

The biological importance of nitric oxide has changed from that of toxic gas to that of an essential cellular signaling agent.1 In many of these processes, the binding of NO to a heme protein and the labilization of the ligand trans to NO or another rearrangement is the significant signaling event. Proposed mechanisms for the heme proteins cytochrome c′ and soluble guanylate cyclase have presented a scenario in which the coordination to heme changes during the physiological cycle.2 Understanding how these coordination events impact already coordinated ligands (and vice versa) is an important step in understanding the heme-NO interaction. At the literal center of these studies is the heme iron, whose motion along the coordinate axes is associated with reactive modes such as the νFe —Im, νFe—NO/δFe—N—O and heme doming.
In this communication, we examine whether ligand orientation and bond distance changes substantially modulate iron dynamics in six-coordinate [Fe(Porph)(1-MeIm)(NO)]3 derivatives. This study has been facilitated by the isolation of two crystalline polymorphs of [Fe(TpFPP)(1-MeIm)(NO)] that display differing room temperature solid-state νN-O (1631 cm-1 and 1640 cm-1). Additional vibrational data for the polymorphic forms is available from nuclear resonance vibrational measurements (NRVS). Briefly, NRVS is a novel technique that provides information on all vibrational frequencies for which there is iron motion.4 Spectra were obtained on powder samples of the two species and are displayed in Figure S1. The powder data clearly shows differences in the overall iron vibrational modes. We have also obtained oriented single crystal NRVS data on both species that provides detailed information on the character of the modes, which allows for a detailed examination of the differences between the two. An analysis of their molecular structures and vibrational data yields a detailed view of how different molecular structure features affect the dynamics of the iron atom.
The crystalline polymorphs are in the triclinic and monoclinic crystal systems; both are isolated from the same crystallization experiments. The two crystal types are illustrated in Figure S2. Figure 1 illustrates the two molecular structures. The FeNO and imidazole planes are within 1° of coplanarity (monoclinic form) and within 25° (triclinic form). The relative orientation of the imidazole N—CH3 bond and the bent FeNO group are of the opposite sense in the two species; the monoclinic form has a ″cisoid″ arrangement and the triclinic a ″transoid″ one. The two NO ligands make angles of 38.5° and 43.2° with the closest Fe—Np vector, so that when the four porphyrin nitrogen atoms are superimposed, the NO ligands are almost superimposed (see Figure S3). Major differences include relative rotations of the two imidazole ligands, the relative sense of NO and imidazole directions, small differences in the trans Fe—NIm bond distance, core conformations and positions of the peripheral p-fluorophenyl groups. The changing peripheral group directions reflect and contribute to the differing crystal packing of the two complexes.
Figure 1.
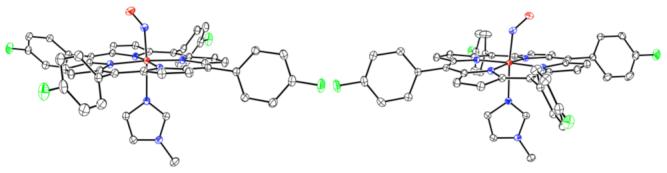
Thermal ellipsoid plot (50% probability) of tri-[Fe(TpFPP)(1-MeIm)(NO)] (left) and mono-[Fe(TpFPP)(1-MeIm)(NO)] (right).5
These two high precision structures strengthen earlier conclusions describing general six-coordinate NO derivatives.6,7 The Fe—NNO vector is indeed distinctly tilted off the heme normal, which leads to an equatorial asymmetry in the Fe—Np bonds; the two that bracket the tilted Fe—NO group are shorter than the opposite pair. The FeNO angles are ∼138° and the iron is displaced from the N4 plane by 0.06 Å towards NO. Selected metrical information for both complexes is given.5
What is the basis for the differing vibrational characteristics of the two polymorphic forms? Although the differing relative orientations of the bent NO group with respect to the imidazole is the most obvious difference in the two structures, we will demonstrate that it plays at most a minor role in vibrational variation; other structural distinctions are dominant. We first consider νN-O. At 293 K, the triclinic crystalline form has νN-O 9 cm-1 greater than the monoclinic form. Temperature-dependent IR has demonstrated that the frequency of νN-O decreases for each compound as the temperature is lowered. At 107 K, the frequency difference between the two is 11 cm-1. At 100 K, the Fe—NIm distance in the triclinic form is 0.038 Å longer than in the monoclinic form. Structures for both forms have also been obtained at 293 K. The Fe—NIm bond distances in both increase by ∼ 0.03 Å,8 and the difference between the two polymorphs remains nearly constant at 0.033 Å.9 Thus the shifts in νN-O are wholly consistent with the temperature dependent changes in the Fe—NIm distances. Will the effects of trans Fe—NIm distance differences also be propagated in the iron out-of-plane (OOP) modes?
DFT calculations were carried out to further explore the effect of ligand orientation and the character of the vibrational modes.11 Calculations carried out on polymorphic [Fe(TpFPP)(1-MeIm)-(NO)], based on X-ray crystal structures, resulted in energy-minimized structures that retained their initial cisoid and transoid configurations. However, other features of the structures became more similar such as the axial and equatorial bond distances. Variation in equatorial bonds with a tilted Fe—NNO vector are also features of both calculations. The predicted vibrational spectra for the cisoid and transoid species show no distinct differences.
NRVS data obtained from single crystals oriented to enhance (and identify) the in-plane and out-of-plane modes are shown in Figure 2. We observe particularly striking vibrational differences between the two polymorphs in the low frequency region for iron motion perpendicular to the porphyrin plane. Modes having frequencies (eFe2 values) of 33 cm-1 (0.06), 140 cm-1 (0.12), and 167 cm-1 (0.11) for tri-[Fe(TpFPP)(1-MeIm)(NO)] shift to 40 cm-1 (0.045), 153 cm-1 (0.07), and 177 cm-1 (0.11) in mono-[Fe(TpFPP)(1-MeIm)(NO)]. DFT calculations suggest that the highest two modes involve iron out-of-plane motion along with rotational and translational motion of the imidazole ligand and have 64% and 44% of the mode kinetic energy (KE) localized on the Fe(1-MeIm)(NO) fragment, but have little resemblance to a two-body Fe—Im oscillator. Figure S4 illustrates the character of the predicted modes, with frequencies (eFe2 values) of 147 cm-1 (0.10) and 166 cm-1 (0.06).15 The significant frequency increases for low frequency modes of the Fe(1-MeIm)(NO) fragment in the cisoid species appear to reflect the 0.04 Å decrease in Fe—NIm. However, the bands associated with the νFe—N(NO) = 432 cm-1 (eFe2 =0.11, cisoid) and 433 cm-1 (eFe2 =0.13, transoid) and with δFe—N—O/νFe—NO = 559 cm-1 (0.05) and 560 cm-1 (0.06) show very small frequency differences consistent with the much smaller (≈ 0.004 Å) change in the Fe—NNO bond distance between these derivatives. The Fe—NIm bond distance variation must change the electron density at iron, which in turn modulates the electron density that iron donates to the NO ligand, but with a smaller effect on the Fe—NNO bond distance.
Figure 2.
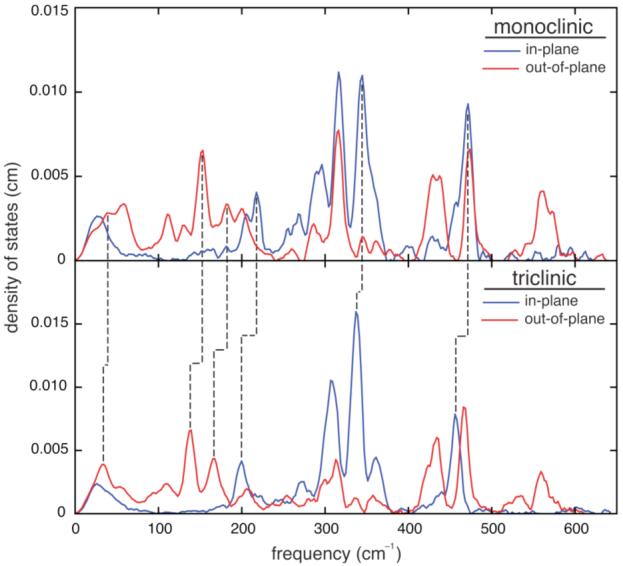
Comparison of the measured VDOS for mono- and tri-[Fe(TpFPP)(1-MeIm)(NO)] with crystals oriented with porphyrin planes aligned either perpendicular to or parallel to the excitation beam.
Earlier work showed the extreme sensitivity of in-plane modes to peripheral substituents.16 The two polymorphs also provide a unique opportunity to explore the systematics of in-plane mode variation. The calculated character of the four prominent modes are shown in Figures S5 and S6. The KE of the 350 cm-1 mode is predominantly located on iron (eFe2 ≈ 0.40), while the modes near 200 and 460 cm-1 are predominately located on the porphyrin (eFe2 ≈ 0.06). DFT calculations suggest that the direction of iron motion in the 350 cm-1 mode is perpendicular to the FeNO plane, not along the Fe—Np directions. This is distinctly different from that of the analogous carbonyl adducts where the iron motion is governed by the porphyrin Fe—Np bonds.17 These two systems suggest that the orientation of imidazole has little effect on the in-plane iron motion. We can thus conclude that the cisoid and transoid configurations are vibrationally indistinguishable since the dihedral angle between the FeNO plane and the closest Np—Fe—Np plane are almost identical. The feature that does lead to the higher frequencies in the monoclinic phase is the systematically shorter Fe—Np bonds (all ∼0.01 Å, despite the equatorial asymmetry).
In conclusion, the two polymorphs of [Fe(TpFPP)(1-MeIm)-(NO)] allow the exploration of the interplay of structural changes and vibrational dynamics. Although DFT calculations suggest that the cisoid and transoid NO/imidazole orientations should be indistinguishable, real experimental differences between the two are found in the X-ray structures and their IR and NRVS spectra. An analysis of the NRVS spectra indicates that the majority of vibrations involving Fe motion perpendicular to the porphyrin plane are sensitive to differences in the Fe—NIm distance trans to NO. The data presented give further evidence of the extremely soft nature of the Fe—NIm bond; changes in the length of this bond are correlated with variation in νN-O. The major in-plane Fe vibrational mode directions are parallel and perpendicular to the FeNO plane and hence oblique to the Fe—Np directions, quite unlike the [Fe(Porph)(R-Im)(CO)] cases where the in-plane iron motions are along Fe—Np bond directions. These differences illustrate the distinct dynamic differences of CO versus NO heme complexes. It remains to be determined whether the dioxygen complexes, geometrically similar to NO, will also show comparable behavior.
Supplementary Material
Acknowledgment
We thank the NIH GM-38401 (WRS) and the NSF PHY-0545787 (JTS) for support; the NSF for X-ray instrumentation support CHE-0443233 and the U.S. Department of Energy, Basic Energy Sciences, Office of Science, for use of the Advanced Photon Source under Contract No. W-31-109-Eng-38.
Footnotes
Supporting Information Available: Complete reference 12. Figures S1-S7. Crystallographic data is available as CIF files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Culotta E, Koshland DE. Science. 1992;258:1862. doi: 10.1126/science.1361684. [DOI] [PubMed] [Google Scholar]
- (2).Andrew CR, George SJ, Lawson DM, Eady RR. Biochemistry. 2002;41:2353. doi: 10.1021/bi011419k. [DOI] [PubMed] [Google Scholar]
- (3).Abbreviations: KE, kinetic energy; Porph, generalized porphyrin dianion;TpFPP, dianion of meso-tetra-para-fluorophenylporphyrin; 1-MeIm, 1-methylimidazole; R-Im, generalized substituted imidazole.
- (4)(a).Scheidt WR, Durbin SM, Sage JT. J. Inorg. Biochem. 2005;99:60. doi: 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]; (b) Sturhahn W. J. Phys.: Condens. Matter. 2006;16:S497. [Google Scholar]
- (5).Metrical information for tri-[Fe(TpFPP)(1-MeIm)(NO)] and mono-[Fe(TpFPP)(1-MeIm)(NO)] [in square brackets]. νNO = 1641 [1632] cm-1, Fe—NNO = 1.7521(9) [1.7481(11)] Å, Fe—NIm = 2.1689(9) [2.1312(11)] Å, <Fe—Np> = 2.011(8) [2.002(14)] Å, <Fe—Np (adj)> = 2.004(3) [1.992(4)] Å, <Fe—Np (opp)> = 2.018(3) [2.012(12)] Å, N-O = 1.1819(12) [1.1808(14)] Å. Fe—NNO tilt = 5.1[7.2]°, Fe-N-O = 138.64(8) [137.27(9)]°, NIm—Fe—NNO = 176.08(4) [175.08(4)]°, N—Fe—N—O = 43.2 [38.5]°.
- (6).Wyllie GRA, Schulz CE, Scheidt WR. Inorg. Chem. 2003;42:5722. doi: 10.1021/ic034473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Scheidt WR, Piciulo PL. J. Am. Chem. Soc. 1976;98:1913. doi: 10.1021/ja00423a044. [DOI] [PubMed] [Google Scholar]
- (8).The change in the Fe—NIm distance as a function of temperature is an unusual structural change in the absence of significant differences in the electronic structure (such as a spin state transition). This is consistent with observed and calculated softness of this bond.
- (9).It has been previously observed that changes in the Fe—NIm bond trans to NO lead to variations in the observed value of νNO. 10
- (10).Scheidt WR, Brinegar AC, Ferro EB, Kirner JF. J. Am. Chem. Soc. 1977;99:7315. [Google Scholar]
- (11).DFT calculations were performed with Gaussian 03,12 employing the B3LYP13 hybrid functional and Ahlrich’s VTZ14 basis set for the Featom and 6-31G* basis set for all other atoms.
- (12).Frisch MJ, et al. Gaussian, Inc. Gaussian 03: Revision C.02. Wallingford, CT: 2004. [Google Scholar]
- (13)(a).Becke AD. J. Chem. Phys. 1993;98:5684. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- (14).Schafer A, Horn H, Ahlrich RJ. Phys. Chem. 1992;97:2571. [Google Scholar]
- (15).Additional changes appear in the 200-220 cm-1 region, but are more difficult to interpret because of overlapping contributions from modes with Fe displacement parallel and perpendicular to the porphyrin plane. The most prominent out-of-plane mode predicted in this region has some Fe—Im stretching character, but only 16% of the mode KE is localized on the Fe(1-MeIm)(NO) fragment and may be less sensitive to the Fe coordination sphere.
- (16).Leu B, Zgierski M, Wyllie GRA, Scheidt WR, Sturhahn W, Alp EE, Durbin SM, Sage JT. J. Am. Chem. Soc. 2004;126:4211. doi: 10.1021/ja038526h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Leu BM, Silvernail NJ, Zgierski MZ, Wyllie GRA, Ellison MK, Scheidt WR, Zhao J, Sturhahn W, Alp EE, Durbin SM, Sage JT. Biophy. J. 2006, in press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.