, and J.C. Cambier. 1996. Molecular mechanisms of signal transduction by B cell receptors. In Weir's handbook of experimental immunology. L.A. Herzenberg, D.M. Weir, L.A. Herzenberg, C. Blackwell, editors. Blackwell Science, Cambridge, MA. 88.1–88.12.
- 25.Wicker LS, Boltz R, Jr, Matt V, Nichols EA, Peterson LB, Sigal NH. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. Eur J Immunol. 1990;20:2277–2283. doi: 10.1002/eji.1830201017. [DOI] [PubMed] [Google Scholar]
- 26.Klaus GG, Choi MS, Holman M. Properties of mouse CD40. Ligation of CD40 activates B cells via a Ca(++)-dependent, FK506-sensitive pathway. Eur J Immunol. 1994;24:3229–3232. doi: 10.1002/eji.1830241248. [DOI] [PubMed] [Google Scholar]
- 27.Lund FE, Yu N, Kim KM, Reth M, Howard MC. Signaling through CD38 augments B cell antigen receptor (BCR) responses and is dependent on BCR expression. J Immunol. 1996;157:1455–1467. [PubMed] [Google Scholar]
- 28.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 29.Choi MS, Brines RD, Holman MJ, Klaus GG. Induction of NF-AT in normal B lymphocytes by anti-immunoglobulin or CD40 ligand in conjunction with IL-4. Immunity. 1994;1:179–187. doi: 10.1016/1074-7613(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 30.Li YY, Baccam M, Waters SB, Pessin JE, Bishop GA, Koretzky GA. CD40 ligation results in protein kinase C-independent activation of ERK and JNK in resting murine splenic B cells. J Immunol. 1996;157:1440–1447. [PubMed] [Google Scholar]
- 31.Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, Fluckiger AC, Witte ON, Kinet JP. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271:822–825. doi: 10.1126/science.271.5250.822. [DOI] [PubMed] [Google Scholar]
- 32.Wahl MI, Fluckiger AC, Kato RM, Park H, Witte ON, Rawlings DJ. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton's tyrosine kinase via alternative receptors. Proc Natl Acad Sci USA. 1997;94:11526–11533. doi: 10.1073/pnas.94.21.11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkhardt AL, Brunswick M, Bolen JB, Mond JJ. Anti-immunoglobulin stimulation of B lymphocytes activates src-related protein-tyrosine kinases. Proc Natl Acad Sci USA. 1991;88:7410–7414. doi: 10.1073/pnas.88.16.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cambier JC, Campbell KS. Membrane immunoglobulin and its accomplices: new lessons from an old receptor. FASEB J. 1992;6:3207–3217. doi: 10.1096/fasebj.6.13.1397843. [DOI] [PubMed] [Google Scholar]
- 35.DeFranco AL, Richards JD, Blum JH, Stevens TL, Law DA, Chan VW, Datta SK, Foy SP, Hourihane SL, Gold MR, et al. Signal transduction by the B-cell antigen receptor. Ann NY Acad Sci. 1995;766:195–201. doi: 10.1111/j.1749-6632.1995.tb26662.x. [DOI] [PubMed] [Google Scholar]
- 36.Yao L, Kawakami Y, Kawakami T. The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc Natl Acad Sci USA. 1994;91:9175–9179. doi: 10.1073/pnas.91.19.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 38.Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science. 1991;251:192–194. doi: 10.1126/science.1702903. [DOI] [PubMed] [Google Scholar]
- 39.van Noesel CJ, Lankester AC, van Schijndel GM, van Lier RA. The CR2/CD19 complex on human B cells contains the src-family kinase Lyn. Int Immunol. 1993;5:699–705. doi: 10.1093/intimm/5.7.699. [DOI] [PubMed] [Google Scholar]
- 40.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 41.Marais RM, Parker PJ. Purification and characterisation of bovine brain protein kinase C isotypes α, β, and γ. Eur J Biochem. 1989;182:129–137. doi: 10.1111/j.1432-1033.1989.tb14809.x. [DOI] [PubMed] [Google Scholar]
- 42.Baixeras E, Kroemer G, Cuende E, Marquez C, Bosca L, Ales JE, Martinez, Martinez C. Signal transduction pathways involved in B-cell induction. Immunol Rev. 1993;132:607–614. doi: 10.1111/j.1600-065x.1993.tb00836.x. [DOI] [PubMed] [Google Scholar]
- 43.Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 44.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]